
Formulation of Piperine Nanoparticles: In Vitro Breast Cancer Cell Line and In Vivo Evaluation

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
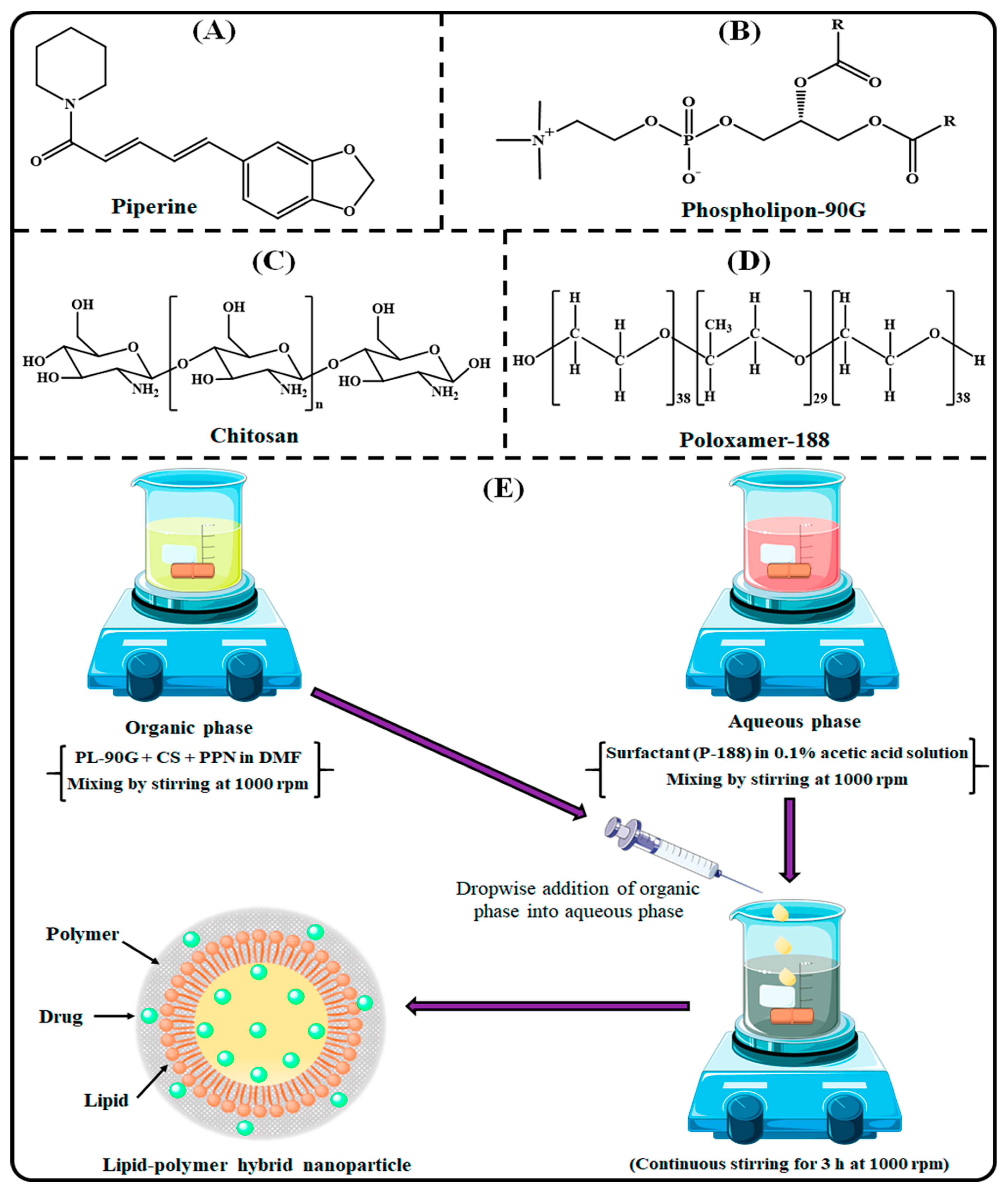
2.1. Materials
2.2. Experimental Design
2.3. Development of PPN-LPHNPs
2.4. Characterization of PPN-LPHNPs
2.4.1. Particles Characterization
2.4.2. Entrapment Efficiency (%EE) and Loading Capacity (%LC)
2.5. Stability Studies
2.5.1. Gastrointestinal Stability
2.5.2. Colloidal Stability
2.6. PPN Release Study
2.7. Bioadhesion Study
2.8. Cytotoxicity Study
2.9. Ex Vivo PPN Permeation Study
2.10. Pharmacokinetic Study
2.11. Statistical Analysis
3. Results and Discussion
3.1. Optimization of PPN-LPHNPs by Statistical Design
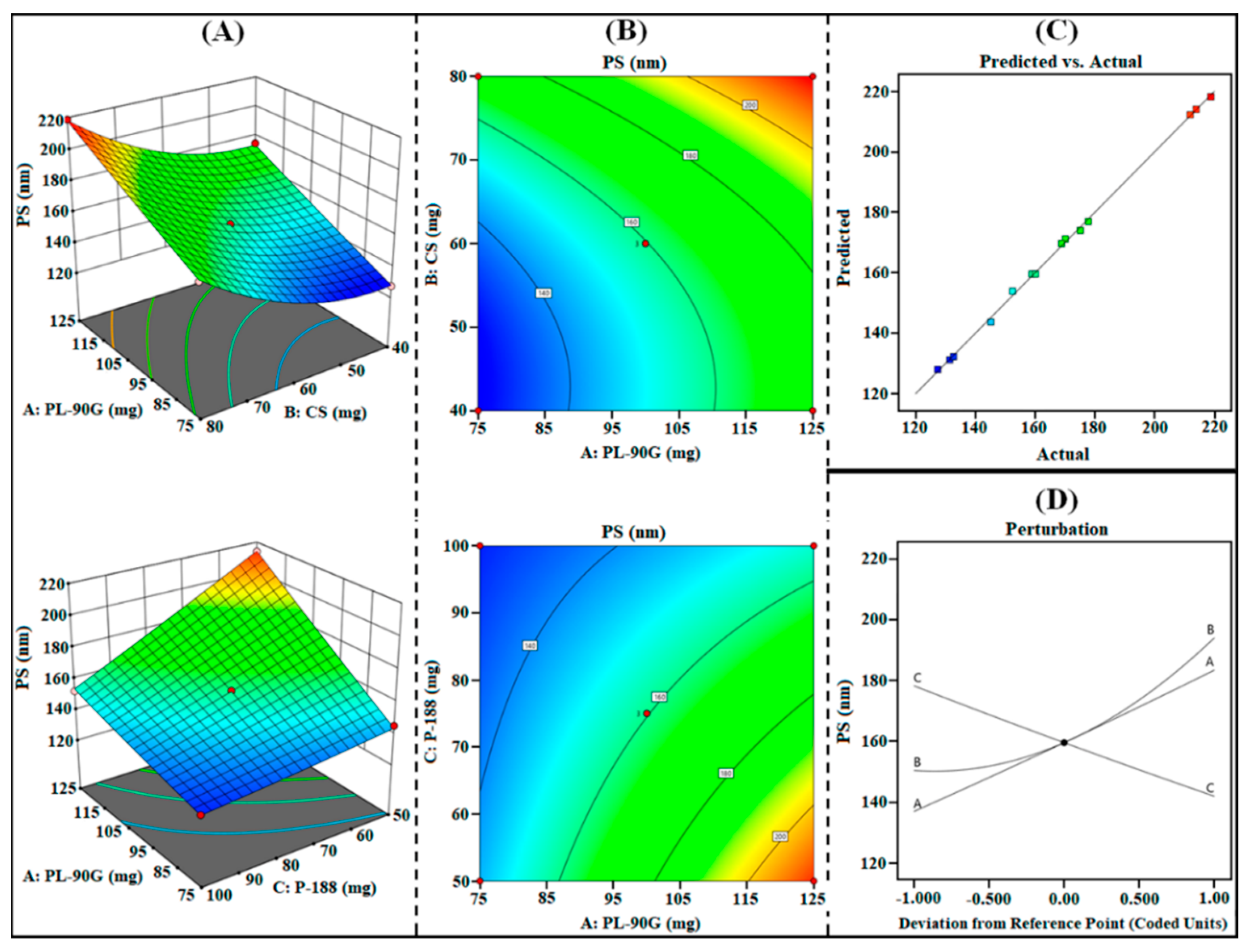
3.1.1. Effect on R1 (PS)
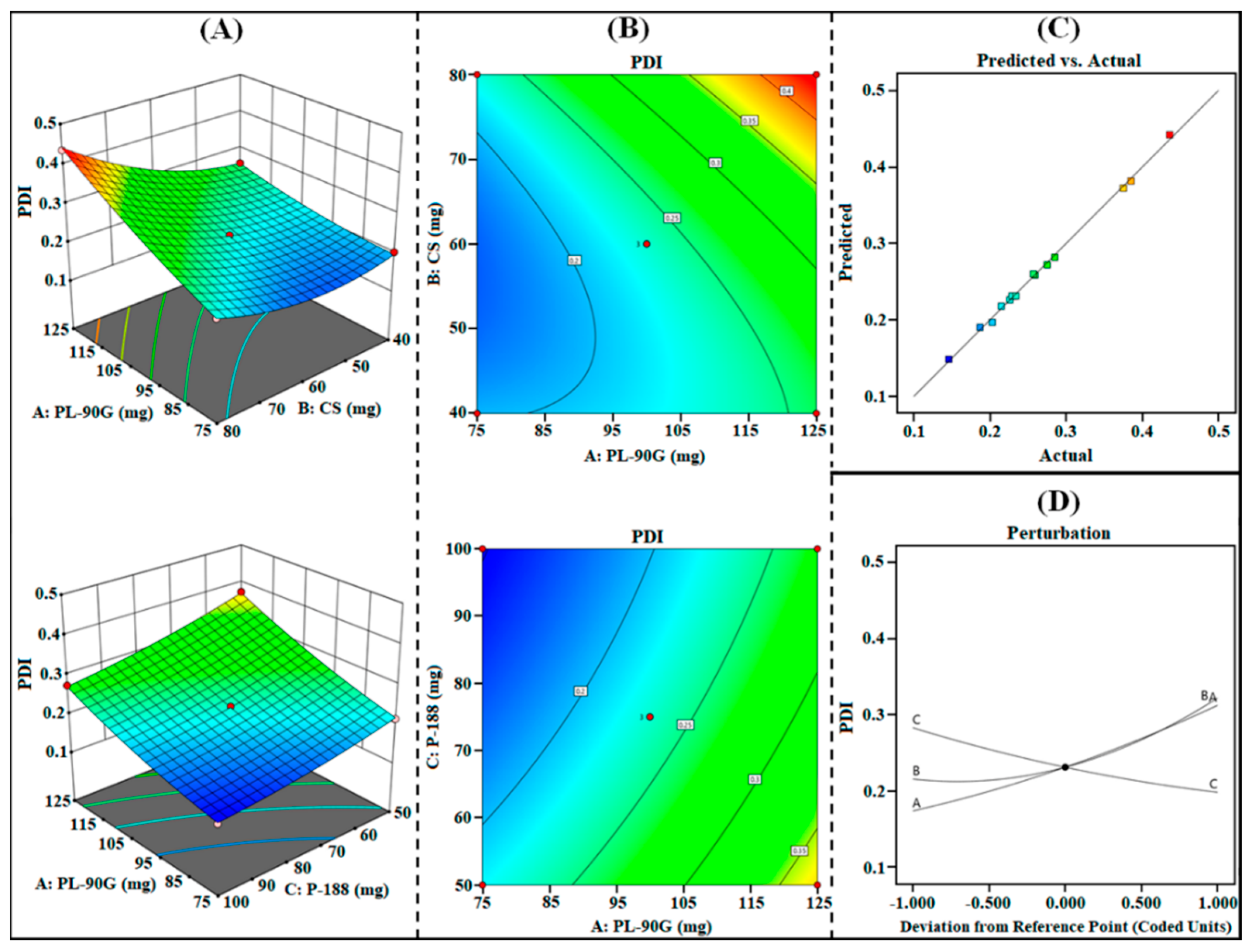
3.1.2. Effect on R2 (PDI)
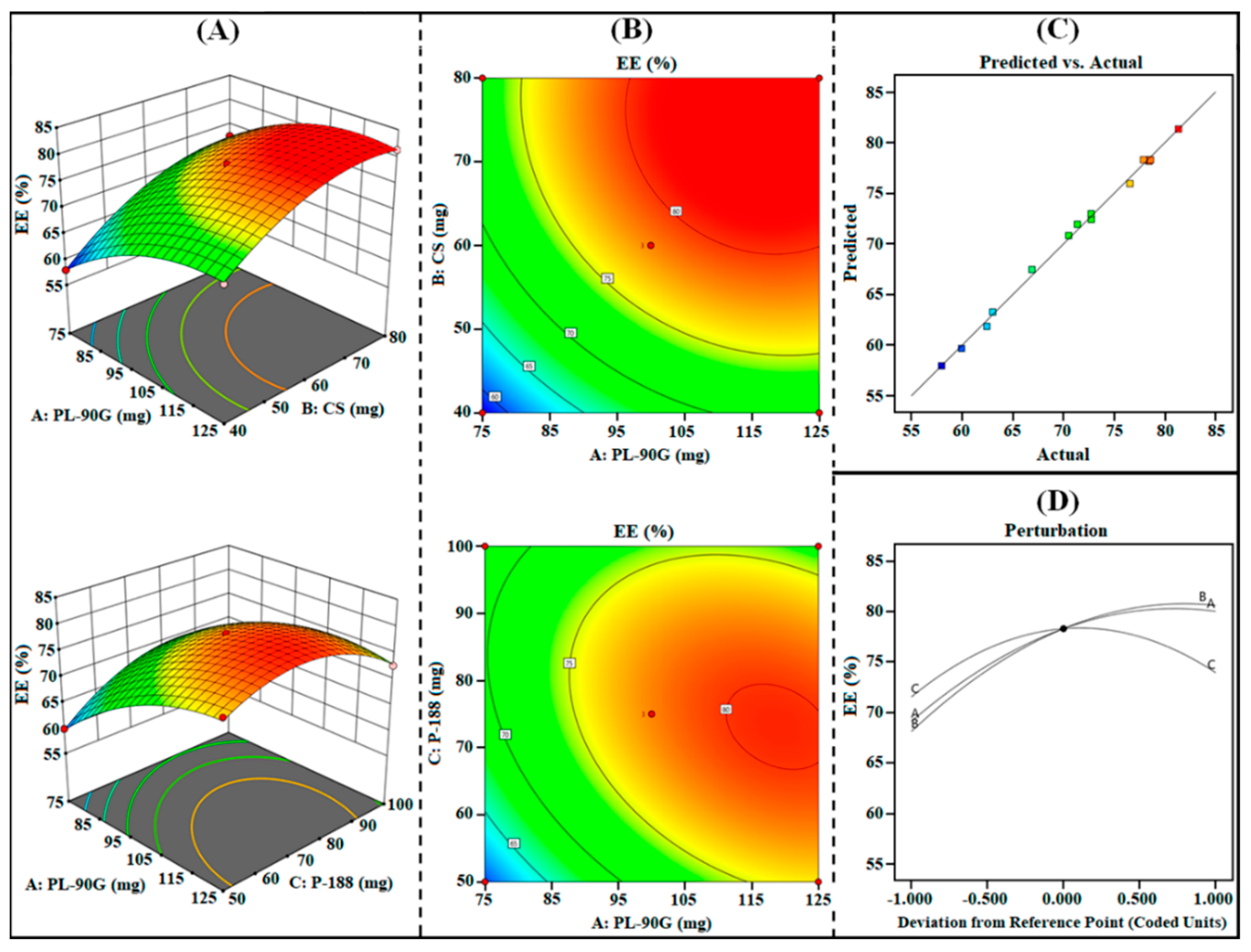
3.1.3. Effect on R3 (EE)
3.1.4. Optimized PPN-LPHNPs
3.2. Characterization of PPN-LPHNPs
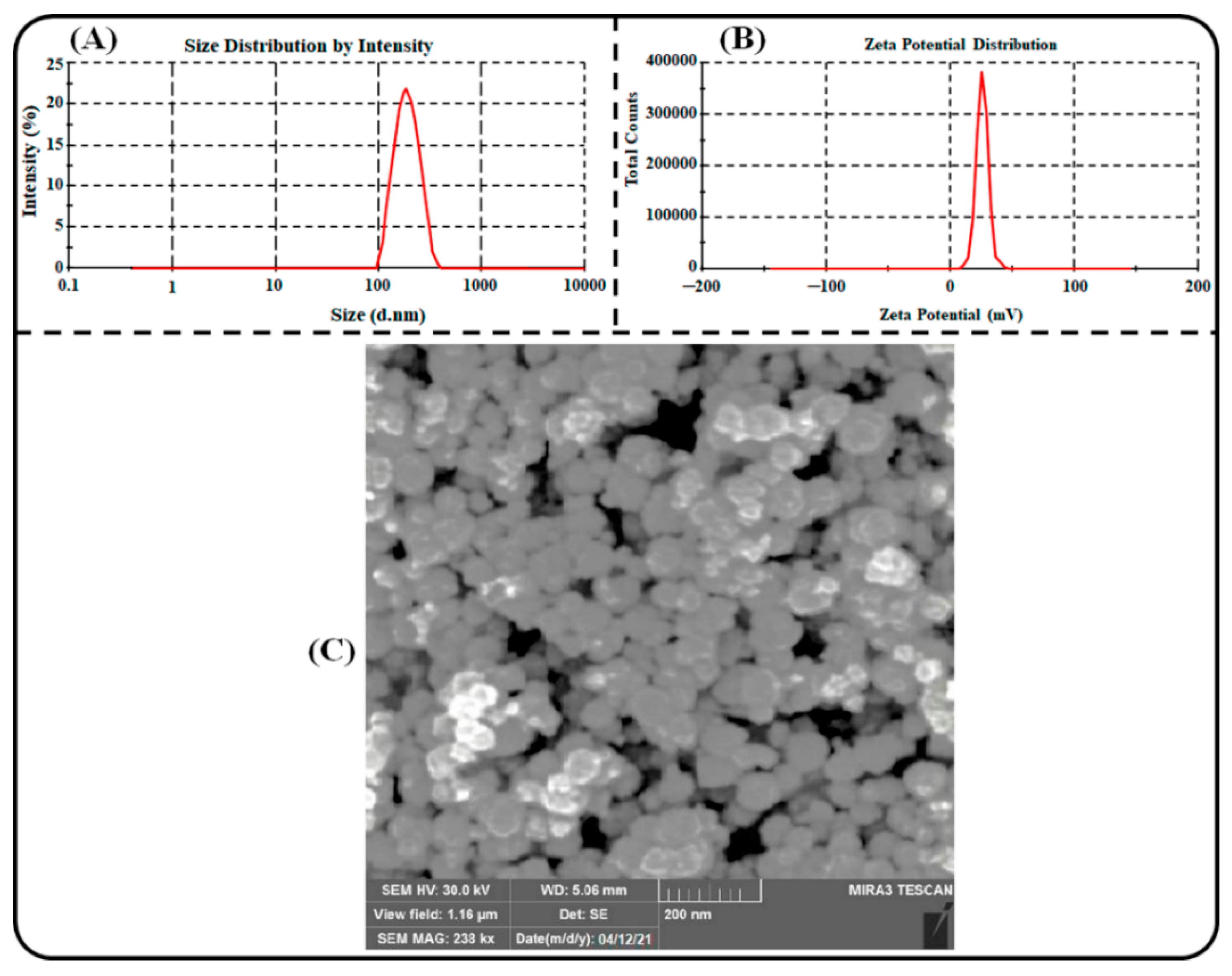
3.2.1. Particles Characterization
3.2.2. %EE and %LC
3.3. Stability Studies
3.3.1. Gastrointestinal Stability
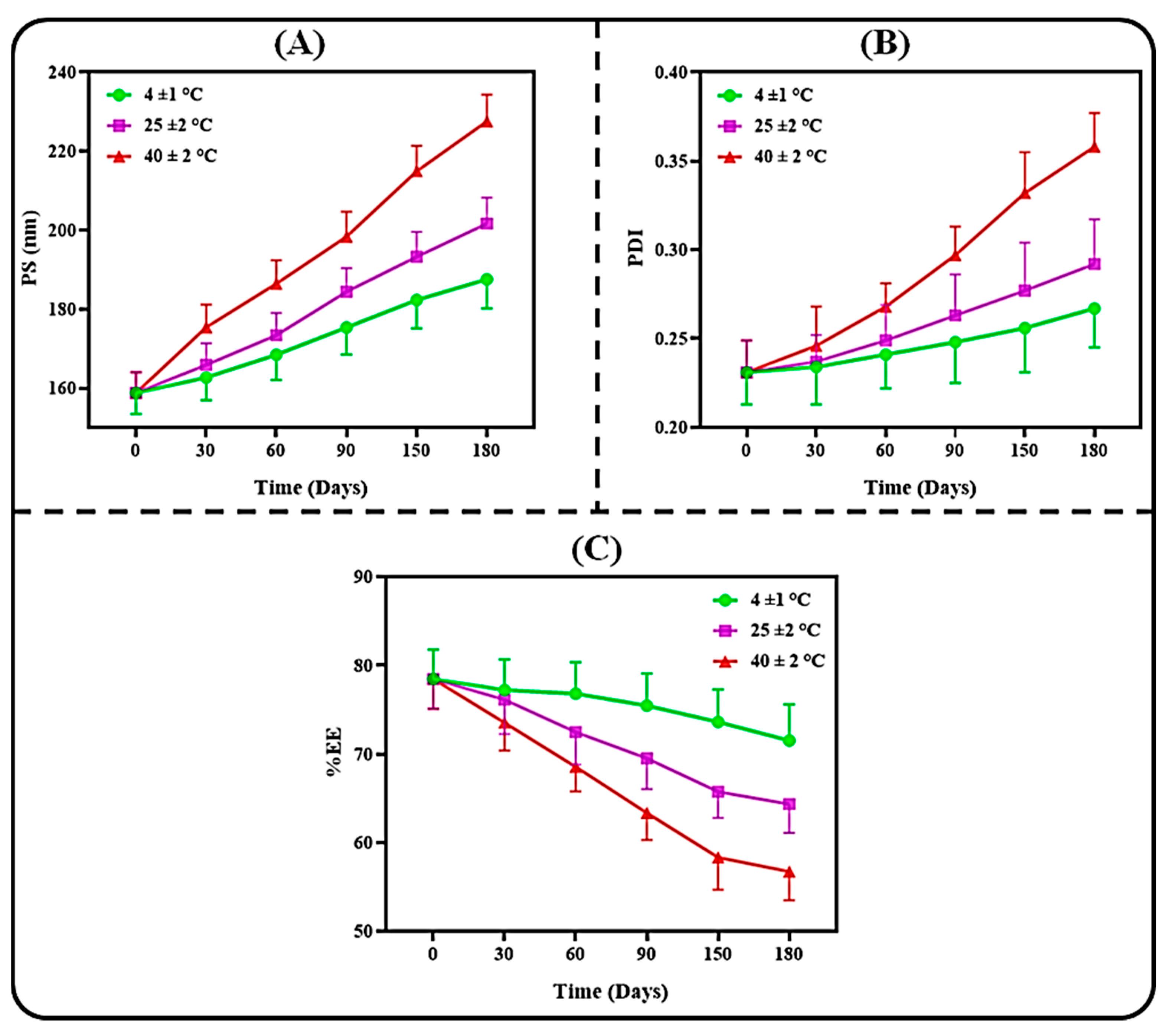
3.3.2. Colloidal Stability
3.4. PPN Release Study
3.5. Bioadhesion Study
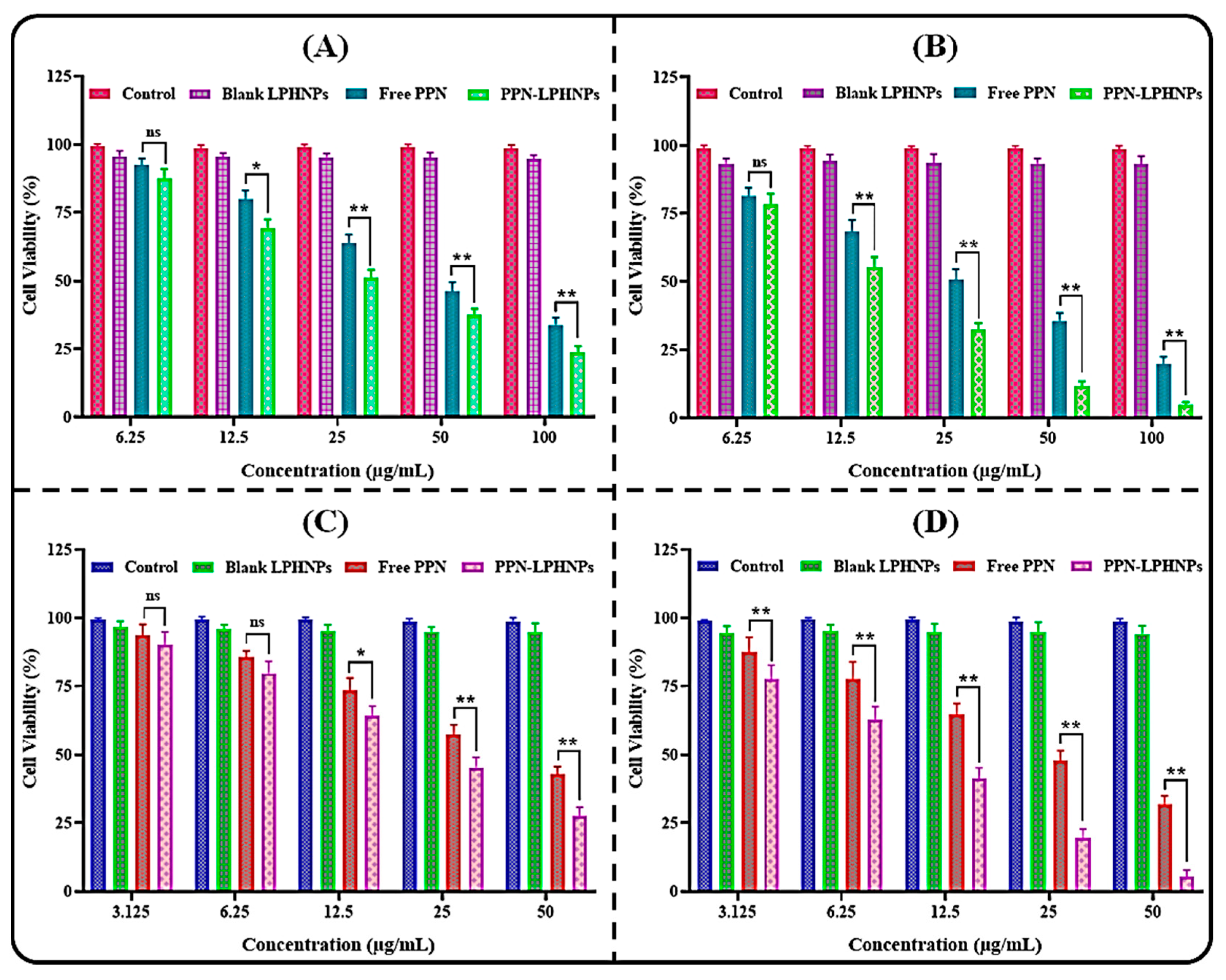
3.6. Cytotoxicity Study
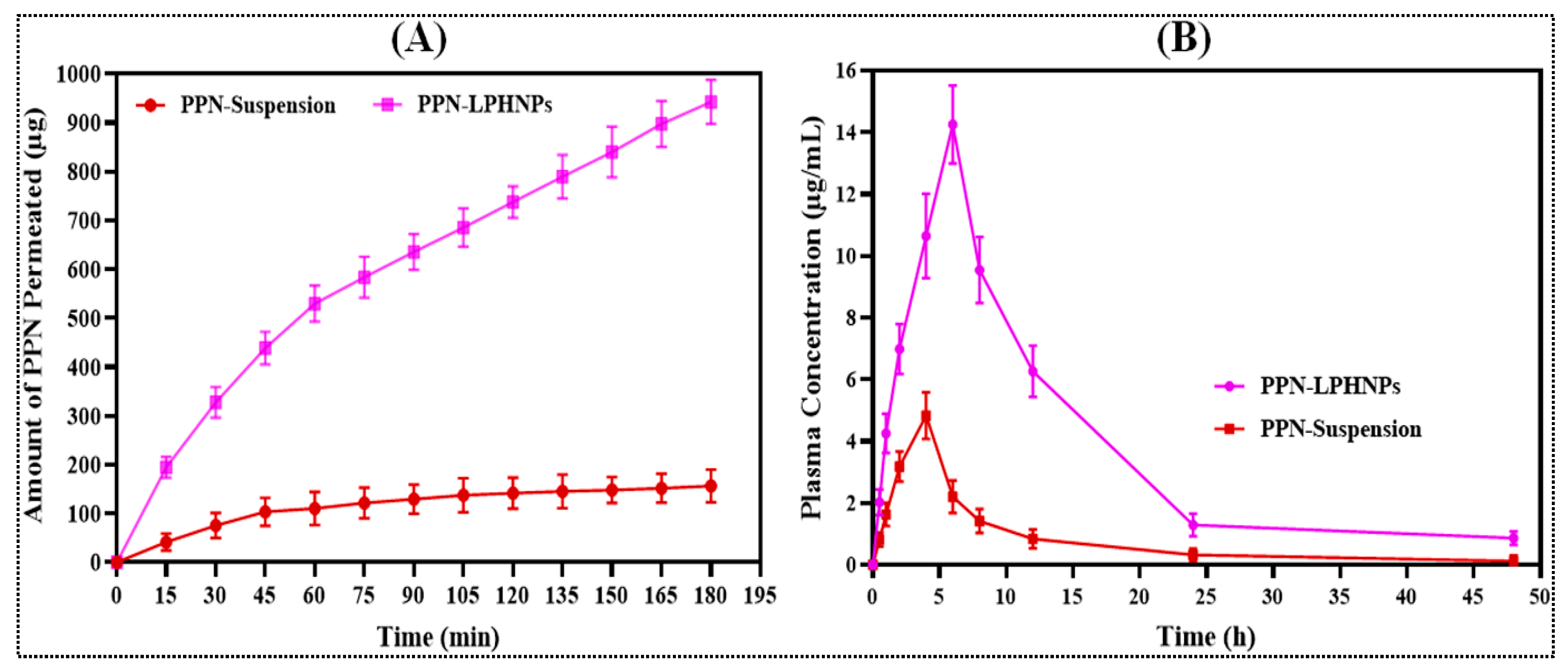
3.7. Ex Vivo PPN Permeation Study
3.8. Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizwanullah, M.; Ahmad, M.Z.; Ghoneim, M.M.; Alshehri, S.; Imam, S.S.; Md, S.; Alhakamy, N.A.; Jain, K.; Ahmad, J. Receptor-mediated targeted delivery of surface-modified nanomedicine in breast cancer: Recent update and challenges. Pharmaceutics 2021, 13, 2039. [Google Scholar] [CrossRef] [PubMed]
- Tharkar, P.; Madani, A.U.; Lasham, A.; Shelling, A.N.; Al-Kassas, R. Nanoparticulate carriers: An emerging tool for breast cancer therapy. J. Drug Target. 2015, 23, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Fisusi, F.A.; Akala, E.O. Drug combinations in breast cancer therapy. Pharm. Nanotechnol. 2019, 7, 3–23. [Google Scholar] [CrossRef]
- Taylor, C.W.; Kirby, A.M. Cardiac side-effects from breast cancer radiotherapy. Clin. Oncol. 2015, 27, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Rizwanullah, M.; Amin, S.; Mir, S.R.; Fakhri, K.U.; Rizvi, M.M.A. Phytochemical based nanomedicines against cancer: Current status and future prospects. J. Drug Target. 2018, 26, 731–752. [Google Scholar] [CrossRef]
- Meghwal, M.; Goswami, T.K. Piper nigrum and piperine: An update. Phytother. Res. 2013, 27, 1121–1130. [Google Scholar] [CrossRef]
- Haq, I.U.; Imran, M.; Nadeem, M.; Tufail, T.; Gondal, T.A.; Mubarak, M.S. Piperine: A review of its biological effects. Phytother. Res. 2021, 35, 680–700. [Google Scholar] [CrossRef]
- Kesarwani, K.; Gupta, R. Bioavailability enhancers of herbal origin: An overview. Asian Pac. J. Trop. Biomed. 2013, 3, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Quijia, C.R.; Araujo, V.H.; Chorilli, M. Piperine: Chemical, biological and nanotechnological applications. Acta Pharm. 2021, 71, 185–213. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Ray, A.K.; Mishra, S.K. Molecular and pharmacological aspects of piperine as a potential molecule for disease prevention and management: Evidence from clinical trials. Beni Suef Univ. J. Basic Appl. Sci. 2022, 11, 16. [Google Scholar] [CrossRef]
- Imam, S.S.; Alshehri, S.; Ghoneim, M.M.; Zafar, A.; Alsaidan, O.A.; Alruwaili, N.K.; Gilani, S.J.; Rizwanullah, M. Recent advancement in chitosan-based nanoparticles for improved oral bioavailability and bioactivity of phytochemicals: Challenges and perspectives. Polymers 2021, 13, 4036. [Google Scholar] [CrossRef] [PubMed]
- Quijia, C.R.; Chorilli, M. Piperine for treating breast cancer: A review of molecular mechanisms, combination with anticancer drugs, and nanosystems. Phytother. Res. 2022, 36, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Cui, C.; Ji, H.; Tang, J.; Wang, Z.; Liu, H.; Qin, M.; Li, X.; Wu, L. Enhanced oral bioavailability of piperine by self-emulsifying drug delivery systems: In vitro, in vivo and in situ intestinal permeability studies. Drug Deliv. 2015, 22, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, K.; Wang, Y.; Gao, F.; Liu, Q.; Zhang, C.; Li, P.; Zhang, J.; Chen, J. Targeting engineered nanoparticles for breast cancer therapy. Pharmaceutics 2021, 13, 1829. [Google Scholar] [CrossRef]
- Xu, Y.; Michalowski, C.B.; Beloqui, A. Advances in lipid carriers for drug delivery to the gastrointestinal tract. Curr. Opin. Colloid Interface Sci. 2021, 52, 101414. [Google Scholar] [CrossRef]
- Obireddy, S.R.; Lai, W.F. Preparation and characterization of 2-hydroxyethyl starch microparticles for co-delivery of multiple bioactive agents. Drug Deliv. 2021, 28, 1562–1568. [Google Scholar] [CrossRef]
- Zu, M.; Ma, Y.; Cannup, B.; Xie, D.; Jung, Y.; Zhang, J.; Yang, C.; Gao, F.; Merlin, D.; Xiao, B. Oral delivery of natural active small molecules by polymeric nanoparticles for the treatment of inflammatory bowel diseases. Adv. Drug Deliv. Rev. 2021, 176, 113887. [Google Scholar] [CrossRef]
- Das, S.S.; Bharadwaj, P.; Bilal, M.; Barani, M.; Rahdar, A.; Taboada, P.; Bungau, S.; Kyzas, G.Z. Stimuli-responsive polymeric nanocarriers for drug delivery, imaging, and theragnosis. Polymers 2020, 12, 1397. [Google Scholar] [CrossRef]
- Rizwanullah, M.; Alam, M.; Mir, S.R.; Rizvi, M.; Amin, S. Polymer-lipid hybrid nanoparticles: A next-generation nanocarrier for targeted treatment of solid tumors. Curr. Pharm. Des. 2020, 26, 1206–1215. [Google Scholar] [CrossRef]
- Sivadasan, D.; Sultan, M.H.; Madkhali, O.; Almoshari, Y.; Thangavel, N. Polymeric Lipid Hybrid Nanoparticles (PLNs) as Emerging Drug Delivery Platform—A Comprehensive Review of Their Properties, Preparation Methods, and Therapeutic Applications. Pharmaceutics 2021, 13, 1291. [Google Scholar] [CrossRef]
- Date, T.; Nimbalkar, V.; Kamat, J.; Mittal, A.; Mahato, R.I.; Chitkara, D. Lipid-polymer hybrid nanocarriers for delivering cancer therapeutics. J. Control. Release 2018, 271, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid-polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hoogevest, P. Review—An update on the use of oral phospholipid excipients. Eur. J. Pharm. Sci. 2017, 108, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, M.S.; Kazi, M.; Alsenaidy, M.A.; Ahmad, M.Z. Advances in oral drug delivery. Front. Pharmacol. 2021, 12, 618411. [Google Scholar] [CrossRef] [PubMed]
- Esim, O.; Bakirhan, N.K.; Sarper, M.; Savaser, A.; Ozkan, S.A.; Ozkan, Y. Influence of emulsifiers on the formation and in vitro anticancer activity of epirubicin loaded PLGA nanoparticles. J. Drug Deliv. Sci. Tech. 2020, 60, 102027. [Google Scholar] [CrossRef]
- Dong, W.; Wang, X.; Liu, C.; Zhang, X.; Zhang, X.; Chen, X.; Kou, Y.; Mao, S. Chitosan based polymer-lipid hybrid nanoparticles for oral delivery of enoxaparin. Int. J. Pharm. 2018, 547, 499–505. [Google Scholar] [CrossRef]
- Rizwanullah, M.; Perwez, A.; Mir, S.R.; Rizvi, M.M.; Amin, S. Exemestane encapsulated polymer-lipid hybrid nanoparticles for improved efficacy against breast cancer: Optimization, in vitro characterization and cell culture studies. Nanotechnology 2021, 32, 415101. [Google Scholar] [CrossRef]
- Thakur, K.; Sharma, G.; Singh, B.; Chhibber, S.; Patil, A.B.; Katare, O.P. Chitosan-tailored lipidic nano constructs of Fusidic acid as a promising vehicle for wound infections: An explorative study. Int. J. Biol. Macromol. 2018, 115, 1012–1025. [Google Scholar] [CrossRef]
- Zeng, C.; Zheng, R.; Yang, X.; Du, Y.; Xing, J.; Lan, W. Improved oral delivery of tilianin through lipid–polymer hybrid nanoparticles to enhance bioavailability. Biochem. Biophys. Res. Commun. 2019, 519, 316–322. [Google Scholar] [CrossRef]
- Patel, R.R.; Khan, G.; Chaurasia, S.; Kumar, N.; Mishra, B. Rationally developed core-shell polymeric-lipid hybrid nanoparticles as a delivery vehicle for cromolyn sodium: Implications of lipid envelop on in vitro and in vivo behavior of nanoparticles upon oral administration. RSC Adv. 2015, 5, 76491–76506. [Google Scholar] [CrossRef]
- Ana, R.; Mendes, M.; Sousa, J.; Pais, A.; Falcão, A.; Fortuna, A.; Vitorino, C. Rethinking carbamazepine oral delivery using polymer-lipid hybrid nanoparticles. Int. J. Pharm. 2019, 554, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Hejjaji, E.M.; Smith, A.M.; Morris, G.A. Evaluation of the mucoadhesive properties of chitosan nanoparticles prepared using different chitosan to tripolyphosphate (CS: TPP) ratios. Int. J. Biol. Macromol. 2018, 120, 1610–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafar, A.; Alruwaili, N.K.; Imam, S.S.; Alsaidan, O.A.; Alharbi, K.S.; Yasir, M.; Elmowafy, M.; Mohammed, E.F.; Al-Oanzi, Z.H. Formulation of chitosan-coated piperine NLCs: Optimization, in vitro characterization, and in vivo preclinical assessment. AAPS Pharm. Sci. Tech. 2021, 22, 231. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, T.E.; Ilbasmis-Tamer, S.; Takka, S. Antitumor activity of gemcitabine hydrochloride loaded lipid polymer hybrid nanoparticles (LPHNs): In vitro and in vivo. Int. J. Pharm. 2020, 580, 119246. [Google Scholar] [CrossRef]
- Gilani, S.J.; Bin-Jumah, M.; Rizwanullah, M.; Imam, S.S.; Imtiyaz, K.; Alshehri, S.; Rizvi, M.M.A. Chitosan coated luteolin nanostructured lipid carriers: Optimization, in vitro-ex vivo assessments and cytotoxicity study in breast cancer cells. Coatings 2021, 11, 158. [Google Scholar] [CrossRef]
- Setyaningsih, D.; Santoso, Y.A.; Hartini, Y.S.; Murti, Y.B.; Hinrichs, W.L.; Patramurti, C. Isocratic high-performance liquid chromatography (HPLC) for simultaneous quantification of curcumin and piperine in a microparticle formulation containing Curcuma longa and Piper nigrum. Heliyon 2021, 7, e06541. [Google Scholar] [CrossRef]
- Jain, A.; Thakur, K.; Sharma, G.; Kush, P.; Jain, U.K. Fabrication, characterization and cytotoxicity studies of ionically cross-linked docetaxel loaded chitosan nanoparticles. Carbohydr. Polym. 2016, 137, 65–74. [Google Scholar] [CrossRef]
- Tahir, N.; Madni, A.; Balasubramanian, V.; Rehman, M.; Correia, A.; Kashif, P.M.; Mäkilä, E.; Salonen, J.; Santos, H.A. Development and optimization of methotrexate-loaded lipid-polymer hybrid nanoparticles for controlled drug delivery applications. Int. J. Pharm. 2017, 533, 156–168. [Google Scholar] [CrossRef]
- Garg, N.K.; Singh, B.; Sharma, G.; Kushwah, V.; Tyagi, R.K.; Jain, S.; Katare, O.P. Development and characterization of single step self-assembled lipid polymer hybrid nanoparticles for effective delivery of methotrexate. RSC Adv. 2015, 5, 62989–62999. [Google Scholar] [CrossRef]
- Mahmood, S.; Kiong, K.C.; Tham, C.S.; Chien, T.C.; Hilles, A.R.; Venugopal, J.R. PEGylated lipid polymeric nanoparticle–encapsulated acyclovir for in vitro controlled release and ex vivo gut sac permeation. AAPS Pharm. Sci. Tech. 2020, 21, 285. [Google Scholar] [CrossRef]
- Yu, F.; Ao, M.; Zheng, X.; Li, N.; Xia, J.; Li, Y.; Li, D.; Hou, Z.; Qi, Z.; Chen, X.D. PEG–lipid–PLGA hybrid nanoparticles loaded with berberine–phospholipid complex to facilitate the oral delivery efficiency. Drug Deliv. 2017, 24, 825–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajra, B.; Dalwadi, C.; Patel, R. Formulation and optimization of itraconazole polymeric lipid hybrid nanoparticles (Lipomer) using box behnken design. DARU J. Pharm. Sci. 2015, 23, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.M.; Madni, A.; Torchilin, V.; Filipczak, N.; Pan, J.; Tahir, N.; Shah, H. Lipid-chitosan hybrid nanoparticles for controlled delivery of cisplatin. Drug Deliv. 2019, 26, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Davarani, F.H.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.N.; Granja, S.; Boni, F.I.; Prezotti, F.G.; Ferreira, L.; Cury, B.S.; Reis, R.M.; Baltazar, F.; Gremião, M.P. Modulating chitosan-PLGA nanoparticle properties to design a co-delivery platform for glioblastoma therapy intended for nose-to-brain route. Drug Deliv. Transl. Res. 2020, 10, 1729–1747. [Google Scholar] [CrossRef]
- Zhu, Y.; Gu, Z.; Liao, Y.; Li, S.; Xue, Y.; Firempong, M.A.; Xu, Y.; Yu, J.; Smyth, H.D.; Xu, X. Improved intestinal absorption and oral bioavailability of astaxanthin using poly (ethylene glycol)-graft-chitosan nanoparticles: Preparation, in vitro evaluation, and pharmacokinetics in rats. J. Sci. Food. Agric. 2022, 102, 1002–1011. [Google Scholar] [CrossRef]
- Khan, S.; Aamir, M.N.; Madni, A.; Jan, N.; Khan, A.; Jabar, A.; Shah, H.; Rahim, M.A.; Ali, A. Lipid poly (ε-caprolactone) hybrid nanoparticles of 5-fluorouracil for sustained release and enhanced anticancer efficacy. Life Sci. 2021, 284, 119909. [Google Scholar] [CrossRef]
- Unagolla, J.M.; Jayasuriya, A.C. Drug transport mechanisms and in vitro release kinetics of vancomycin encapsulated chitosan-alginate polyelectrolyte microparticles as a controlled drug delivery system. Eur. J. Pharm. Sci. 2018, 114, 199–209. [Google Scholar] [CrossRef]
- Pauluk, D.; Padilha, A.K.; Khalil, N.M.; Mainardes, R.M. Chitosan-coated zein nanoparticles for oral delivery of resveratrol: Formation, characterization, stability, mucoadhesive properties and antioxidant activity. Food Hydrocoll. 2019, 94, 411–417. [Google Scholar] [CrossRef]
- Chen, M.C.; Mi, F.L.; Liao, Z.X.; Hsiao, C.W.; Sonaje, K.; Chung, M.F.; Hsu, L.W.; Sung, H.W. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv. Drug Deliv. Rev. 2013, 65, 865–879. [Google Scholar] [CrossRef]
- Rao, S.; Prestidge, C.A. Polymer-lipid hybrid systems: Merging the benefits of polymeric and lipid-based nanocarriers to improve oral drug delivery. Expert Opin. Drug Deliv. 2016, 13, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Qian, Y.; Fang, G. Development of lipid–polymer hybrid nanoparticles for improving oral absorption of enoxaparin. Pharmaceutics 2020, 12, 607. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, H.; Xu, E.; Zhang, X.; Guan, J.; Zhao, R.; Mao, S. Exploring the potential of functional polymer-lipid hybrid nanoparticles for enhanced oral delivery of paclitaxel. Asian J. Pharm. Sci. 2021, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, N.; Song, X. Bioadhesive polymer/lipid hybrid nanoparticles as oral delivery system of raloxifene with enhancive intestinal retention and bioavailability. Drug Deliv. 2021, 28, 252–260. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Levels Used, Actual (Coded Factor) | ||
|---|---|---|---|
| Independent variables | Low (−1) | Medium (0) | High (+1) |
| A = Concentration of PL-90G (mg) | 75 | 100 | 125 |
| B = Concentration of CS (mg) | 40 | 60 | 80 |
| C = Concentration of P-188 (mg) | 50 | 75 | 100 |
| Dependent variables | Goal | ||
| R1 = Particle size (PS; nm) | Minimize | ||
| R2 = Polydispersity index (PDI) | Minimize | ||
| R3 = Entrapment efficiency (EE; %) | Maximize | ||
| Formulations | Independent Variables | Dependent Variables | ||||
|---|---|---|---|---|---|---|
| A | B | C | R1 | R2 | R3 | |
| F1 | 75 | 60 | 100 | 131.47 | 0.146 | 66.89 |
| F2 | 125 | 40 | 75 | 175.11 | 0.259 | 70.51 |
| F3 | 125 | 60 | 100 | 152.47 | 0.275 | 72.73 |
| F4 | 100 | 60 | 75 | 159.72 | 0.231 | 78.46 |
| F5 | 75 | 60 | 50 | 145.21 | 0.215 | 59.94 |
| F6 | 100 | 80 | 100 | 177.81 | 0.285 | 78.43 |
| F7 | 100 | 40 | 100 | 132.71 | 0.187 | 62.44 |
| F8 | 125 | 80 | 75 | 218.76 | 0.436 | 81.34 |
| F9 | 125 | 60 | 50 | 213.84 | 0.375 | 76.55 |
| F10 | 100 | 40 | 50 | 168.83 | 0.257 | 63.02 |
| F11 | 75 | 40 | 75 | 127.47 | 0.203 | 57.98 |
| F12 | 100 | 80 | 50 | 211.83 | 0.385 | 71.37 |
| F13 | 75 | 80 | 75 | 170.13 | 0.226 | 72.74 |
| F14 | 100 | 60 | 75 | 160.12 | 0.234 | 78.58 |
| F15 | 100 | 60 | 75 | 158.94 | 0.229 | 77.89 |
| Model | R2 | Adjusted R2 | Predicted R2 | SD | Remark |
|---|---|---|---|---|---|
| Response (R1) | |||||
| Linear | 0.9018 | 0.8751 | 0.8034 | 10.32 | – |
| 2F1 | 0.9495 | 0.9116 | 0.7883 | 8.68 | – |
| Quadratic | 0.9992 | 0.9976 | 0.9878 | 1.39 | Suggested |
| Response (R2) | |||||
| Linear | 0.8606 | 0.8225 | 0.7353 | 0.0334 | – |
| 2F1 | 0.9334 | 0.8834 | 0.7878 | 0.0270 | – |
| Quadratic | 0.9981 | 0.9948 | 0.9722 | 0.0057 | Suggested |
| Response (R3) | |||||
| Linear | 0.6967 | 0.6140 | 0.5351 | 4.71 | – |
| 2F1 | 0.7557 | 0.5725 | 0.4750 | 4.96 | – |
| Quadratic | 0.9974 | 0.9927 | 0.9631 | 0.6462 | Suggested |
| Model | Source | PS | PDI | %EE |
|---|---|---|---|---|
| Regression analysis | ||||
| Quadratic | Sum of Squares | 11,920.87 | 0.0876 | 802.50 |
| df | 9 | 9 | 9 | |
| Mean Square | 1324.54 | 0.0097 | 89.17 | |
| F- Value | 681.38 | 297.92 | 213.55 | |
| p-value, Prob > F | <0.0001 | <0.0001 | <0.0001 | |
| Remark | Suggested, significant | |||
| Lack of fit tests | ||||
| Quadratic | Sum of Squares | 9.00 | 0.0002 | 1.82 |
| df | 3 | 3 | 3 | |
| Mean Square | 3.00 | 0.0001 | 0.6053 | |
| F- Value | 8.33 | 7.93 | 4.45 | |
| p-value, Prob > F | 0.1091 | 0.1140 | 0.1888 | |
| Remark | Suggested, not significant | |||
| Parameters | SGF (pH 1.2) | SIF (pH 6.8) | ||
|---|---|---|---|---|
| Initial | Final | Initial | Final | |
| PS (nm) | 158.72 ± 5.27 | 174.26 ± 5.71 | 158.72 ± 5.27 | 169.67 ± 4.57 |
| PDI | 0.231 ± 0.01 | 0.253 ± 0.02 | 0.231 ± 0.01 | 0.235 ± 0.01 |
| EE (%) | 78.46 ± 3.34 | 74.37 ± 4.17 | 78.46 ± 3.34 | 72.63 ± 3.54 |
| ZP (mV) | +24.31 ± 2.41 | +21.07 ± 2.14 | +24.31 ± 2.41 | +22.72 ± 2.36 |
| Parameters | PPN-Suspension | PPN-LPHNPs |
|---|---|---|
| (µg/mL) | 4.83 | 14.25 * |
| (h) | 4 | 6 |
| (µh/mL) | 38.842 | 176.82 * |
| (µh/mL) | 40.679 | 196.206 * |
| (µh2/mL) | 402.527 | 2169.082 * |
| (µh2/mL) | 522.736 | 3531.615 * |
| MRT (h) | 10.363 | 12.267 |
| (h) | 10.769 | 15.445 |
| (h−1) | 0.064 | 0.044 |
| – | 4.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazmi, I.; Al-Abbasi, F.A.; Imam, S.S.; Afzal, M.; Nadeem, M.S.; Altayb, H.N.; Alshehri, S. Formulation of Piperine Nanoparticles: In Vitro Breast Cancer Cell Line and In Vivo Evaluation. Polymers 2022, 14, 1349. https://doi.org/10.3390/polym14071349
Kazmi I, Al-Abbasi FA, Imam SS, Afzal M, Nadeem MS, Altayb HN, Alshehri S. Formulation of Piperine Nanoparticles: In Vitro Breast Cancer Cell Line and In Vivo Evaluation. Polymers. 2022; 14(7):1349. https://doi.org/10.3390/polym14071349
Chicago/Turabian StyleKazmi, Imran, Fahad A. Al-Abbasi, Syed Sarim Imam, Muhammad Afzal, Muhammad Shahid Nadeem, Hisham N. Altayb, and Sultan Alshehri. 2022. "Formulation of Piperine Nanoparticles: In Vitro Breast Cancer Cell Line and In Vivo Evaluation" Polymers 14, no. 7: 1349. https://doi.org/10.3390/polym14071349
APA StyleKazmi, I., Al-Abbasi, F. A., Imam, S. S., Afzal, M., Nadeem, M. S., Altayb, H. N., & Alshehri, S. (2022). Formulation of Piperine Nanoparticles: In Vitro Breast Cancer Cell Line and In Vivo Evaluation. Polymers, 14(7), 1349. https://doi.org/10.3390/polym14071349