
Probing Oxygen-to-Hydrogen Peroxide Electro-Conversion at Electrocatalysts Derived from Polyaniline

 ,
,  , ,
, , 
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental
2.1. Materials and Chemicals
2.2. Synthesis of Polyaniline-Based Materials by Oxidative Polymerization and Thermal Treatments
2.3. Physicochemical Characterization
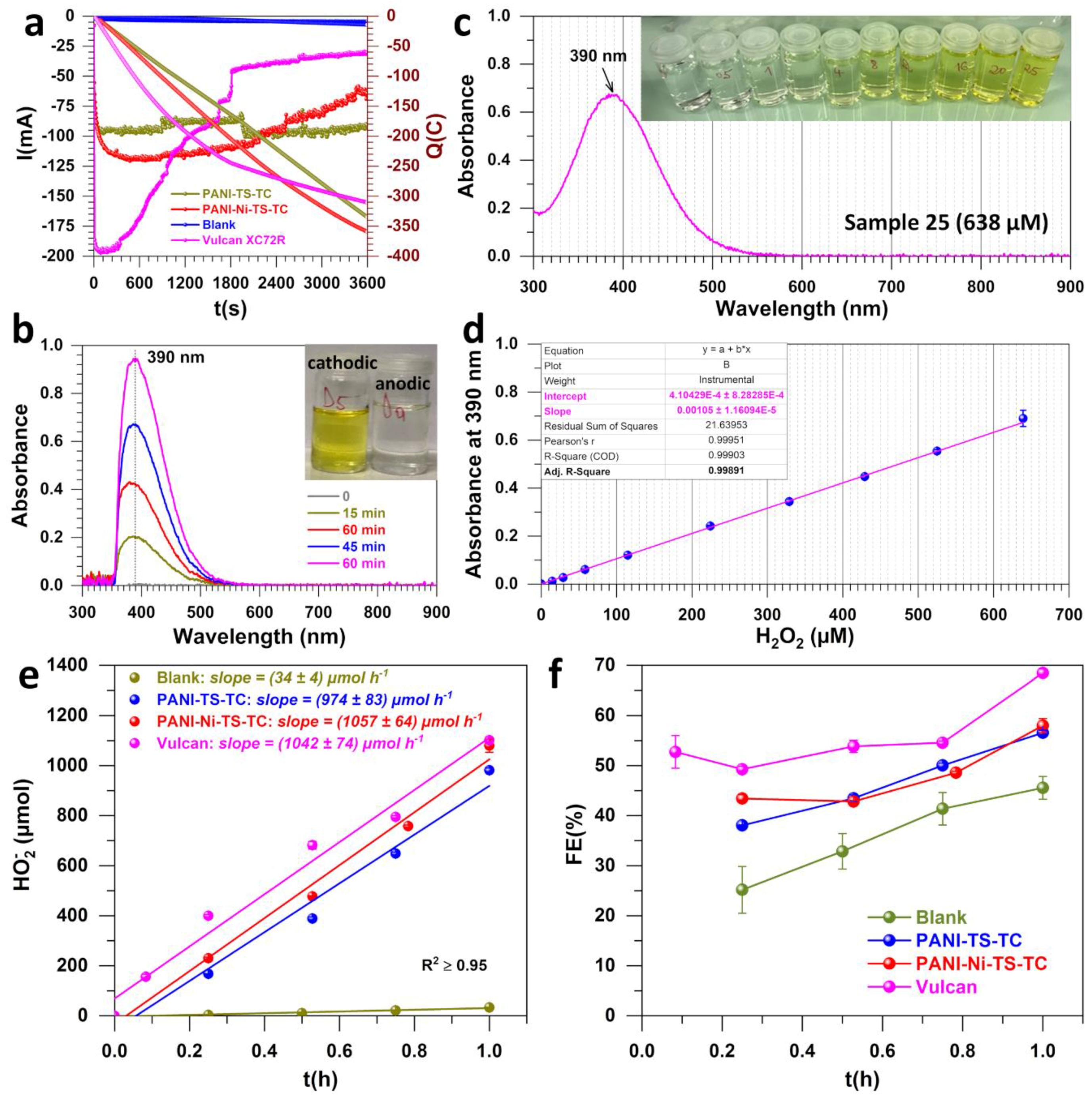
2.4. Electrochemical Measurements, Bulk Electrolysis and UV-Vis Assays
3. Results and Discussion
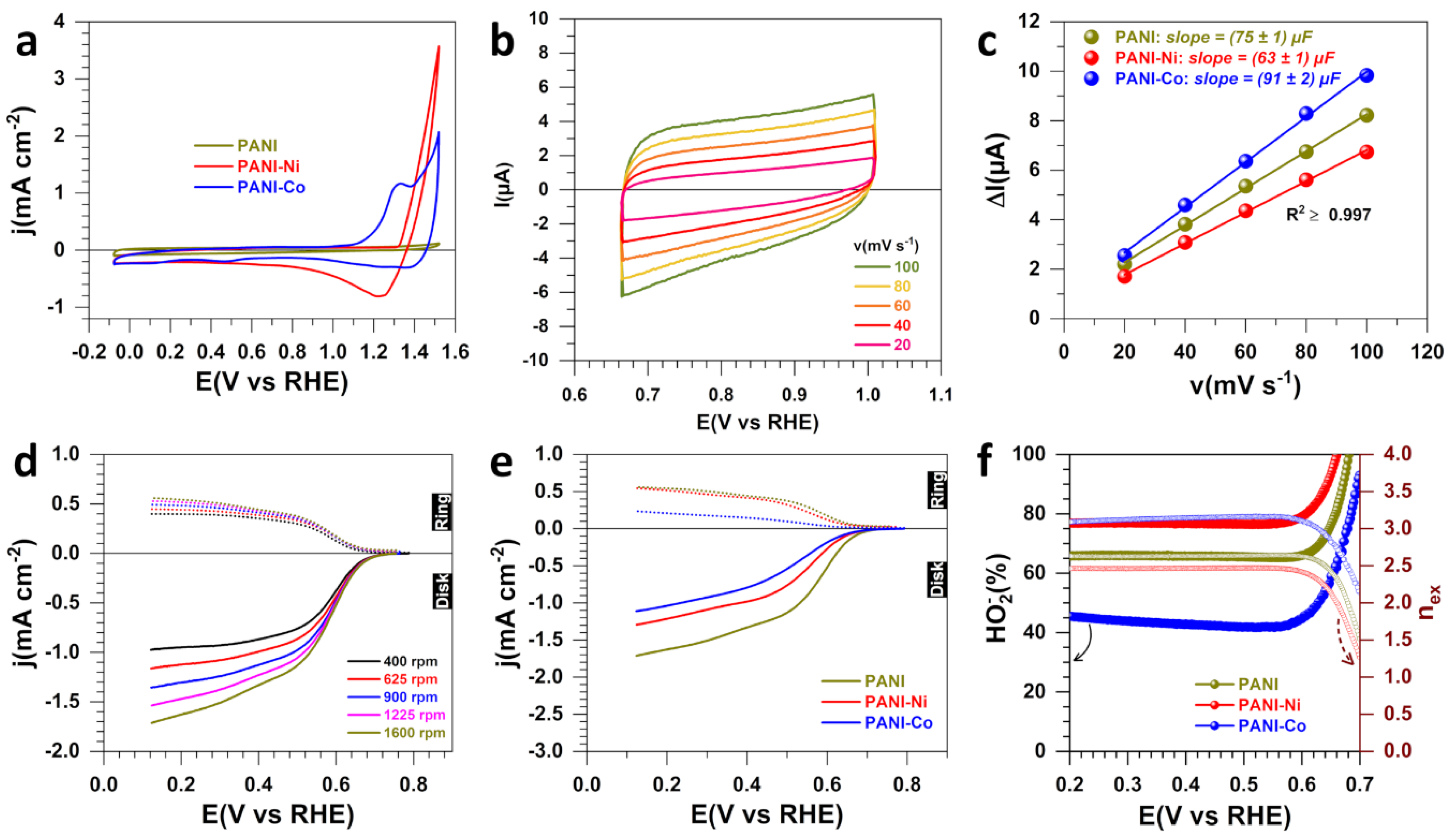
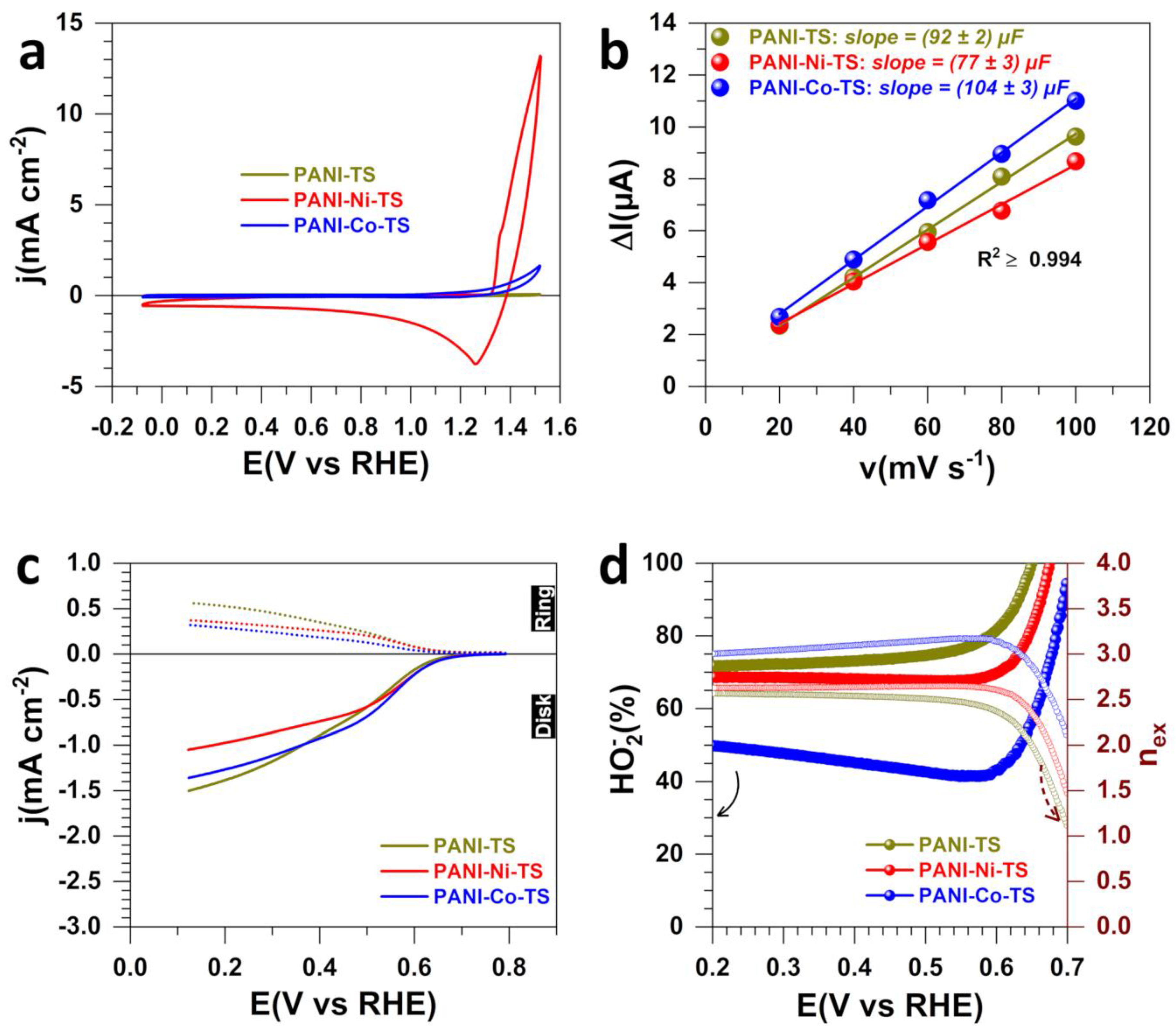
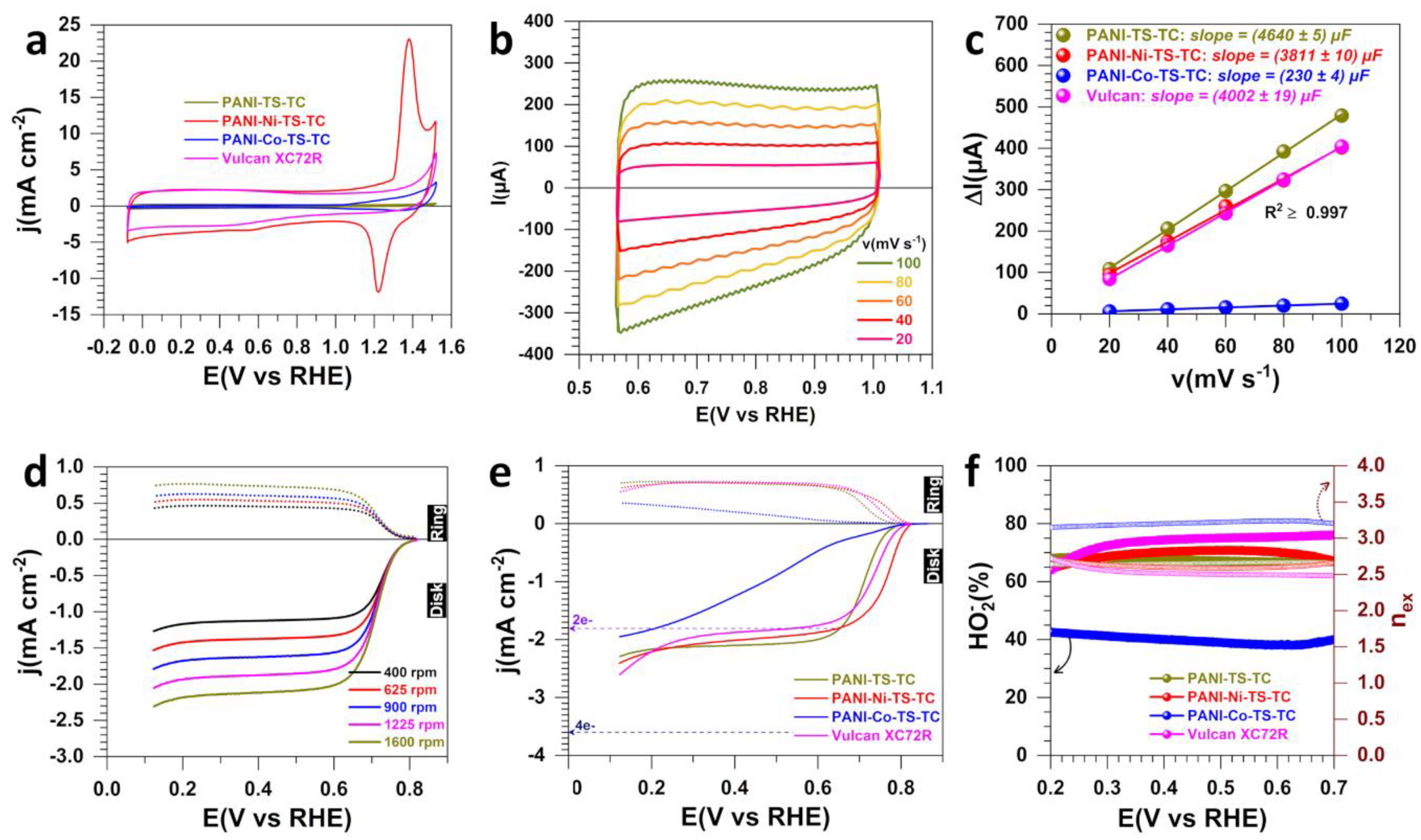
3.1. Electrochemical Analysis
- (i)
- The existence of a mass transport process that is the rate-determining step (rds),
- (ii)
- The reaction is of a first order reaction with respect to the electroreactive species (O2).
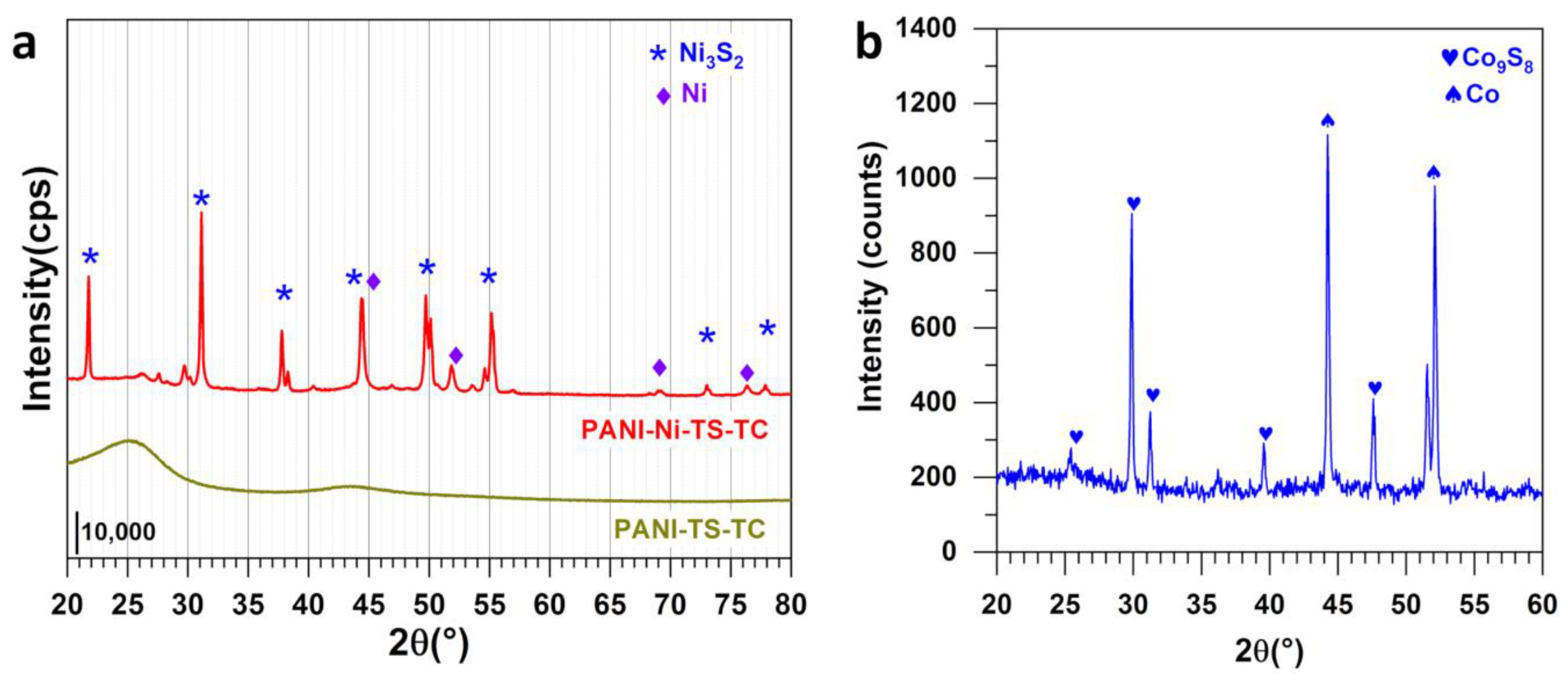
3.2. Structural Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.C.; Mavrikis, S.; Wang, L.; Ponce de León, C. Future perspectives for the advancement of electrochemical hydrogen peroxide production. Curr. Opin. Electrochem. 2021, 30, 100792. [Google Scholar] [CrossRef]
- Jung, E.; Shin, H.; Hooch Antink, W.; Sung, Y.-E.; Hyeon, T. Recent Advances in Electrochemical Oxygen Reduction to H2O2: Catalyst and Cell Design. ACS Energy Lett. 2020, 5, 1881–1892. [Google Scholar] [CrossRef]
- Yang, S.; Verdaguer-Casadevall, A.; Arnarson, L.; Silvioli, L.; Čolić, V.; Frydendal, R.; Rossmeisl, J.; Chorkendorff, I.; Stephens, I.E.L. Toward the Decentralized Electrochemical Production of H2O2: A Focus on the Catalysis. ACS Catal. 2018, 8, 4064–4081. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Pan, Z.; Hu, S.; Kim, J.-H. Cathodic Hydrogen Peroxide Electrosynthesis Using Anthraquinone Modified Carbon Nitride on Gas Diffusion Electrode. ACS Appl. Energy Mater. 2019, 2, 7972–7979. [Google Scholar] [CrossRef]
- Siahrostami, S.; Verdaguer-Casadevall, A.; Karamad, M.; Deiana, D.; Malacrida, P.; Wickman, B.; Escudero-Escribano, M.; Paoli, E.A.; Frydendal, R.; Hansen, T.W.; et al. Enabling direct H2O2 production through rational electrocatalyst design. Nat. Mater. 2013, 12, 1137–1143. [Google Scholar] [CrossRef] [Green Version]
- Zakaria, M.B.; Li, C.; Pramanik, M.; Tsujimoto, Y.; Hu, M.; Malgras, V.; Tominaka, S.; Yamauchi, Y. Nanoporous Mn-based electrocatalysts through thermal conversion of cyano-bridged coordination polymers toward ultra-high efficiency hydrogen peroxide production. J. Mater. Chem. A 2016, 4, 9266–9274. [Google Scholar] [CrossRef]
- Xia, C.; Xia, Y.; Zhu, P.; Fan, L.; Wang, H. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 2019, 366, 226–231. [Google Scholar] [CrossRef]
- Morandi, P.; Flaud, V.; Tingry, S.; Cornu, D.; Holade, Y. Tartaric acid regulated the advanced synthesis of bismuth-based materials with tunable performance towards the electrocatalytic production of hydrogen peroxide. J. Mater. Chem. A 2020, 8, 18840–18855. [Google Scholar] [CrossRef]
- Sun, Y.; Silvioli, L.; Sahraie, N.R.; Ju, W.; Li, J.; Zitolo, A.; Li, S.; Bagger, A.; Arnarson, L.; Wang, X.; et al. Activity-Selectivity Trends in the Electrochemical Production of Hydrogen Peroxide over Single-Site Metal-Nitrogen-Carbon Catalysts. J. Am. Chem. Soc. 2019, 141, 12372–12381. [Google Scholar] [CrossRef]
- Mase, K.; Yoneda, M.; Yamada, Y.; Fukuzumi, S. Efficient Photocatalytic Production of Hydrogen Peroxide from Water and Dioxygen with Bismuth Vanadate and a Cobalt(II) Chlorin Complex. ACS Energy Lett. 2016, 1, 913–919. [Google Scholar] [CrossRef]
- Yu, T.; Breslin, C.B. Review—2D Graphene and Graphene-Like Materials and Their Promising Applications in the Generation of Hydrogen Peroxide. J. Electrochem. Soc. 2020, 167, 126502. [Google Scholar] [CrossRef]
- Jung, E.; Shin, H.; Lee, B.-H.; Efremov, V.; Lee, S.; Lee, H.S.; Kim, J.; Hooch Antink, W.; Park, S.; Lee, K.-S.; et al. Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production. Nat. Mater. 2020, 19, 436–442. [Google Scholar] [CrossRef]
- Sheng, H.; Hermes, E.D.; Yang, X.; Ying, D.; Janes, A.N.; Li, W.; Schmidt, J.R.; Jin, S. Electrocatalytic Production of H2O2 by Selective Oxygen Reduction Using Earth-Abundant Cobalt Pyrite (CoS2). ACS Catal. 2019, 9, 8433–8442. [Google Scholar] [CrossRef]
- Zhu, Q.; Hinkle, M.; Kim, D.J.; Kim, J.-H. Modular Hydrogen Peroxide Electrosynthesis Cell with Anthraquinone-Modified Polyaniline Electrocatalyst. ACS EST Eng. 2021, 1, 446–455. [Google Scholar] [CrossRef]
- Isaji, T.; Abe, M.; Amaya, T.; Hirao, T. Hydrogen Peroxide Generation Using Polyaniline/Transition Metal Nanohybrid Electrodes. J. Inorg. Organomet. Polym. Mater. 2015, 25, 855–859. [Google Scholar] [CrossRef]
- Rabl, H.; Wielend, D.; Tekoglu, S.; Seelajaroen, H.; Neugebauer, H.; Heitzmann, N.; Apaydin, D.H.; Scharber, M.C.; Sariciftci, N.S. Are Polyaniline and Polypyrrole Electrocatalysts for Oxygen (O2) Reduction to Hydrogen Peroxide (H2O2)? ACS Appl. Energy Mater. 2020, 3, 10611–10618. [Google Scholar] [CrossRef]
- Quílez-Bermejo, J.; Morallón, E.; Cazorla-Amorós, D. Polyaniline-Derived N-Doped Ordered Mesoporous Carbon Thin Films: Efficient Catalysts towards Oxygen Reduction Reaction. Polymers 2020, 12, 2382. [Google Scholar] [CrossRef] [PubMed]
- Djara, R.; Holade, Y.; Merzouki, A.; Masquelez, N.; Cot, D.; Rebiere, B.; Petit, E.; Huguet, P.; Canaff, C.; Morisset, S.; et al. Insights from the Physicochemical and Electrochemical Screening of the Potentiality of the Chemically Synthesized Polyaniline. J. Electrochem. Soc. 2020, 167, 066503. [Google Scholar] [CrossRef]
- Djara, R.; Holade, Y.; Merzouki, A.; Lacour, M.-A.; Masquelez, N.; Flaud, V.; Cot, D.; Rebiere, B.; van der Lee, A.; Cambedouzou, J.; et al. Nanostructured Carbon-Nitrogen-Sulfur-Nickel Networks Derived from Polyaniline as Bifunctional Catalysts for Water Splitting. Front. Chem. 2020, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Holade, Y.; Morais, C.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Enhancing the available specific surface area of carbon supports to boost the electroactivity of nanostructured Pt catalysts. Phys. Chem. Chem. Phys. 2014, 16, 25609–25620. [Google Scholar] [CrossRef]
- Shinozaki, K.; Zack, J.W.; Richards, R.M.; Pivovar, B.S.; Kocha, S.S. Oxygen Reduction Reaction Measurements on Platinum Electrocatalysts Utilizing Rotating Disk Electrode Technique: I. Impact of Impurities, Measurement Protocols and Applied Corrections. J. Electrochem. Soc. 2015, 162, F1144–F1158. [Google Scholar] [CrossRef]
- Cassani, A.; Tuleushova, N.; Wang, Q.; Guesmi, H.; Bonniol, V.; Cambedouzou, J.; Tingry, S.; Bechelany, M.; Cornu, D.; Holade, Y. Fe-Modified Pd as an Effective Multifunctional Electrocatalyst for Catalytic Oxygen Reduction and Glycerol Oxidation Reactions in Alkaline Media. ACS Appl. Energy Mater. 2021, 4, 9944–9960. [Google Scholar] [CrossRef]
- Garsany, Y.; Singer, I.L.; Swider-Lyons, K.E. Impact of film drying procedures on RDE characterization of Pt/VC electrocatalysts. J. Electroanal. Chem. 2011, 662, 396–406. [Google Scholar] [CrossRef]
- Garsany, Y.; Baturina, O.A.; Swider-Lyons, K.E.; Kocha, S.S. Experimental Methods for Quantifying the Activity of Platinum Electrocatalysts for the Oxygen Reduction Reaction. Anal. Chem. 2010, 82, 6321–6328. [Google Scholar] [CrossRef] [PubMed]
- Kosimaningrum, W.E.; Le, T.X.H.; Holade, Y.; Bechelany, M.; Tingry, S.; Buchari, B.; Noviandri, I.; Innocent, C.; Cretin, M. Surfactant- and Binder-Free Hierarchical Platinum Nanoarrays Directly Grown onto a Carbon Felt Electrode for Efficient Electrocatalysis. ACS Appl. Mater. Interfaces 2017, 9, 22476–22489. [Google Scholar] [CrossRef] [Green Version]
- Patterson, B.D.; MacRae, E.A.; Ferguson, I.B. Estimation of hydrogen peroxide in plant extracts using titanium(IV). Anal. Biochem. 1984, 139, 487–492. [Google Scholar] [CrossRef]
- Bonét-Maury, P. Titrage photocolorimétrique de faibles quantités d’eau oxygénée. C. R. Acad. Sci. 1944, 218, 117–119. [Google Scholar]
- Lim, J.; Hoffmann, M.R. Substrate oxidation enhances the electrochemical production of hydrogen peroxide. Chem. Eng. J. 2019, 374, 958–964. [Google Scholar] [CrossRef]
- Djara, R.; Lacour, M.-A.; Merzouki, A.; Cambedouzou, J.; Cornu, D.; Tingry, S.; Holade, Y. Iridium and Ruthenium Modified Polyaniline Polymer Leads to Nanostructured Electrocatalysts with High Performance Regarding Water Splitting. Polymers 2021, 13, 190. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Trasatti, S.; Petrii, O.A. Real surface area measurements in electrochemistry. Pure Appl. Chem. 1991, 63, 711–734. [Google Scholar] [CrossRef]
- Holade, Y.; Sahin, N.; Servat, K.; Napporn, T.; Kokoh, K. Recent Advances in Carbon Supported Metal Nanoparticles Preparation for Oxygen Reduction Reaction in Low Temperature Fuel Cells. Catalysts 2015, 5, 310–348. [Google Scholar] [CrossRef] [Green Version]
- Acres, G.J.K.; Frost, J.C.; Hards, G.A.; Potter, R.J.; Ralph, T.R.; Thompsett, D.; Burstein, G.T.; Hutchings, G.J. Electrocatalysts for fuel cells. Catal. Today 1997, 38, 393–400. [Google Scholar] [CrossRef]
- Chai, G.-L.; Boero, M.; Hou, Z.; Terakura, K.; Cheng, W. Indirect Four-Electron Oxygen Reduction Reaction on Carbon Materials Catalysts in Acidic Solutions. ACS Catal. 2017, 7, 7908–7916. [Google Scholar] [CrossRef]
- Gileadi, E. Electrods Kinetics for Chemists, Chemical Engineers, and Materials Scientists; John Wiley & Sons, Ltd.: New York, NY, USA, 1993; p. 616. [Google Scholar]
- Napporn, T.W.; Holade, Y.; Kokoh, B.; Mitsushima, S.; Mayer, K.; Eichberger, B.; Hacker, V. Chapter 9—Electrochemical Measurement Methods and Characterization on the Cell Level. In Fuel Cells and Hydrogen: From Fundamentals to Applied Research; Hacker, V., Mitsushima, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 175–214. [Google Scholar]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2017, 95, 197–206. [Google Scholar] [CrossRef]
- Vork, F.T.A.; Barendrecht, E. The reduction of dioxygen at polypyrrole-modified electrodes with incorporated Pt particles. Electrochim. Acta 1990, 35, 135–139. [Google Scholar] [CrossRef] [Green Version]
- Houache, M.S.E.; Cossar, E.; Ntais, S.; Baranova, E.A. Electrochemical modification of nickel surfaces for efficient glycerol electrooxidation. J. Power Sources 2018, 375, 310–319. [Google Scholar] [CrossRef]
- Hajjar, P.; Lacour, M.-A.; Masquelez, N.; Cambedouzou, J.; Tingry, S.; Cornu, D.; Holade, Y. Insights on the Electrocatalytic Seawater Splitting at Heterogeneous Nickel-Cobalt Based Electrocatalysts Engineered from Oxidative Aniline Polymerization and Calcination. Molecules 2021, 26, 5926. [Google Scholar] [CrossRef]
- Favaro, M.; Perini, L.; Agnoli, S.; Durante, C.; Granozzi, G.; Gennaro, A. Electrochemical behavior of N and Ar implanted highly oriented pyrolytic graphite substrates and activity toward oxygen reduction reaction. Electrochim. Acta 2013, 88, 477–487. [Google Scholar] [CrossRef]
- Tuci, G.; Zafferoni, C.; Rossin, A.; Milella, A.; Luconi, L.; Innocenti, M.; Truong Phuoc, L.; Duong-Viet, C.; Pham-Huu, C.; Giambastiani, G. Chemically Functionalized Carbon Nanotubes with Pyridine Groups as Easily Tunable N-Decorated Nanomaterials for the Oxygen Reduction Reaction in Alkaline Medium. Chem. Mater. 2014, 26, 3460–3470. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095–14107. [Google Scholar] [CrossRef] [Green Version]
- Padya, B.; Kalita, D.; Jain, P.K.; Padmanabham, G.; Ravi, M.; Bhat, K.S. Self-organized growth of bamboo-like carbon nanotube arrays for field emission properties. Appl. Nanosci. 2012, 2, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Vikkisk, M.; Kruusenberg, I.; Joost, U.; Shulga, E.; Tammeveski, K. Electrocatalysis of oxygen reduction on nitrogen-containing multi-walled carbon nanotube modified glassy carbon electrodes. Electrochim. Acta 2013, 87, 709–716. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Li, H.; Li, X.; Zhao, B.; Yang, J. Electrocatalysis of oxygen reduction on carbon nanotubes with different surface functional groups in acid and alkaline solutions. Int. J. Hydrog. Energy 2014, 39, 16964–16975. [Google Scholar] [CrossRef]
- Yang, H.-H.; McCreery, R.L. Elucidation of the Mechanism of Dioxygen Reduction on Metal-Free Carbon Electrodes. J. Electrochem. Soc. 2000, 147, 3420. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.; Hahn, C.; Xiang, C.; Jaramillo, T.F.; Weber, A.Z. Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm. ACS Energy Lett. 2018, 4, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Schröder, J.; Mints, V.A.; Bornet, A.; Berner, E.; Fathi Tovini, M.; Quinson, J.; Wiberg, G.K.H.; Bizzotto, F.; El-Sayed, H.A.; Arenz, M. The Gas Diffusion Electrode Setup as Straightforward Testing Device for Proton Exchange Membrane Water Electrolyzer Catalysts. JACS Au 2021, 1, 247–251. [Google Scholar] [CrossRef]
- San Roman, D.; Krishnamurthy, D.; Garg, R.; Hafiz, H.; Lamparski, M.; Nuhfer, N.T.; Meunier, V.; Viswanathan, V.; Cohen-Karni, T. Engineering Three-Dimensional (3D) Out-of-Plane Graphene Edge Sites for Highly Selective Two-Electron Oxygen Reduction Electrocatalysis. ACS Catal. 2020, 10, 1993–2008. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Sinev, I.; Ju, W.; Bergmann, A.; Dresp, S.; Kühl, S.; Spöri, C.; Schmies, H.; Wang, H.; Bernsmeier, D.; et al. Efficient Electrochemical Hydrogen Peroxide Production from Molecular Oxygen on Nitrogen-Doped Mesoporous Carbon Catalysts. ACS Catal. 2018, 8, 2844–2856. [Google Scholar] [CrossRef]
- Chang, Q.; Zhang, P.; Mostaghimi, A.H.B.; Zhao, X.; Denny, S.R.; Lee, J.H.; Gao, H.; Zhang, Y.; Xin, H.L.; Siahrostami, S.; et al. Promoting H2O2 production via 2-electron oxygen reduction by coordinating partially oxidized Pd with defect carbon. Nat. Commun. 2020, 11, 2178. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Chen, Z.; Wu, H.; Wang, Y.; Cao, E.; Lu, X.; Wu, Y.; Ren, Z. Phytic acid-guided ultra-thin N,P co-doped carbon coated carbon nanotubes for efficient all-pH electrocatalytic hydrogen evolution. Nanoscale 2019, 11, 23027–23034. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Wang, Y.; Jiang, H.; Yuan, S. MWCNT Decorated Rich N-Doped Porous Carbon with Tunable Porosity for CO2 Capture. Molecules 2021, 26, 3451. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Xie, N.; Zhao, B.; Zhong, L.; Zhang, Y.; Niu, L. Optimizing Surface N-Doping of Fe-N-C Catalysts Derived from Fe/Melamine-Decorated Polyaniline for Oxygen Reduction Electrocatalysis. Adv. Mater. Interfaces 2021, 8, 2100197. [Google Scholar] [CrossRef]
- Dong, Y.; Zhou, M.; Tu, W.; Zhu, E.; Chen, Y.; Zhao, Y.; Liao, S.; Huang, Y.; Chen, Q.; Li, Y. Hollow Loofah-Like N, O-Co-Doped Carbon Tube for Electrocatalysis of Oxygen Reduction. Adv. Funct. Mater. 2019, 29, 1900015. [Google Scholar] [CrossRef]
- Xiang, X.; Li, X.; Huang, Z.; Gao, T.; Yuan, H.; Xiao, D. Sphere-and-Flake-Structured Cu, N Co-Doped Carbon Catalyst Designed by a Template-Free Method for Robust Oxygen Reduction Reaction. ChemElectroChem 2019, 6, 1078–1087. [Google Scholar] [CrossRef]
- Wagner, R.S.; Ellis, W.C. Vapor-liquid-solid mechanism of single crystal growth. Appl. Phys. Lett. 1964, 4, 89–90. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, J.; Zhao, H.; Jiang, R.; Tian, F.; Zhang, R. Tremella-like Ni3S2/MnS with ultrathin nanosheets and abundant oxygen vacancies directly used for high speed overall water splitting. Appl. Catal. B Environ. 2019, 257, 117899. [Google Scholar] [CrossRef]
- Wang, N.; Han, G.; Chang, Y.; Hou, W.; Xiao, Y.; Li, H. Preparing Ni3S2 composite with neural network-like structure for high-performance flexible asymmetric supercapacitors. Electrochim. Acta 2019, 317, 322–332. [Google Scholar] [CrossRef]
- Zhou, W.; Cao, X.; Zeng, Z.; Shi, W.; Zhu, Y.; Yan, Q.; Liu, H.; Wang, J.; Zhang, H. One-step synthesis of Ni3S2 nanorod@Ni(OH)2 nanosheet core-shell nanostructures on a three-dimensional graphene network for high-performance supercapacitors. Energy Environ. Sci. 2013, 6, 2216–2221. [Google Scholar] [CrossRef] [Green Version]
- Ou, X.; Gan, L.; Luo, Z. Graphene-templated growth of hollow Ni3S2 nanoparticles with enhanced pseudocapacitive performance. J. Mater. Chem. A 2014, 2, 19214–19220. [Google Scholar] [CrossRef]
- Tian, D.; Chen, S.; Zhu, W.; Wang, C.; Lu, X. Metal-organic framework derived hierarchical Ni/Ni3S2 decorated carbon nanofibers for high-performance supercapacitors. Mater. Chem. Front. 2019, 3, 1653–1660. [Google Scholar] [CrossRef]
- Jiang, N.; Tang, Q.; Sheng, M.; You, B.; Jiang, D.-E.; Sun, Y. Nickel sulfides for electrocatalytic hydrogen evolution under alkaline conditions: A case study of crystalline NiS, NiS2, and Ni3S2 nanoparticles. Catal. Sci. Technol. 2016, 6, 1077–1084. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holade, Y.; Knani, S.; Lacour, M.-A.; Cambedouzou, J.; Tingry, S.; Napporn, T.W.; Cornu, D. Probing Oxygen-to-Hydrogen Peroxide Electro-Conversion at Electrocatalysts Derived from Polyaniline. Polymers 2022, 14, 607. https://doi.org/10.3390/polym14030607
Holade Y, Knani S, Lacour M-A, Cambedouzou J, Tingry S, Napporn TW, Cornu D. Probing Oxygen-to-Hydrogen Peroxide Electro-Conversion at Electrocatalysts Derived from Polyaniline. Polymers. 2022; 14(3):607. https://doi.org/10.3390/polym14030607
Chicago/Turabian StyleHolade, Yaovi, Sarra Knani, Marie-Agnès Lacour, Julien Cambedouzou, Sophie Tingry, Teko W. Napporn, and David Cornu. 2022. "Probing Oxygen-to-Hydrogen Peroxide Electro-Conversion at Electrocatalysts Derived from Polyaniline" Polymers 14, no. 3: 607. https://doi.org/10.3390/polym14030607
APA StyleHolade, Y., Knani, S., Lacour, M.-A., Cambedouzou, J., Tingry, S., Napporn, T. W., & Cornu, D. (2022). Probing Oxygen-to-Hydrogen Peroxide Electro-Conversion at Electrocatalysts Derived from Polyaniline. Polymers, 14(3), 607. https://doi.org/10.3390/polym14030607