
Chitosan–Cu Catalyzed Novel Ferrocenated Spiropyrrolidines: Green Synthesis, Single Crystal X-ray Diffraction, Hirshfeld Surface and Antibacterial Studies



 ,
,  ,
,
Abstract
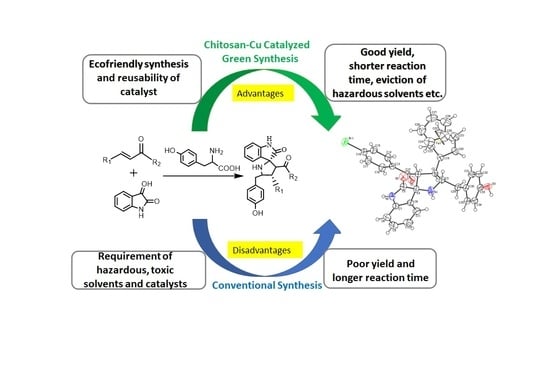
1. Introduction
2. Materials and Methods
2.1. General
2.2. Synthesis of Catalyst, Chitosan–Cu
2.3. Synthesis of Spiropyyrolidines (3a–b)
2.3.1. General Procedure for Conventional Synthesis of Spiropyyrolidines (3a–b)
2.3.2. Green Method for Synthesis of Spiropyyrolidines (3a–b)
2.3.3. Synthesis of Spiropyyrolidines under Different Solvent Assisted Condition Using Chitosan–Cu Catalyst
2.3.4. Synthesis of Spiropyyrolidines under Solvent-Free Condition Using Different Catalytic System
2.3.5. 3′-(4-Bromobenzoyl)-5′-(4-hydroxybenzyl)-4′-ferrocenylspiro[indoline-3,2′-pyrrolidin]-2 One (3a)
2.3.6. 3′-(4-Bromobenzoyl)-4′-ferrocenylspiro[indoline-3,2′-pyrrolidin]-2-one (3b)
2.4. Recycling and Reusage of Chitosan–Cu Catalyst
2.5. Single Crystal X-ray Crystallography
2.6. Antibacterial Study
3. Results and Discussion
3.1. Crystal Structure Description
3.2. Hirshfeld Surface Analysis
3.3. Antibacterial Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asad, M.; Arshad, M.N.; Oves, M.; Khalid, M.; Khan, S.A.; Asiri, A.M.; Rehan, M.; Dzudzevic-Cancar, H. N-Trifluoroacetylated pyrazolines: Synthesis, characterization and antimicrobial studies. Bioorg. Chem. 2020, 99, 103842. [Google Scholar] [CrossRef] [PubMed]
- Asad, M.; Siddiqui, Z.N.; Asiri, A.M.; Mahmood, A. Synthesis, DFT and biological studies of novel 3,6-dimethyl-1,8-diphenyl-diazocino[3,4-c:7,8-c’]bispyrazole. J. Mol. Struct. 2019, 1176, 66–72. [Google Scholar] [CrossRef]
- Pandey, G.; Banerjee, P.; Gadre, S.R. Construction of Enantiopure Pyrrolidine Ring System via Asymmetric [3+2]-Cycloaddition of Azomethine Ylides. Chem. Rev. 2006, 106, 4484–4517. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Ziegelmeyer, E.C.; Farinde, O.N.; Truong, J.T.; Huynh, M.M.; Li, W. Visible light bromide catalysis for oxazoline, pyrrolidine, and dihydrooxazine syntheses via C sp 3 –H functionalizations. Chem. Commun. 2021, 57, 10387–10390. [Google Scholar] [CrossRef]
- Kasturi, S.; Surarapu, S.; Uppalanchi, S.; Anireddy, J.S.; Dwivedi, S.; Anantaraju, H.S.; Perumal, Y.; Sigalapalli, D.K.; Babu, B.N.; Ethiraj, K.S. Synthesis and α-glucosidase inhibition activity of dihydroxy pyrrolidines. Bioorg. Med. Chem. Lett. 2017, 27, 2818–2823. [Google Scholar] [CrossRef]
- Arun, Y.; Saranraj, K.; Balachandran, C.; Perumal, P.T. Novel spirooxindole–pyrrolidine compounds: Synthesis, anticancer and molecular docking studies. Eur. J. Med. Chem. 2014, 74, 50–64. [Google Scholar] [CrossRef]
- Toumi, A.; Boudriga, S.; Hamden, K.; Sobeh, M.; Cheurfa, M.; Askri, M.; Knorr, M.; Strohmann, C.; Brieger, L. Synthesis, antidiabetic activity and molecular docking study of rhodanine-substitued spirooxindole pyrrolidine derivatives as novel α-amylase inhibitors. Bioorg. Chem. 2021, 106, 104507. [Google Scholar] [CrossRef]
- Gaudernak, E.; Seipelt, J.; Triendl, A.; Grassauer, A.; Kuechler, E. Antiviral Effects of Pyrrolidine Dithiocarbamate on Human Rhinoviruses. J. Virol. 2002, 76, 6004–6015. [Google Scholar] [CrossRef]
- Hong, C.Y.; Kim, Y.K.; Chang, J.H.; Kim, S.H.; Choi, H.; Nam, D.H.; Kim, Y.Z.; Kwak, J.H. Novel Fluoroquinolone Antibacterial Agents Containing Oxime-Substituted (Aminomethyl) pyrrolidines: Synthesis and Antibacterial Activity of 7-(4-(Aminomethyl)-3-(methoxyimino)pyrrolidin-1-yl)-1-cyclopropyl-6-fluoro- 4-oxo-1,4-dihydro[1,8]naphthyridine-3-ca. J. Med. Chem. 1997, 40, 3584–3593. [Google Scholar] [CrossRef]
- Kumar, R.R.; Perumal, S.; Senthilkumar, P.; Yogeeswari, P.; Sriram, D. A highly atom economic, chemo-, regio- and stereoselective synthesis, and discovery of spiro-pyrido-pyrrolizines and pyrrolidines as antimycobacterial agents. Tetrahedron 2008, 64, 2962–2971. [Google Scholar] [CrossRef]
- Asad, M.; Khan, S.A.; Arshad, M.N.; Asiri, A.M.; Rehan, M. Design and synthesis of novel pyrazoline derivatives for their spectroscopic, single crystal X-ray and biological studies. J. Mol. Struct. 2021, 1234, 130131. [Google Scholar] [CrossRef]
- Asad, M.; Arshad, M.N.; Khan, S.A.; Oves, M.; Khalid, M.; Asiri, A.M.; Braga, A.A.C. Cyclization of chalcones into N-propionyl pyrazolines for their single crystal X-ray, computational and antibacterial studies. J. Mol. Struct. 2020, 1201, 127186. [Google Scholar] [CrossRef]
- Khan, S.A.; Asiri, A.M.; Basisi, H.M.; Asad, M.; Zayed, M.E.M.; Sharma, K.; Wani, M.Y. Synthesis and evaluation of Quinoline-3-carbonitrile derivatives as potential antibacterial agents. Bioorg. Chem. 2019, 88, 102968. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Sharma, D.; Srivastava, A.K.; Varma, A.; Jayadev, M.S.K.; Joshi, R.K. Evaluation of anticancer activity of ferrocene based benzothiazole and β-ketooxothioacetal. J. Organomet. Chem. 2022, 979, 122500. [Google Scholar] [CrossRef]
- Madhavan, N.; Jones, C.W.; Weck, M. Rational Approach to Polymer-Supported Catalysts: Synergy between Catalytic Reaction Mechanism and Polymer Design. Acc. Chem. Res. 2008, 41, 1153–1165. [Google Scholar] [CrossRef]
- Yin, L.; Liebscher, J. Carbon−Carbon Coupling Reactions Catalyzed by Heterogeneous Palladium Catalysts. Chem. Rev. 2007, 107, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, M.; Yao, C.; Shi, Y.; Ye, D.; Wu, J.; Zhao, D. A facile strategy for the preparation of well-dispersed bimetal oxide CuFe2O4 nanoparticles supported on mesoporous silica. J. Mater. Chem. A 2013, 1, 6742. [Google Scholar] [CrossRef]
- Gu, L.; Ma, D.; Yao, S.; Wang, C.; Shen, W.; Bao, X. Structured zeolites catalysts with hierarchical channel structure. Chem. Comm. 2010, 46, 1733. [Google Scholar] [CrossRef] [PubMed]
- Polshettiwar, V.; Luque, R.; Fihri, A.; Zhu, H.; Bouhrara, M.; Basset, J.-M. Magnetically Recoverable Nanocatalysts. Chem. Rev. 2011, 111, 3036–3075. [Google Scholar] [CrossRef]
- Zhang, J.; Han, D.; Zhang, H.; Chaker, M.; Zhao, Y.; Ma, D. In situ recyclable gold nanoparticles using CO2-switchable polymers for catalytic reduction of 4-nitrophenol. Chem. Commun. 2012, 48, 11510. [Google Scholar] [CrossRef]
- Di Carlo, G.; Curulli, A.; Toro, R.G.; Bianchini, C.; De Caro, T.; Padeletti, G.; Zane, D.; Ingo, G.M. Green Synthesis of Gold–Chitosan Nanocomposites for Caffeic Acid Sensing. Langmuir 2012, 28, 5471–5479. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Xu, J.; Yu, W.; Zhang, P. A highly active and easily recoverable chitosan@copper catalyst for the C–S coupling and its application in the synthesis of zolimidine. Green Chem. 2014, 16, 3007–3012. [Google Scholar] [CrossRef]
- Sun, W.; Xia, C.G.; Wang, H.W. Efficient heterogeneous catalysts for the cyclopropanation of olefins. New J. Chem. 2002, 26, 755–758. [Google Scholar] [CrossRef]
- Baig, R.N.; Varma, R.S. Organic synthesis via magnetic attraction: Benign and sustainable protocols using magnetic nanoferrites. Green Chem. 2013, 15, 1834. [Google Scholar] [CrossRef]
- Chiessi, E.; Branca, M.; Palleschi, A.; Pispisa, B. Copper (II) complexes immobilized on a polymeric matrix. thermodynamics, spectroscopy, and molecular modeling. Inorg. Chem. 1995, 34, 2600–2609. [Google Scholar] [CrossRef]
- Kaur, N. Polycyclic Six-Membered N -Heterocycles: Microwave-Assisted Synthesis. Synth. Commun. 2015, 45, 35–69. [Google Scholar] [CrossRef]
- Suresh Babu, A.R.; Raghunathan, R. Ultrasonic assisted-silica mediated [3+2] cycloaddition of azomethine ylides—A facile multicomponent one-pot synthesis of novel dispiroheterocycles. Tetrahedron Lett. 2007, 48, 6809–6813. [Google Scholar] [CrossRef]
- Jain, R.; Sharma, K.; Kumar, D. Ionic liquid mediated 1,3-dipolar cycloaddition of azomethine ylides: A facile and green synthesis of novel dispiro heterocycles. Tetrahedron Lett. 2012, 53, 1993–1997. [Google Scholar] [CrossRef]
- Agilent. CrysAlisPRO.; Agilent Technologies Ltd.: Yarnton, UK, 2014. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sec. A 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON—A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2005. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Cruickshank, R.; Duguid, J.P.; Marmion, B.P.; Swain, R.H.A. Medicinal Microbiology, II, 12th ed.; Churchill Livingstone: London, UK, 1975; pp. 196–202. [Google Scholar]
- Siddiqui, Z.N.; Mohammed Musthafa, T.N.; Ahmad, A.; Khan, A.U. Thermal solvent-free synthesis of novel pyrazolyl chalcones and pyrazolines as potential antimicrobial agents. Bioorgan. Med. Chem. Lett. 2011, 21, 2860–2865. [Google Scholar] [CrossRef]
- Collins, A.H. Microbiological Methods, 2nd ed.; Butterworth: London, UK, 1976. [Google Scholar]
- Sarrafi, Y.; Hamzehloueian, M.; Alimohammadi, K.; Yeganegi, S. Experimental and theoretical approaches to [1,5]-prototropic generation of an azomethine ylide and a 1,3-dipolar cycloaddition for novel spiropyrrolidine oxindoles synthesis. J. Mol. Struct. 2012, 1030, 168–176. [Google Scholar] [CrossRef]
- Asad, M.; Arshad, M.N.; Asiri, A.M.; Khan, S.A.; Rehan, M.; Oves, M. Synthesis, Characterization, Molecular Docking and Antimicrobial Activity of Novel Spiropyrrolidine Derivatives. Polycycl. Aromat. Compd. 2022, 42, 5385–5397. [Google Scholar] [CrossRef]
- Asad, M.; Arshad, M.N.; Asiri, A.M.; Musthafa, T.N.M.; Khan, S.A.; Rehan, M.; Oves, M. Synthesis of N-Methylspiropyrrolidine Hybrids for Their Structural Characterization, Biological and Molecular Docking Studies. Polycycl. Aromat. Compd. 2022. [Google Scholar] [CrossRef]
- Azum, N.; Arshad, M.A.; Birinji, A.S.; Al-Amry, K.A.; Asiri, A.M.; Taib, L.A.; Angari, Y.M.A.; Khan, S.B.; Facchetti, A. Crystal Structure and Electrochemical Properties of 1-(4-Bromophenyl)-Ferrocene-Prop-2-en-1-One and 1-(3-(4-Bromophenyl)-5-(Ferrocene)-4.5-Dihydropyrazol-1-yl) Ethenone. J. Electrochem. Sci. 2019, 14, 8355–8370. [Google Scholar] [CrossRef]
- Ahmed, M.; Abdul Qadir, M.; Imtiaz Shafiq, M.; Muddassar, M.; Hameed, A.; Nadeem Arshad, M.; Asiri, A.M. Curcumin: Synthesis optimization and in silico interaction with cyclin dependent kinase. Acta Pharm. 2017, 67, 385–395. [Google Scholar] [CrossRef]
- Hussain, M.M.; Rahman, M.M.; Arshad, M.N.; Asiri, A.M. Electrochemical Detection of Ni 2+ Ions Using Synthesized (E)-N′-Chlorobenzylidene-4-methylbenzenesulfonohydrazide Derivatives Modified with a Nafion Matrix. ChemistrySelect 2017, 2, 7455–7464. [Google Scholar] [CrossRef]
- Ahmed, M.; Qadir, M.A.; Hameed, A.; Arshad, M.N.; Asiri, A.M.; Muddassar, M. Azomethines, isoxazole, N-substituted pyrazoles and pyrimidine containing curcumin derivatives: Urease inhibition and molecular modeling studies. Biochem. Biophys. Res. Commun. 2017, 490, 434–440. [Google Scholar] [CrossRef]
- Arshad, M.N.; Şahin, O.; Zia-ur-Rehman, M.; Shafiq, M.; Khan, I.U.; Asiri, A.M.; Khan, S.B.; Alamry, K.A. Crystallographic Studies of Dehydration Phenomenon in Methyl 3-hydroxy-2-methyl-1,1,4-trioxo-1,2,3,4-tetrahydro-1λ 6-benzo[e][1,2]thiazine-3-carboxylate. J. Chem. Crystallogr. 2013, 43, 671–676. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B Struct. Sci. 1990, 46, 256. [Google Scholar] [CrossRef] [PubMed]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Conventional Method (Refluxing in Methanol) | Solvent-Free Heating Method with Chitosan–Cu Catalyst | ||
|---|---|---|---|---|
| Time | Yield (%) | Time | Yield (%) | |
| 3a | 5 h | 71 | 15 min | 91 |
| 3b | 5 h | 65 | 20 min | 88 |
| Entry | Solvents | Temperature | Time | Yield (%) |
|---|---|---|---|---|
| 1 | Methanol | Reflux | 50 min | 75 |
| 2 | Chloroform | Reflux | 1.5 h | 64 |
| 3 | PEG-400 | Reflux | 1 h | 65 |
| 4 | Water | Reflux | 50 min | 78 |
| 5 | Solvent-free | 60 °C | 15 min | 91 |
| Entry | Catalysts | Time | Yield |
|---|---|---|---|
| 1 | Na(CH3CO2) | 80 min | 55% |
| 2 | Cu(CH3CO2)2 | 40 min | 56% |
| 3 | Chitosan | 90 min | 45% |
| 4 | Chitosan–Cu | 15 min | 91% |
| Entry | No. of Cycles | Yield (%) | Time (min) |
|---|---|---|---|
| 1 | I | 91 | 15 |
| 2 | II | 91 | 15 |
| 3 | III | 91 | 15 |
| 4 | IV | 91 | 15 |
| 5 | V | 88 | 15 |
| Designed Code | 19015 |
| CCDC No. | 2215469 |
| Compound’s empirical formula | C35H29N2O3BrFe |
| Formula weight (F. wt.) | 661.381 |
| Temp./K | 296.15 |
| Crystal system | monoclinic |
| Space group | P21/c |
| Length of side a/Å | 16.7910 (13) |
| Length of side b/Å | 7.8298 (6) |
| Length of side c/Å | 23.1312 (16) |
| (Alpha Angle) α/° | 90 |
| (Beta Angle) β/° | 106.316 (8) |
| (Gamma angle) γ/° | 90 |
| Volume/Å3 | 2918.6 (4) |
| Z | 4 |
| ρcalc g/cm3 | 1.505 |
| μ/mm−1 | 1.925 |
| F(000) | 1353.2 |
| Crystal size/mm3 | 0.42 × 0.35 × 0.33 |
| Radiation used | Mo Kα (λ = 0.71073) |
| 2θ/° | 5.78 to 58.54 (range for data collection) |
| Index ranges for collection (h,k,l) | −15 ≤ h ≤ 22, −9 ≤ k ≤ 9, −28 ≤ l ≤ 31 |
| Reflections collected | 16408 |
| Independent reflections | 6913 [Rint = 0.0379, Rsigma = 0.0540] |
| Data | 6913 |
| Restraints | 0 |
| Parameters | 380 |
| Goodness-of-fit on F2 | 1.055 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0453, wR2 = 0.1011 |
| Final R indexes [all data] | R1 = 0.0929, wR2 = 0.1279 |
| D | H | A | d(D-H)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° |
|---|---|---|---|---|---|---|
| O3 | H3a | O2 1 | 0.820 | 1.833 (4) | 2.650 (3) | 173.9 (2) |
| C15 | H15 | O1 2 | 0.930 | 2.486 (5) | 3.401 (4) | 167.7 (1) |
| O3 | H1O | O2 3 | 0.820 | 1.833 (4) | 2.650 (3) | 173.9 (4) |
| Diameter of Zone of Inhibition (mm) | |||||
|---|---|---|---|---|---|
| Gram-Positive Bacteria | Gram-Negative Bacteria | ||||
| S. pyogenes | S. aureus | K. pneumoniae | P. aeruginosa | E. coli | |
| 3a | 14.4 ± 0.4 | 13.1 ± 0.5 | 15.7 ± 0.4 | 15.2 ± 0.4 | 14.1 ± 0.5 |
| 3b | 17.4 ± 0.5 | 16.3 ± 0.4 | 16.2 ± 0.5 | 18.1 ± 0.4 | 17.5 ± 0.5 |
| standard | 23.0 ± 0.2 | 22.0 ± 0.2 | 19.0 ± 0.2 | 32.0 ± 0.3 | 27.0 ± 0.2 |
| DMSO | - | - | - | - | - |
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S. pyogenes | MRSA * | K. pneumoniae | P. aeruginosa | E. coli | ||||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 3a | 25 | >100 | 25 | 100 | 50 | 100 | 50 | 50 | 50 | 50 |
| 3b | 25 | 100 | 25 | >100 | 50 | >100 | 100 | >100 | 50 | >100 |
| Standard | 12.5 | 12.5 | 6.25 | 12.5 | 6.25 | 25 | 12.5 | 25 | 6.25 | 12.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asad, M.; Arshad, M.N.; Asiri, A.M.; Rahman, M.M.; Kumaran, S.; Thorakkattil Neerankuzhiyil, M.M. Chitosan–Cu Catalyzed Novel Ferrocenated Spiropyrrolidines: Green Synthesis, Single Crystal X-ray Diffraction, Hirshfeld Surface and Antibacterial Studies. Polymers 2023, 15, 429. https://doi.org/10.3390/polym15020429
Asad M, Arshad MN, Asiri AM, Rahman MM, Kumaran S, Thorakkattil Neerankuzhiyil MM. Chitosan–Cu Catalyzed Novel Ferrocenated Spiropyrrolidines: Green Synthesis, Single Crystal X-ray Diffraction, Hirshfeld Surface and Antibacterial Studies. Polymers. 2023; 15(2):429. https://doi.org/10.3390/polym15020429
Chicago/Turabian StyleAsad, Mohammad, Muhammad Nadeem Arshad, Abdullah M. Asiri, Mohammed M. Rahman, Snigdha Kumaran, and Mohammed Musthafa Thorakkattil Neerankuzhiyil. 2023. "Chitosan–Cu Catalyzed Novel Ferrocenated Spiropyrrolidines: Green Synthesis, Single Crystal X-ray Diffraction, Hirshfeld Surface and Antibacterial Studies" Polymers 15, no. 2: 429. https://doi.org/10.3390/polym15020429
APA StyleAsad, M., Arshad, M. N., Asiri, A. M., Rahman, M. M., Kumaran, S., & Thorakkattil Neerankuzhiyil, M. M. (2023). Chitosan–Cu Catalyzed Novel Ferrocenated Spiropyrrolidines: Green Synthesis, Single Crystal X-ray Diffraction, Hirshfeld Surface and Antibacterial Studies. Polymers, 15(2), 429. https://doi.org/10.3390/polym15020429