
Material and Environmental Properties of Natural Polymers and Their Composites for Packaging Applications—A Review



Abstract
1. Introduction
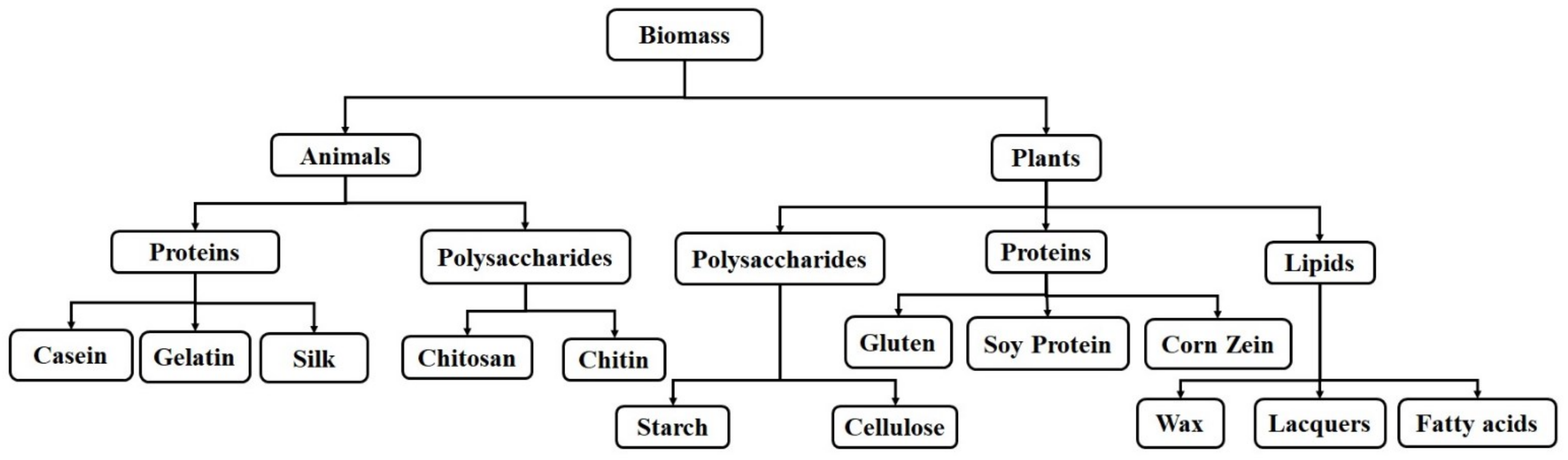
2. Natural Polymers
2.1. Protein-Based Natural Polymers
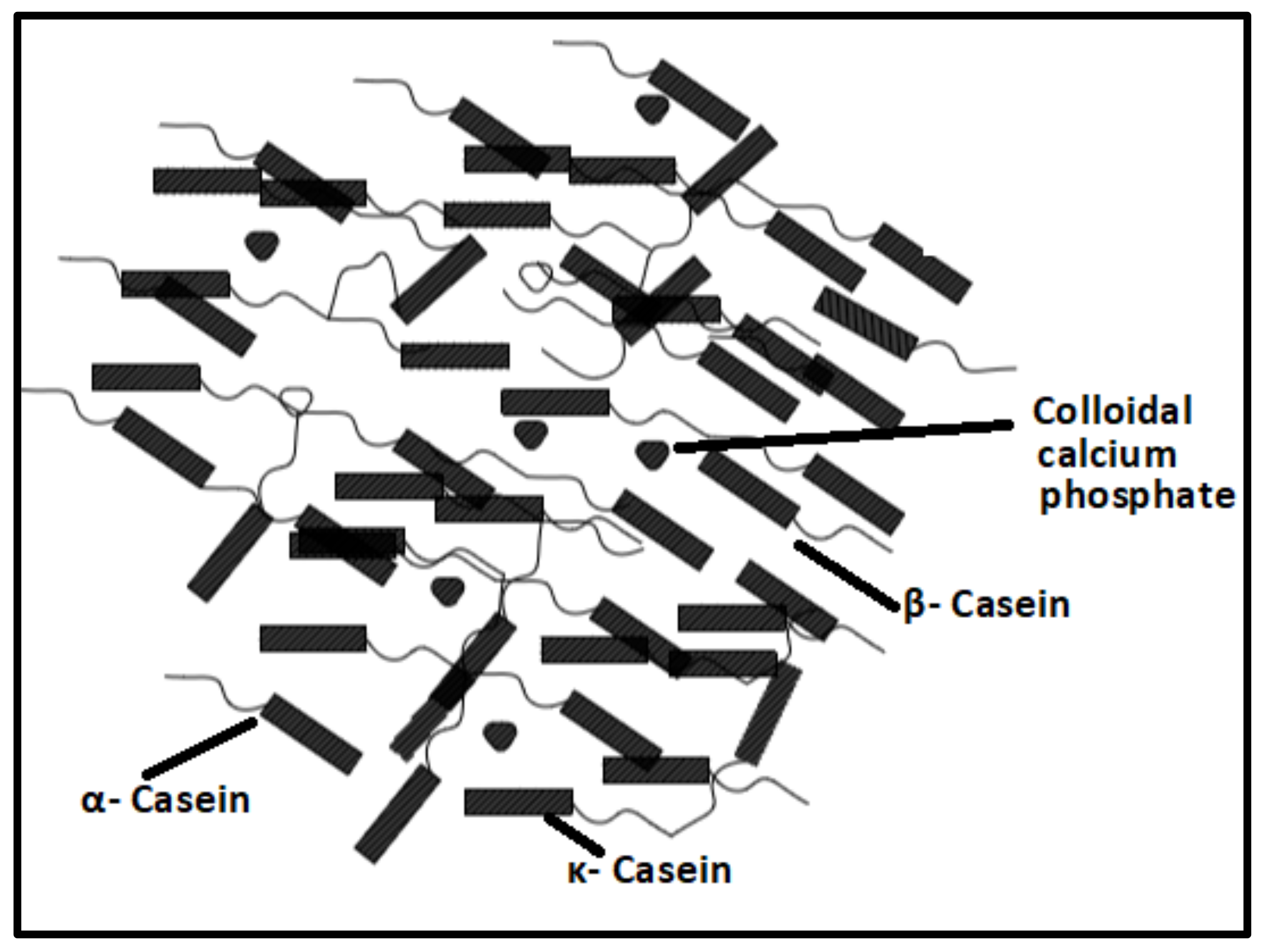
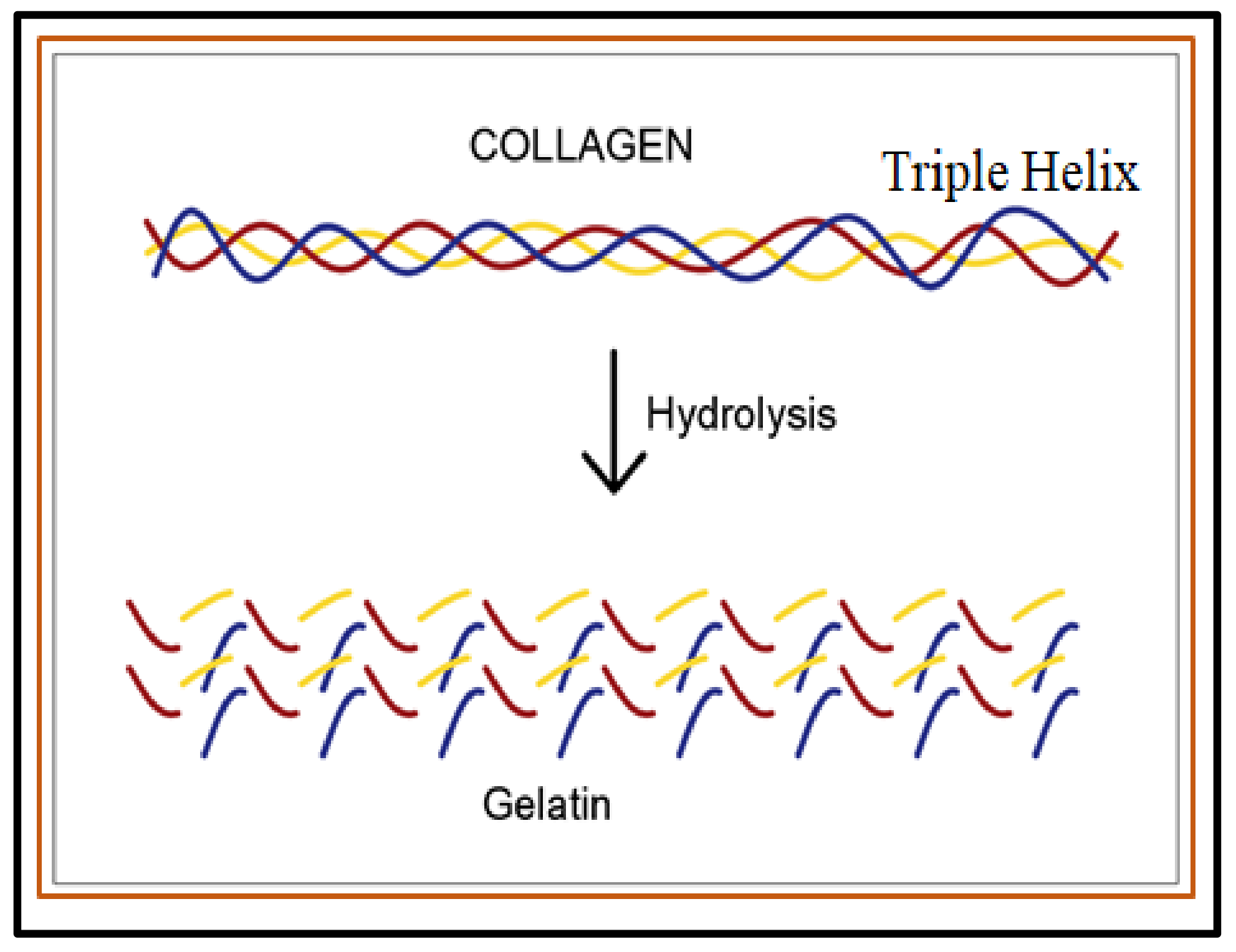
2.1.1. Proteins-Based Natural Polymers from an Animal Resource
2.1.2. Proteins-Based Natural Polymers from Plant Resources
2.2. Polysaccharide-Based Natural Polymers
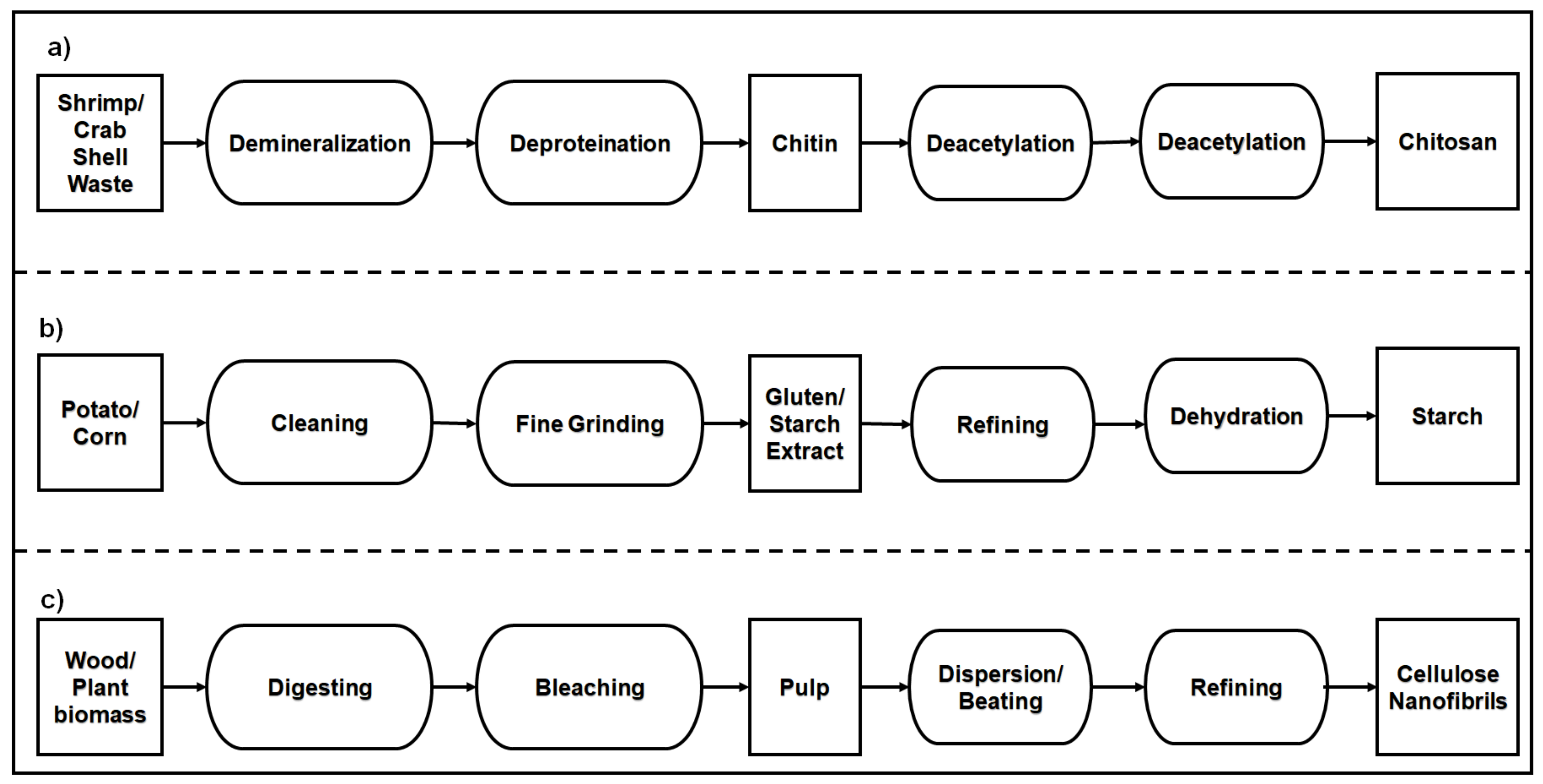
2.2.1. Polysaccharide-Based Natural Polymers from an Animal Resource
2.2.2. Polysaccharide-Based Natural Polymers from Plant Resource
2.3. Lipid-Based Natural Polymers
3. Natural Polymer Nanocomposites
3.1. Protein—CNF Composite Films
3.2. Polysaccharides–CNF/CNC Composite Films
4. Environmental Impact Assessment of Natural Polymers and Their Nanocomposites
A Generic Approach for LCA of Natural Polymer Nanocomposite Packaging Product
- Natural polymer matrix and reinforcement materials production.
- Natural polymer nanocomposite production.
- Packaging product manufacturing.
- Packaging product end-of-life management.
5. Overview of LCA Studies on Natural Polymers and Their Nanocomposites
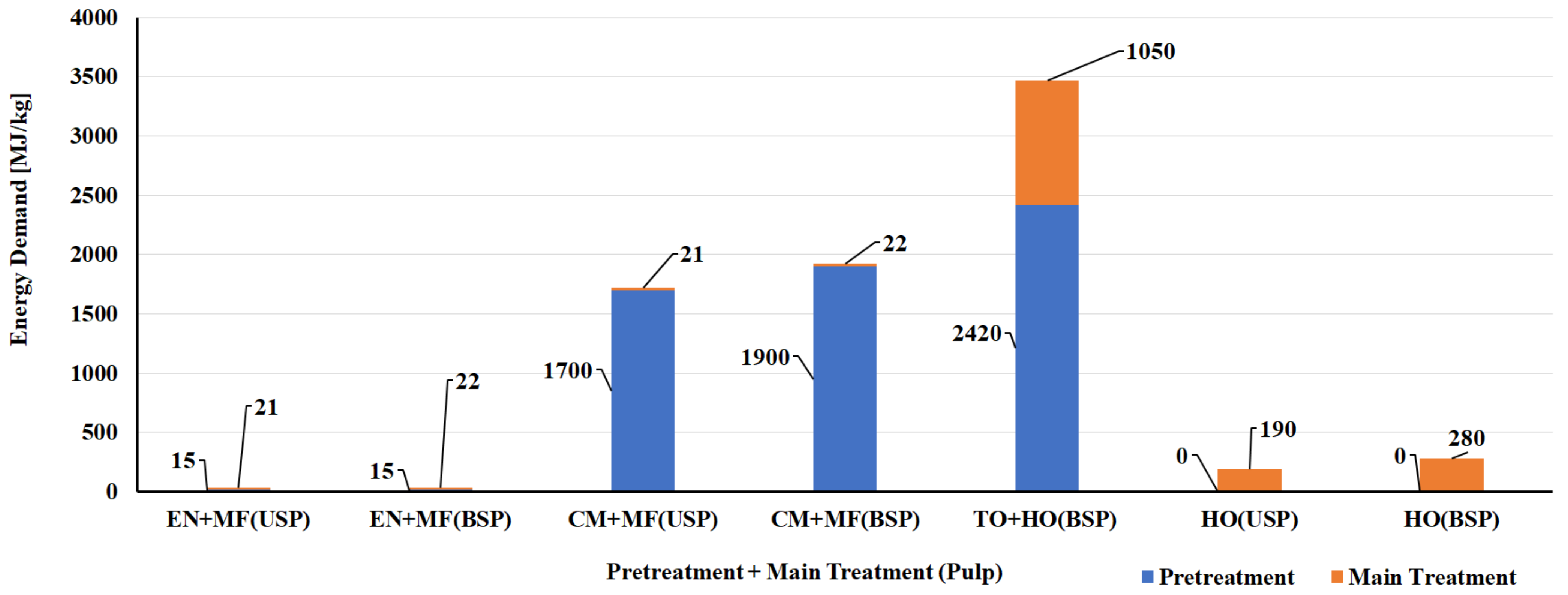
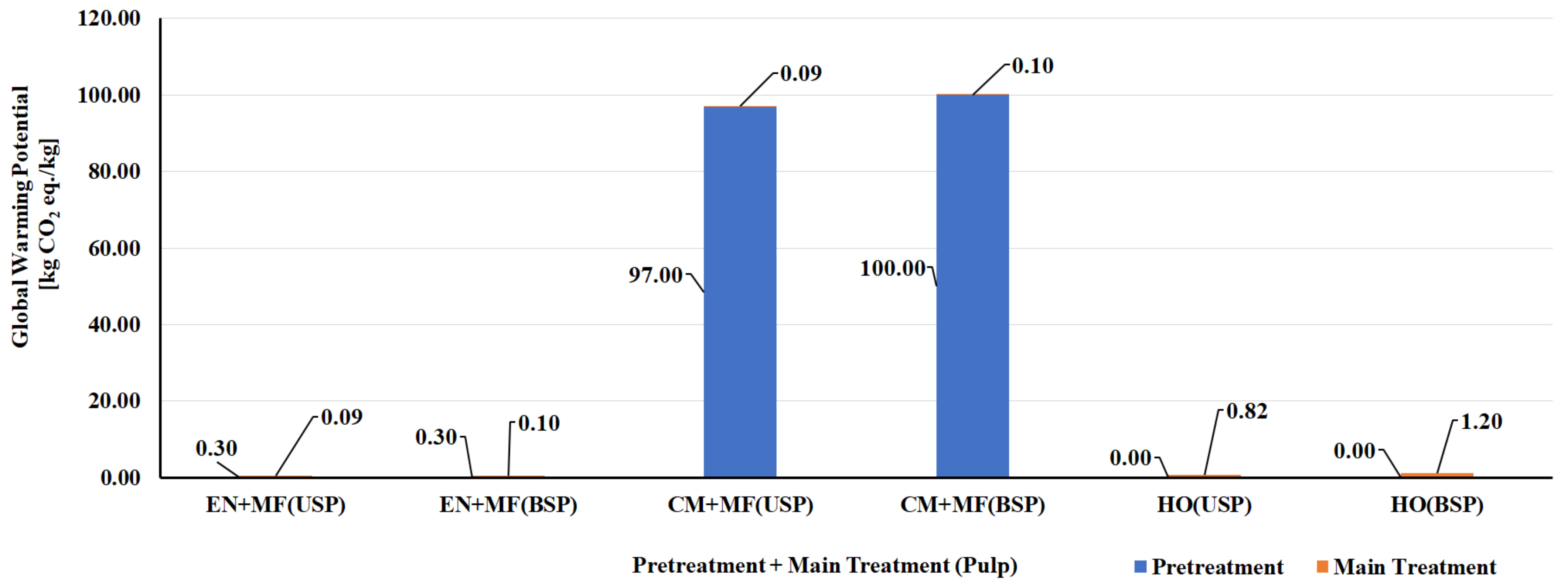
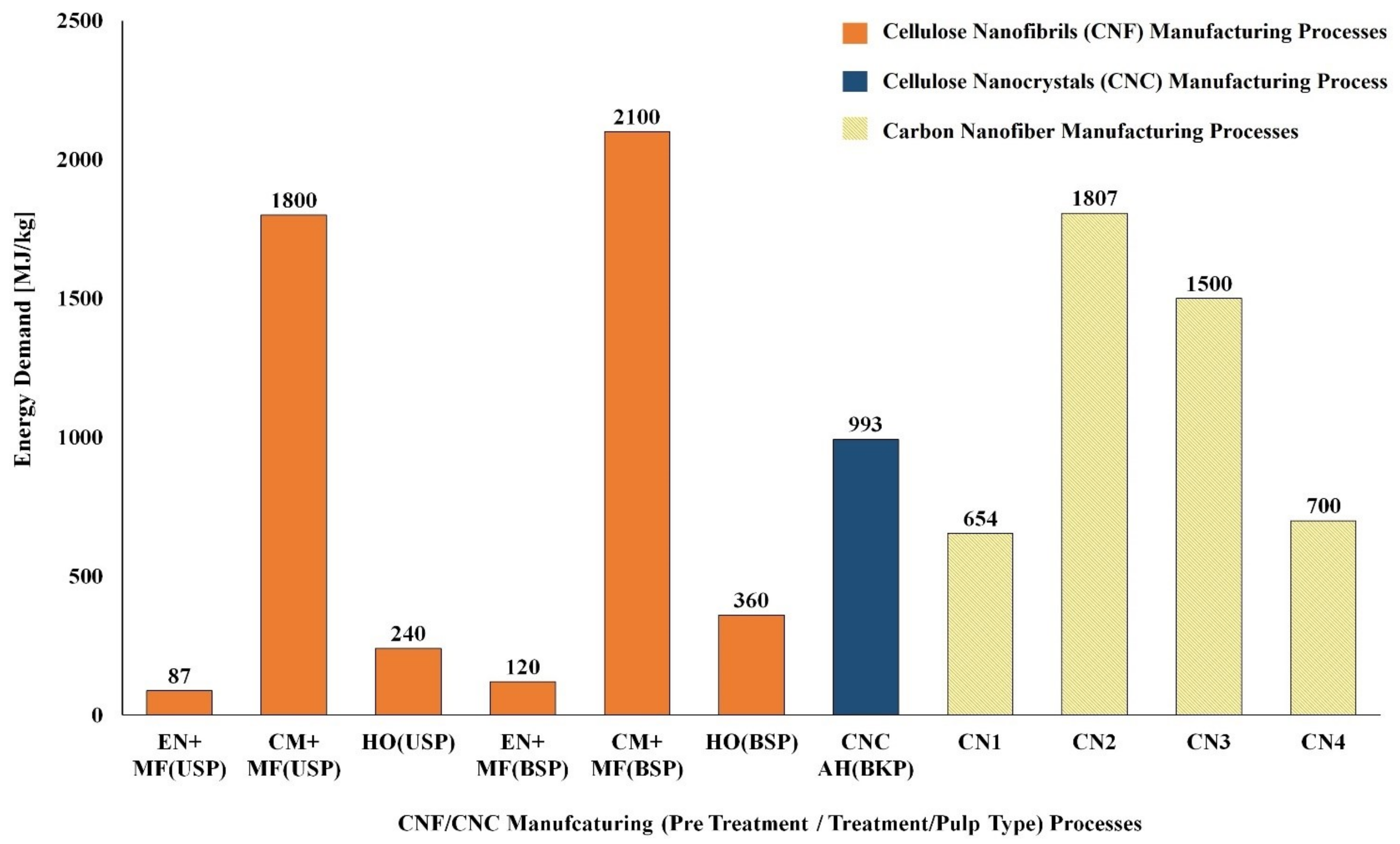
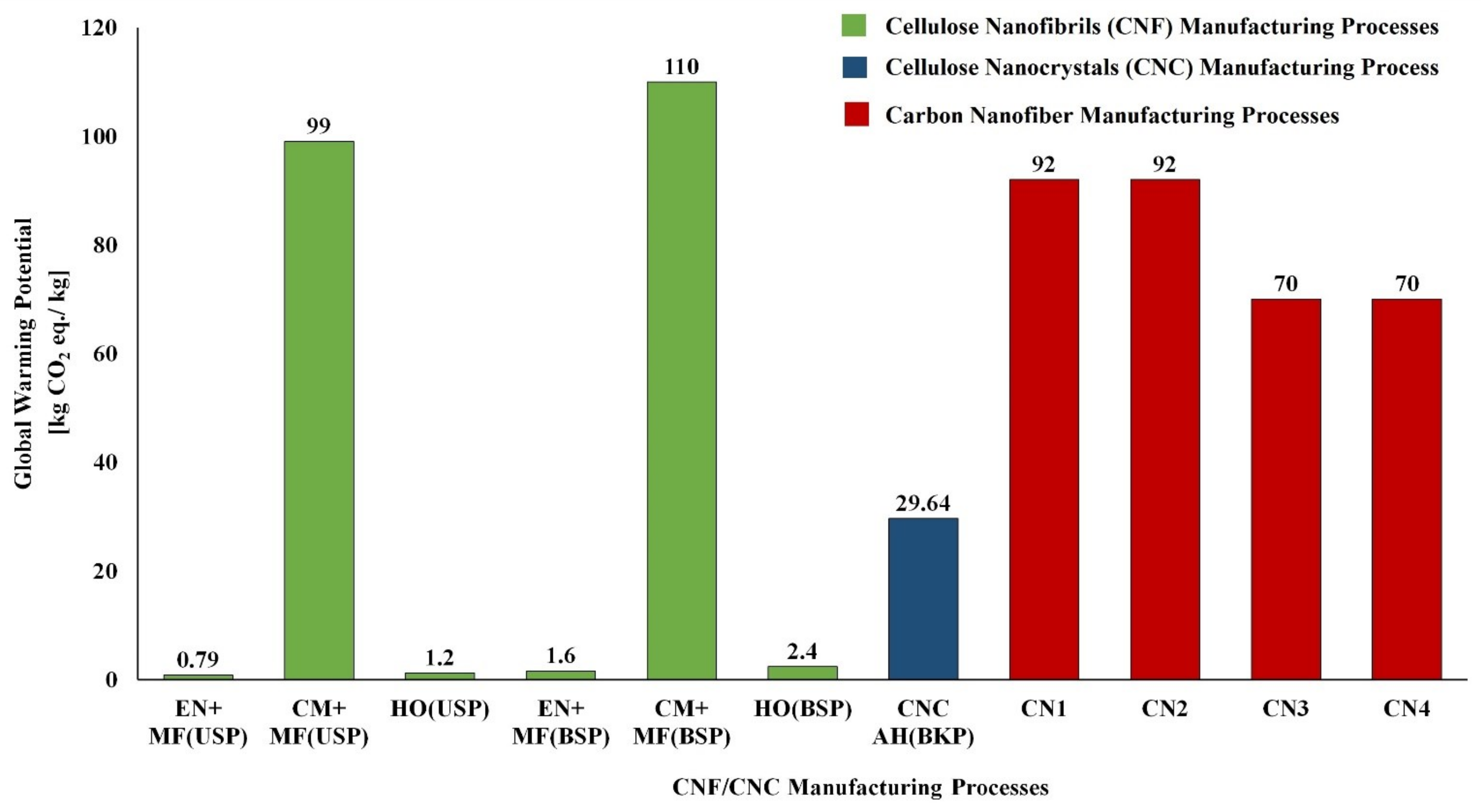
5.1. LCA of CNF and CNC Natural Polymer Manufacturing
5.2. LCA of Chitosan Natural Polymer Manufacturing Process
5.3. LCA of the Thermoplastic Starch Polymer Manufacturing Process
6. Current Knowledge on and Opportunities for Packaging Films Made of Natural Polymers and Their Composites
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Walker, T.; Gramlich, D.; Dumont-Bergeron, A. The case for a plastic tax: A review of its benefits and disadvantages within a circular economy. In Sustainability; Emerald Publishing Limited: Bingley, UK, 2020. [Google Scholar]
- Europe, P. Plastics—The Facts 2016. An analysis of European plastics production, demand and waste data. In Proceedings of the K-2016: Trade Fair for Plastics and Rubber, Düsseldorf, Germany, 19–26 October 2016. [Google Scholar]
- Hamman, C.W. Energy for Plastic, Coursework for Physics. Available online: https://large.stanford.edu/courses/2010/ph240/hamman1/ (accessed on 18 September 2022).
- Kutz, M. Applied Plastics Engineering Handbook Processing and Materials, 1st ed.; Kutz, M., Ed.; William Andrew, An Imprint of Elsevier: Waltham, MA, USA, 2011. [Google Scholar]
- Liepins, R.; Pearce, E.M. Chemistry and Toxicity of Flame Retardants for Plastics. Environ. Health Perspect. 1976, 17, 55–63. [Google Scholar] [CrossRef]
- Andrady, L.A.; Rajapakse, N. Additives and chemicals in plastics. In Hazardous Chemicals Associated with Plastics in the Marine Environment; Takada, H., Karapanagioti, H.K., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–17. [Google Scholar]
- Lithner, D.; Larsson, Å.; Dave, G. Environmental and health hazard ranking and assessment of plastic polymers based on chemical composition. Sci. Total Environ. 2011, 409, 3309–3324. [Google Scholar] [CrossRef] [PubMed]
- Halden, R.U. Plastics and health risks. Annu. Rev. Public Health 2010, 31, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.M.; Calafat, A.M. Human body burdens of chemicals used in plastic manufacture. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.J. Understanding plastics pollution: The role of economic development and technological research. Environ. Pollut. 2019, 249, 812–821. [Google Scholar] [CrossRef]
- The U.S. Environmental Protection Agency (EPA). Advancing Sustainable Materials Management: 2015 Fact Sheet. Available online: https://www.epa.gov/sites/default/files/2018-07/documents/2015_smm_msw_factsheet_07242018_fnl_508_002.pdf (accessed on 18 September 2022).
- Napper, I.E.; Thompson, R.C. Chapter 22—Marine plastic pollution: Other than microplastic. In Waste, 2nd ed.; Letcher, T.M., Vallero, D.A., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 425–442. [Google Scholar]
- Avio, C.G.; Gorbi, S.; Regoli, F. Plastics and microplastics in the oceans: From emerging pollutants to emerged threat. Mar. Environ. Res. 2017, 128, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Jambeck, J.R.; Geyer, R.; Wilcox, C.; Siegler, T.R.; Perryman, M.; Andrady, A.; Narayan, R.; Law, K.L. Plastic waste inputs from land into the ocean. Science 2015, 347, 768–771. [Google Scholar] [CrossRef]
- Ng, E.L.; Lwanga, E.H.; Eldridge, S.M.; Johnston, P.; Hu, H.W.; Geissen, V.; Chen, D. An overview of microplastic and nanoplastic pollution in agroecosystems. Sci. Total Environ. 2018, 627, 1377–1388. [Google Scholar] [CrossRef]
- Barnes, D.K.; Galgani, F.; Thompson, R.C.; Barlaz, M. Accumulation and fragmentation of plastic debris in global environments. Philos. Trans. R. Soc. B: Biol. Sci. 2009, 364, 1985–1998. [Google Scholar] [CrossRef]
- Derraik, J.G.B. The pollution of the marine environment by plastic debris: A review. Mar. Pollut. Bull. 2002, 44, 842–852. [Google Scholar] [CrossRef]
- Lehner, R.; Weder, C.; Petri-Fink, A.; Rothen-Rutishauser, B. Emergence of nanoplastic in the environment and possible impact on human health. Environ. Sci. Technol. 2019, 53, 1748–1765. [Google Scholar] [CrossRef]
- Wright, S.L.; Kelly, F.J. Plastic and human health: A micro issue? Environ. Sci. Technol. 2017, 51, 6634–6647. [Google Scholar] [CrossRef]
- Allwood, J.M.; Cullen, J.M.; Milford, R.L. Options for achieving a 50% cut in industrial carbon emissions by 2050. Environ. Sci. Technol. 2010, 44, 1888–1894. [Google Scholar] [CrossRef]
- UNEP. Marine Plastic Debris and Microplastics: Global Lessons and Research to Inspire Action and Guide Policy Change; UN: San Francisco, CA, USA, 2016. [Google Scholar]
- MacArthur, D.E.; Waughray, D.; Stuchtey, M. The New Plastics Economy, Rethinking the Future of Plastics; World Economic Forum: Geneva, Switzerland, 2016. [Google Scholar]
- Foschi, E.; Bonoli, A. The commitment of packaging industry in the framework of the European strategy for plastics in a circular economy. Adm. Sci. 2019, 9, 18. [Google Scholar] [CrossRef]
- Mozaffari, N.; Kholdebarin, A. A review: Investigation of plastics effect on the environment, bioplastic global market share and its future perspectives. Sci. Tech. J. Technogen. Ecol. Saf. 2019, 5, 47–54. [Google Scholar]
- Rudin, A.; Choi, P. Chapter 13—Biopolymers. In The Elements of Polymer Science & Engineering, 3rd ed.; Rudin, A., Choi, P., Eds.; Academic Press: Boston, MA, USA, 2013; pp. 521–535. [Google Scholar]
- Petersen, K.; Nielsen, P.V.; Bertelsen, G.; Lawther, M.; Olsen, M.B.; Nilsson, N.H.; Mortensen, G. Potential of biobased materials for food packaging. Trends Food Sci. Technol. 1999, 10, 52–68. [Google Scholar] [CrossRef]
- Sangroniz, A.; Zhu, J.B.; Tang, X.; Etxeberria, A.; Chen, E.Y.X.; Sardon, H. Packaging materials with desired mechanical and barrier properties and full chemical recyclability. Nat. Commun. 2019, 10, 3559. [Google Scholar] [CrossRef]
- Ahmed, S. Bio-Based Materials for Food Packaging Green and Sustainable Advanced Packaging Materials: Green and Sustainable Advanced Packaging Materials; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Vartiainen, J.; Vähä-Nissi, M.; Harlin, A. Biopolymer films and coatings in packaging applications—A review of recent developments. Mater. Sci. Appl. 2014, 05, 708–718. [Google Scholar] [CrossRef]
- Peelman, N.; Ragaert, P.; De Meulenaer, B.; Adons, D.; Peeters, R.; Cardon, L.; Van Impe, F.; Devlieghere, F. Application of bioplastics for food packaging. Trends Food Sci. Technol. 2013, 32, 128–141. [Google Scholar] [CrossRef]
- Shankar, S.; Rhim, J.-W. Bionanocomposite films for food packaging applications. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Rhim, J.-W.; Ng, P.K.W. Natural biopolymer-based nanocomposite films for packaging applications. Crit. Rev. Food Sci. Nutr. 2007, 47, 411–433. [Google Scholar] [CrossRef]
- Cha, D.S.; Chinnan, M.S. Biopolymer-based antimicrobial packaging: A review. Crit. Rev. Food Sci. Nutr. 2004, 44, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.L. Introduction to biopolymers from renewable resources. In Biopolymers from Renewable Resources; Kaplan, D.L., Ed.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 1–29. [Google Scholar]
- Khwaldia, K.; Perez, C.; Banon, S.; Desobry, S.; Hardy, J. Milk proteins for edible films and coatings. Crit. Rev. Food Sci. Nutr. 2004, 44, 239–251. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Agriculture, E.R.S. Available online: https://www.ers.usda.gov/data-products/ (accessed on 18 September 2022).
- Mitchell, H.H.; Zimmerman, R.L.; Hamilton, T.S. Determination of the amount of connective tissue in meat. J. Anim. Sci. 1927, 1927, 257. [Google Scholar] [CrossRef]
- Domenek, S.; Feuilloley, P.; Gratraud, J.; Morel, M.H.; Guilbert, S. Biodegradability of wheat gluten based bioplastics. Chemosphere 2004, 54, 551–559. [Google Scholar] [CrossRef]
- Wang, J.; Chen, P.; Wang, D.; Shannon, G.; Zeng, A.; Orazaly, M.; Wu, C. Identification and mapping of stable QTL for protein content in soybean seeds. Mol. Breed. 2015, 35, 1–10. [Google Scholar] [CrossRef]
- Rouf Shah, T.; Prasad, K.; Kumar, P. Maize—A potential source of human nutrition and health: A review. Cogent Food Agric. 2016, 2, 1166995. [Google Scholar] [CrossRef]
- Robertson, T.M.; Alzaabi, A.Z.; Robertson, M.D.; Fielding, B.A. Starchy carbohydrates in a healthy diet: The role of the humble potato. Nutrients 2018, 10, 1764. [Google Scholar] [CrossRef]
- Muñoz, I.; Rodríguez, C.; Gillet, D.; Moerschbacher, B.M. Life cycle assessment of chitosan production in India and Europe. Int. J. Life Cycle Assess. 2018, 23, 1151–1160. [Google Scholar] [CrossRef]
- Lowther, A.; Liddel, M.; Yencho, M. Fisheries of the United States 2018: Current Fishery Statistics no. 2018; National Oceanic and Atmospheric Administration: Silver Spring, MD, USA, 2020.
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Langholtz, M.; Stokes, B.; Eaton, L. 2016 Billion-ton report: Advancing domestic resources for a thriving bioeconomy (executive summary). Ind. Biotechnol. 2016, 12, 282–289. [Google Scholar] [CrossRef]
- Chen, H. Functional properties and applications of edible films made of milk proteins. J. Dairy Sci. 1995, 78, 2563–2583. [Google Scholar] [CrossRef]
- Dalgleish, D. Milk proteins. Chemistry and physics. In Food Proteins; Fox, P.F., Condon, J.J., Eds.; Applied Science Publishers: London, UK, 1982; pp. 155–178. [Google Scholar]
- Metzger, W. Method of Producing Casein Film. U.S. Patent No. 5,681,517, 28 October 1997. [Google Scholar]
- Patel, S. Emerging trends in nutraceutical applications of whey protein and its derivatives. J. Food Sci. Technol. 2015, 52, 6847–6858. [Google Scholar] [CrossRef]
- Horne, D.S. Casein structure, self-assembly and gelation. Curr. Opin. Colloid Interface Sci. 2002, 7, 456–461. [Google Scholar] [CrossRef]
- Sothornvit, R.; Krochta, J.M. Plasticizer effect on mechanical properties of β-lactoglobulin films. J. Food Eng. 2001, 50, 149–155. [Google Scholar] [CrossRef]
- Hristov, P.; Mitkov, I.; Sirakova, D.; Mehandgiiski, I.; Radoslavov, G. Measurement of casein micelle size in raw dairy cattle milk by dynamic light scattering. In Milk Proteins–From Structure to Biological Properties and Health Aspects; Intech: Rijeka, Croatia, 2016. [Google Scholar]
- Sibilla, S.; Godfrey, M.; Brewer, S.; Budh-Raja, A.; Genovese, L. An overview of the beneficial effects of hydrolysed collagen as a nutraceutical on skin properties: Scientific background and clinical studies. Open Nutraceuticals J. 2015, 8, 29–42. [Google Scholar] [CrossRef]
- Schmidt, M.M.; Dornelles, R.C.P.; Mello, R.O.; Kubota, E.H.; Mazutti, M.A.; Kempka, A.P.; Demiate, I.M. Collagen extraction process. Int. Food Res. J. 2016, 23, 913–922. [Google Scholar]
- Bigi, A.; Panzavolta, S.; Rubini, K. Relationship between triple-helix content and mechanical properties of gelatin films. Biomaterials 2004, 25, 5675–5680. [Google Scholar] [CrossRef]
- Etxabide, A.; Leceta, I.; Cabezudo, S.; Guerrero, P.; de la Caba, K. Sustainable fish gelatin films: From food processing waste to compost. ACS Sustain. Chem. Eng. 2016, 4, 4626–4634. [Google Scholar] [CrossRef]
- Ramos, M.; Valdés, A.; Beltran, A.; Garrigós, M.C. Gelatin-based films and coatings for food packaging applications. Coatings 2016, 6, 41. [Google Scholar] [CrossRef]
- Dash, R.; Mukherjee, S.; Kundu, S. Isolation, purification and characterization of silk protein sericin from cocoon peduncles of tropical tasar silkworm, Antheraea mylitta. Int. J. Biol. Macromol. 2006, 38, 255–258. [Google Scholar] [CrossRef]
- Sothornvit, R.; Chollakup, R.; Potjanart, S. Extracted sericin from silk waste for film formation. Songklanakarin J. Sci. Technol. 2010, 32, 17–22. [Google Scholar]
- Zhang, Y.-Q. Applications of natural silk protein sericin in biomaterials. Biotechnol. Adv. 2002, 20, 91–100. [Google Scholar] [CrossRef]
- Yildirim, M.; Hettiarachchy, N.S. Properties of films produced by cross-linking whey proteins and 11s globulin using transglutaminase. J. Food Sci. 1998, 63, 248–252. [Google Scholar] [CrossRef]
- Motoki, M.; Aso, H.; Seguro, K.; Nio, N. ALPHA.s1-Casein film prepared using transglutaminase. Agric. Biol. Chem. 1987, 51, 993–996. [Google Scholar] [CrossRef]
- Ghosh, A.; Ali, M.A.; Dias, G.J. Effect of cross-linking on microstructure and physical performance of casein protein. Biomacromolecules 2009, 10, 1681–1688. [Google Scholar] [CrossRef]
- McHugh, T.H.; Krochta, J.M. Sorbitol- vs glycerol-plasticized whey protein edible films: Integrated oxygen permeability and tensile property evaluation. J. Agric. Food Chem. 1994, 42, 841–845. [Google Scholar] [CrossRef]
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119. [Google Scholar] [CrossRef]
- Shewry, P.R.; Tatham, A.S.; Forde, J.; Kreis, M.; Miflin, B.J. The classification and nomenclature of wheat gluten proteins: A reassessment. J. Cereal Sci. 1986, 4, 97–106. [Google Scholar] [CrossRef]
- Mojumdar, S.C.; Moresoli, C.; Simon, L.C.; Legge, R.L. Edible wheat gluten (WG) protein films. J. Therm. Anal. Calorim. 2011, 104, 929–936. [Google Scholar] [CrossRef]
- Micard, V.; Belamri, R.; Morel, M.H.; Guilbert, S. Properties of chemically and physically treated wheat gluten films. J. Agric. Food Chem. 2000, 48, 2948–2953. [Google Scholar] [CrossRef]
- Ma, C.Y. Soybean|soy concentrates and isolates. In Reference Module in Food Science; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Wang, Y.; Padua, G.W. Nanoscale characterization of zein self-assembly. Langmuir 2012, 28, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Paraman, I.; Lamsal, B.P. Recovery and characterization of α-zein from corn fermentation coproducts. J. Agric. Food Chem. 2011, 59, 3071–3077. [Google Scholar] [CrossRef] [PubMed]
- Parris, N.; Dickey, L.C. Extraction and solubility characteristics of zein proteins from dry-milled corn. J. Agric. Food Chem. 2001, 49, 3757–3760. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Muñoz, P.; Kanavouras, A.; Ng, P.K.; Gavara, R. Development and characterization of biodegradable films made from wheat gluten protein fractions. J. Agric. Food Chem. 2003, 51, 7647–7654. [Google Scholar] [CrossRef] [PubMed]
- Rhim, J.-W.; Lee, J.H.; Ng, P.K.W. Mechanical and barrier properties of biodegradable soy protein isolate-based films coated with polylactic acid. LWT—Food Sci. Technol. 2007, 40, 232–238. [Google Scholar] [CrossRef]
- Shi, K.; Yu, H.; Lakshmana Rao, S.; Lee, T.C. Improved mechanical property and water resistance of zein films by plasticization with tributyl citrate. J. Agric. Food Chem. 2012, 60, 5988–5993. [Google Scholar] [CrossRef]
- Kurita, K. Chitin and Chitosan: Functional biopolymers from marine crustaceans. Mar. Biotechnol. 2006, 8, 203. [Google Scholar] [CrossRef]
- Kabalak, M.; Aracagök, D.; Torun, M. Extraction, characterization and comparison of chitins from large bodied four Coleoptera and Orthoptera species. Int. J. Biol. Macromol. 2019, 145, 402–409. [Google Scholar] [CrossRef]
- Aranday-García, R.; Saimoto, H.; Shirai, K.; Ifuku, S. Chitin biological extraction from shrimp wastes and its fibrillation for elastic nanofiber sheets preparation. Carbohydr. Polym. 2019, 213, 112–120. [Google Scholar] [CrossRef]
- Agbaje, O.B.A.; Shir, I.B.; Zax, D.B.; Schmidt, A.; Jacob, D.E. Biomacromolecules within bivalve shells: Is chitin abundant? Acta Biomater. 2018, 80, 176–187. [Google Scholar] [CrossRef]
- King, C.; Shamshina, J.L.; Gurau, G.; Berton, P.; Khan, N.F.A.F.; Rogers, R.D. A platform for more sustainable chitin films from an ionic liquid process. Green Chem. 2016, 19, 117–126. [Google Scholar] [CrossRef]
- Madeleine-Perdrillat, C.; Karbowiak, T.; Raya, J.; Gougeon, R.; Bodart, P.R.; Debeaufort, F. Water-induced local ordering of chitosan polymer chains in thin layer films. Carbohydr. Polym. 2015, 118, 107–114. [Google Scholar] [CrossRef]
- Ma, X.; Qiao, C.; Wang, X.; Yao, J.; Xu, J. Structural characterization and properties of polyols plasticized chitosan films. Int. J. Biol. Macromol. 2019, 135, 240–245. [Google Scholar] [CrossRef]
- Guerrero, P.; Muxika, A.; Zarandona, I.; De La Caba, K. Crosslinking of chitosan films processed by compression molding. Carbohydr. Polym. 2019, 206, 820–826. [Google Scholar] [CrossRef]
- Lörcks, J. Properties and applications of compostable starch-based plastic material. Polym. Degrad. Stab. 1998, 59, 245–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Rempel, C.; McLaren, D. Chapter 16—Thermoplastic starch. In Innovations in Food Packaging, 2nd ed.; Han, J.H., Ed.; Academic Press: San Diego, CA, USA, 2014; pp. 391–412. [Google Scholar]
- Talja, R.A.; Helén, H.; Roos, Y.H.; Jouppila, K. Effect of various polyols and polyol contents on physical and mechanical properties of potato starch-based films. Carbohydr. Polym. 2007, 67, 288–295. [Google Scholar] [CrossRef]
- Jimenez, A.; Fabra, M.J.; Talens, P.; Chiralt, A. Edible and biodegradable starch films: A review. Food Bioprocess Technol. 2012, 5, 2058–2076. [Google Scholar] [CrossRef]
- O’Sullivan, A.C. Cellulose: The structure slowly unravels. Cellulose 1997, 4, 173–207. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Habibi, Y.; Rojas, O.J.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films from wood pulps: Mechanical processing and physical properties. Bioresour. Technol. 2010, 101, 5961–5968. [Google Scholar] [CrossRef]
- Zhao, Y.; Moser, C.; Lindström, M.E.; Henriksson, G.; Li, J. Cellulose nanofibers from softwood, hardwood, and tunicate: Preparation–structure–film performance interrelation. ACS Appl. Mater. Interfaces 2017, 9, 13508–13519. [Google Scholar] [CrossRef]
- Saucedo-Pompa, S.; Rojas-Molina, R.; Aguilera-Carbó, A.F.; Saenz-Galindo, A.; de La Garza, H.; Jasso-Cantú, D.; Aguilar, C.N. Edible film based on candelilla wax to improve the shelf life and quality of avocado. Food Res. Int. 2009, 42, 511–515. [Google Scholar] [CrossRef]
- Donhowe, G.; Fennema, O. Water vapor and oxygen permeability of wax films. J. Am. Oil Chem. Soc. 1993, 70, 867–873. [Google Scholar] [CrossRef]
- Melvin, C.; Jewell, E.; de Vooys, A.; Lammers, K.; Murray, N.M. Surface and adhesion characteristics of current and next generation steel packaging materials. J. Packag. Technol. Res. 2018, 2, 93–103. [Google Scholar] [CrossRef]
- Grassino, A.; Pezzani, A.; Squitieri, G. Characterisation of different types of lacquers used in food packaging: Lacquer adhesion tests. Acta Aliment. —Acta Aliment. 2007, 36, 27–37. [Google Scholar]
- Allman, A.; Jewell, E.; de Vooys, A.; Hayes, R. Inter-layer adhesion performance of steel packaging materials for food cans under retort conditions. J. Packag. Technol. Res. 2018, 2, 115–124. [Google Scholar] [CrossRef]
- Zuo, H.; Cao, Z.; Shu, J.; Xu, D.; Zhong, J.; Zhao, J.; Wang, T.; Chen, Y.; Gao, F.; Shen, L. Effect of structure on the properties of ambient-cured coating films prepared via a Michael addition reaction based on an acetoacetate-modified castor oil prepared by thiol-ene coupling. Prog. Org. Coat. 2019, 135, 27–33. [Google Scholar] [CrossRef]
- Ruiz, M.M.; Schroeder, W.F.; Hoppe, C.E. The use of a fatty acid/β-Hydroxyester blend to enhance the surface hydrophilicity of crosslinked poly(ethylene glycol) coatings. Prog. Org. Coat. 2019, 135, 313–320. [Google Scholar] [CrossRef]
- Swain, S.K.; Pattanayak, A.J.; Sahoo, A.P. Functional biopolymer composites. In Functional Biopolymers; Thakur, V.K., Thakur, M.K., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 159–182. [Google Scholar]
- Vinson, J.R.; Sierakowski, R.L. Strength and failure theories, In The Behavior of Structures Composed of Composite Materials; Vinson, J.R., Sierakowski, R.L., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 303–332. [Google Scholar]
- Wagner, H.D. Reinforcement. In Encyclopedia of Polymer Science and Technology, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar] [CrossRef]
- Simari, C.; Potsi, G.; Policicchio, A.; Perrotta, I.; Nicotera, I. Clay–carbon nanotubes hybrid materials for nanocomposite membranes: Advantages of branched structure for proton transport under low humidity conditions in PEMFCs. J. Phys. Chem. C 2016, 120, 2574–2584. [Google Scholar] [CrossRef]
- Gu, D.; Meng, G.; Li, C.; Meiners, W.; Poprawe, R. Selective laser melting of TiC/Ti bulk nanocomposites: Influence of nanoscale reinforcement. Scr. Mater. 2012, 67, 185–188. [Google Scholar] [CrossRef]
- Goettler, L.A.; Lee, K.Y.; Thakkar, H. Layered silicate reinforced polymer nanocomposites: Development and applications. Polym. Rev. 2007, 47, 291–317. [Google Scholar] [CrossRef]
- Ci, L.; Bai, J. Novel micro/nanoscale hybrid reinforcement: Multiwalled carbon nanotubes on SiC particles. Adv. Mater. 2004, 16, 2021–2024. [Google Scholar] [CrossRef]
- Sandler, J.; Werner, P.; Shaffer, M.S.; Demchuk, V.; Altstädt, V.; Windle, A.H. Carbon-nanofibre-reinforced poly(ether ether ketone) composites. Compos. Part A Appl. Sci. Manuf. 2002, 33, 1033–1039. [Google Scholar] [CrossRef]
- Sukyai, P.; Anongjanya, P.; Bunyahwuthakul, N.; Kongsin, K.; Harnkarnsujarit, N.; Sukatta, U.; Sothornvit, R.; Chollakup, R.J.F.R.I. Effect of cellulose nanocrystals from sugarcane bagasse on whey protein isolate-based films. Food Res. Int. 2018, 107, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Qazanfarzadeh, Z.; Kadivar, M. Properties of whey protein isolate nanocomposite films reinforced with nanocellulose isolated from oat husk. Int. J. Biol. Macromol. 2016, 91, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Pereda, M.; Amica, G.; Rácz, I.; Marcovich, N.E. Structure and properties of nanocomposite films based on sodium caseinate and nanocellulose fibers. J. Food Eng. 2011, 103, 76–83. [Google Scholar] [CrossRef]
- Mondragon, G.; Peña-Rodriguez, C.; González, A.; Eceiza, A.; Arbelaiz, A. Bionanocomposites based on gelatin matrix and nanocellulose. Eur. Polym. J. 2015, 62, 1–9. [Google Scholar] [CrossRef]
- Kwak, H.W.; Lee, H.; Lee, M.E.; Jin, H.J. Facile and green fabrication of silk sericin films reinforced with bamboo-derived cellulose nanofibrils. J. Clean. Prod. 2018, 200, 1034–1042. [Google Scholar] [CrossRef]
- Rafieian, F.; Shahedi, M.; Keramat, J.; Simonsen, J. Mechanical, thermal and barrier properties of nano-biocomposite based on gluten and carboxylated cellulose nanocrystals. Ind. Crops Prod. 2014, 53, 282–288. [Google Scholar] [CrossRef]
- Han, Y.; Yu, M.; Wang, L. Soy protein isolate nanocomposites reinforced with nanocellulose isolated from licorice residue: Water sensitivity and mechanical strength. Ind. Crops Prod. 2018, 117, 252–259. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, X.; Zhang, L. Effects of cellulose whiskers on properties of soy protein thermoplastics. Macromol. Biosci. 2006, 6, 524–531. [Google Scholar] [CrossRef]
- Azeredo, H.M.; Mattoso, L.H.C.; Avena-Bustillos, R.J.; Filho, G.C.; Munford, M.L.; Wood, D.; McHugh, T.H. Nanocellulose reinforced chitosan composite films as affected by nanofiller loading and plasticizer content. J. Food Sci. 2010, 75, N1–N7. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, J.; Zhang, L. Structure and properties of the nanocomposite films of chitosan reinforced with cellulose whiskers. J. Polym. Sci. Part B- Polym. Phys. 2009, 47, 1069–1077. [Google Scholar] [CrossRef]
- Savadekar, N.R.; Mhaske, S.T. Synthesis of nano cellulose fibers and effect on thermoplastics starch based films. Carbohydr. Polym. 2012, 89, 146–151. [Google Scholar] [CrossRef]
- Li, M.; Tian, X.; Jin, R.; Li, D. Preparation and characterization of nanocomposite films containing starch and cellulose nanofibers. Ind. Crops Prod. 2018, 123, 654–660. [Google Scholar] [CrossRef]
- Lu, Y.; Weng, L.; Cao, X. Biocomposites of plasticized starch reinforced with cellulose crystallites from cottonseed linter. Macromol. Biosci. 2005, 5, 1101–1107. [Google Scholar] [CrossRef]
- Khattab, A.; Liu, C.; Chirdon, W.; Hebert, C. Mechanical and thermal characterization of carbon nanofiber reinforced low-density polyethylene composites. J. Thermoplast. Compos. Mater. 2013, 26, 954–967. [Google Scholar] [CrossRef]
- Khoo, H.H.; Tan, R.B.H.; Chng, K.W.L. Environmental impacts of conventional plastic and bio-based carrier bags. Int. J. Life Cycle Assess. 2010, 15, 284–293. [Google Scholar] [CrossRef]
- Miller, S.A.; Landis, A.E.; Theis, T.L. Feature: Environmental trade-offs of biobased production. Environ. Sci. Technol. 2007, 41, 5176–5182. [Google Scholar] [CrossRef]
- Finnveden, G.; Moberg, Å. Environmental systems analysis tools—an overview. J. Clean. Prod. 2005, 13, 1165–1173. [Google Scholar] [CrossRef]
- Hottle, T.A.; Bilec, M.M.; Landis, A.E. Sustainability assessments of bio-based polymers. Polym. Degrad. Stab. 2013, 98, 1898–1907. [Google Scholar] [CrossRef]
- Li, Q.; McGinnis, S.; Wong, A.; Renneckar, S. Nanocellulose life cycle assessment. ACS Sustain. Chem. Eng. 2013, 1, 919–928. [Google Scholar] [CrossRef]
- Arvidsson, R.; Nguyen, D.; Svanström, M. Life cycle assessment of cellulose nanofibrils production by mechanical treatment and two different pretreatment processes. Environ. Sci. Technol. 2015, 49, 6881–6890. [Google Scholar] [CrossRef] [PubMed]
- Piccinno, F.; Hischier, R.; Seeger, S.; Som, C. Life cycle assessment of a new technology to extract, functionalize and orient cellulose nanofibers from food waste. ACS Sustain. Chem. Eng. 2015, 3, 1047–1055. [Google Scholar] [CrossRef]
- de Figueirêdo, M.C.B.; de Freitas Rosa, M.; Ugaya, C.M.L.; de Souza, M.D.S.M.; da Silva Braid, A.C.C.; de Melo, L.F.L. Life cycle assessment of cellulose nanowhiskers. J. Clean. Prod. 2012, 35, 130–139. [Google Scholar] [CrossRef]
- Gu, H.; Reiner, R.; Bergman, R.; Rudie, A. LCA study for pilot scale production of cellulose nano crystals (CNC) from wood pulp. In Proceedings of the LCA XV Conference, Vancouver, BC, Canada, 6–8 October 2015. [Google Scholar]
- Khannaa, V.; Bakshi, B.R.; Lee, L.J. Life cycle energy analysis and environmental life cycle assessment of carbon nanofibers production. In Proceedings of the 2007 IEEE International Symposium on Electronics and the Environment, Orlando, FL, USA, 7–10 May 2007; IEEE: Piscataway, NJ, USA, 2007. [Google Scholar]
- Dinkel, F.; Pohl, C.; Ros, M.; Waldeck, B. Ökobilanz Stärkehaltiger Kunststoffe (Nr. 271); B. Study prepared by Carbotech, for the Bundesamt für Umwelt und Landschaft (BUWAL); Federal Office for the Environment: Bern, Switzerland, 1996. [Google Scholar]
- Patel, M.; Bastioli, C.; Marini, L.; Würdinger, E. Environmental Assessment of Bio-Based Polymers and Natural Fibers; Ultrecht University: Ultrecht, The Netherlands, 2002. [Google Scholar]
- Augustine, R.; Rajendran, R.; Cvelbar, U.; Mozetič, M.; George, A. Biopolymers for Health, Food, and Cosmetic Applications. In Handbook of Biopolymer-Based Materials; Wiley: Weinheim, Germany, 2013; pp. 801–849. [Google Scholar]
- Baranwal, J.; Barse, B.; Fais, A.; Delogu, G.L.; Kumar, A. Biopolymer: A Sustainable Material for Food and Medical Applications. Polymers 2022, 14, 983. [Google Scholar] [CrossRef]
- Cencha, L.G.; Allasia, M.; Ronco, L.I.; Luque, G.C.; Picchio, M.L.; Minari, R.J.; Gugliotta, L.M. Proteins as Promising Biobased Building Blocks for Preparing Functional Hybrid Protein/Synthetic Polymer Nanoparticles. Ind. Eng. Chem. Res. 2021, 60, 4745–4765. [Google Scholar] [CrossRef]
- Vieira, M.G.A.; da Silva, M.A.; dos Santos, L.O.; Beppu, M.M. Natural-based plasticizers and biopolymer films: A review. Eur. Polym. J. 2011, 47, 254–263. [Google Scholar] [CrossRef]
- Božič, M.; Vivod, V.; Kavčič, S.; Leitgeb, M.; Kokol, V. New findings about the lipase acetylation of nanofibrillated cellulose using acetic anhydride as acyl donor. Carbohydr. Polym. 2015, 125, 340–351. [Google Scholar] [CrossRef]
- Huang, F.; Wu, X.; Yu, Y.; Lu, Y.; Chen, Q. Acylation of cellulose nanocrystals with acids/trifluoroacetic anhydride and properties of films from esters of CNCs. Carbohydr. Polym. 2017, 155, 525–534. [Google Scholar] [CrossRef]
- Trinh, B.M.; Mekonnen, T. Hydrophobic esterification of cellulose nanocrystals for epoxy reinforcement. Polymer 2018, 155, 64–74. [Google Scholar] [CrossRef]
- Shrestha, S.; Chowdhury, R.A.; Toomey, M.D.; Betancourt, D.; Montes, F.; Youngblood, J.P. Surface hydrophobization of TEMPO-oxidized cellulose nanofibrils (CNFs) using a facile, aqueous modification process and its effect on properties of epoxy nanocomposites. Cellulose 2019, 26, 9631–9643. [Google Scholar] [CrossRef]
- Mah, A.H.; Laws, T.; Li, W.; Mei, H.; Brown, C.C.; Ievlev, A.; Kumar, R.; Verduzco, R.; Stein, G.E. Entropic and Enthalpic Effects in Thin Film Blends of Homopolymers and Bottlebrush Polymers. Macromolecules 2019, 52, 1526–1535. [Google Scholar] [CrossRef]
- Mei, H.; Laws, T.S.; Mahalik, J.P.; Li, J.; Mah, A.H.; Terlier, T.; Bonnesen, P.; Uhrig, D.; Kumar, R.; Stein, G.E.; et al. Entropy and Enthalpy Mediated Segregation of Bottlebrush Copolymers to Interfaces. Macromolecules 2019, 52, 8910–8922. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, N.-H.; Peri, S.; Foster, M.D.; Majkrzak, C.F.; Hu, R.; Wu, D.T. Surface segregation driven by molecular architecture asymmetry in polymer blends. Phys. Rev. Lett. 2014, 113, 225702. [Google Scholar] [CrossRef]
- Miyagi, K.; Mei, H.; Terlier, T.; Stein, G.E.; Verduzco, R. Analysis of Surface Segregation of Bottlebrush Polymer Additives in Thin Film Blends with Attractive Intermolecular Interactions. Macromolecules 2020, 53, 6720–6730. [Google Scholar] [CrossRef]
- Ponnusamy, P.G.; Sundaram, J.; Mani, S. Preparation and characterization of citric acid crosslinked chitosan-cellulose nanofibrils composite films for packaging applications. J. Appl. Polym. Sci. 2022, 139, 52017. [Google Scholar] [CrossRef]
- Udoetok, I.A.; Wilson, L.D.; Headley, J.V. Self-Assembled and Cross-Linked Animal and Plant-Based Polysaccharides: Chitosan–Cellulose Composites and Their Anion Uptake Properties. ACS Appl. Mater. Interfaces 2016, 8, 33197–33209. [Google Scholar] [CrossRef] [PubMed]
- Falamarzpour, P.; Behzad, T.; Zamani, A. Preparation of nanocellulose reinforced chitosan films, cross-linked by adipic acid. Int. J. Mol. Sci. 2017, 18, 396. [Google Scholar] [CrossRef]
- Ponnusamy, P.G.; Sharma, S.; Mani, S. Cotton noil based cellulose microfibers reinforced polylactic acid composite films for improved water vapor and ultraviolet light barrier properties. J. Appl. Polym. Sci. 2022, 139, 52329. [Google Scholar] [CrossRef]
- Ponnusamy, P.G.; Mani, S. Life cycle assessment of manufacturing cellulose nanofibril-reinforced chitosan composite films for packaging applications. Int. J. Life Cycle Assess. 2022, 27, 380–394. [Google Scholar] [CrossRef]
- Technavio. Biopolymers Market by End-User, Type, and Geography—Forecast and Analysis 2021–2025. Available online: https://www.technavio.com/report/biopolymers-market-industry-analysis#utm_source=T5&utm_campaign=Media&utm_medium=BW (accessed on 18 September 2022).
- Market Data Forecast. Asia-Pacific Biopolymers Market by Type (Non-biodegradable, Biodegradable), by Country—(India, China, Japan, South Korea, Australia, New Zealand, Thailand, Malaysia, Vietnam, Philippines, Indonesia, Singapore and Rest of APAC), by End-Use Industry (Packaging, Consumer Goods, Automotive, Textiles, Agriculture)—Analysis, Size, Growth, Investment and Forecast (2022 to 2027). Available online: https://www.marketdataforecast.com/market-reports/asia-pacific-biopolymers-market (accessed on 18 September 2022).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Renewable Resources | Natural Polymer Type and Compositions | Production Volume (Million Metric Tons) | Current Use | Reference |
|---|---|---|---|---|
| Milk | Contains 33 g of protein/L. 80% casein and 20% whey protein | 97.76 | Used as a fat substitute. Butter, dry skim milk, cheese, whey, whey protein concentrate, and lactose are produced from milk. | [35,36] |
| Pork & Beef | More than 29% gelatin is available in pig skin. In beef meat, 10.6~21.9% of gelatin protein available in rib and shank | 11.91 | Used as meat. By-products such as skin, bones, and connective tissues are used to produce gelatin | [36,37] |
| Wheat | Contains 76.5% starch | 47.38 | Used for the production of food products | [36,38] |
| Soybeans | Contains 31.7 to 58.9% protein | 120.07 | Source for animal protein and vegetable oil | [36,39] |
| Corn grain | Contains about 70–72% starch | 371.10 | Source for corn meal, starch, oil, bioethanol, syrup, sugar, and feed grain | [36,40] |
| Potato | Contains 20% of potato dry matter with 60–80% of starch | 22.91 | Source for food products and starch | [36,41] |
| Crustaceans (Shrimp and Crab) | Crab shell contains 9.6% chitin and shrimp shell contains 4% chitin | 0.32 | Source for seafood and compost | [42,43] |
| Forestry biomass resources | 40~50% cellulose | 139.71 | Biofuels, wood products such as timber, lumber, etc. | [44,45] |
| Agricultural biomass resources | 25~40% cellulose | 130.64 | Source for bioenergy, biofuels, and bioproducts | [44,45] |
| Waste (Agricultural wastes, forestry wastes) | 25~50% cellulose | 61.69 | Source for compost, bioenergy | [44,45] |
| Natural Polymer | Plasticizer/ Crosslinker | TS a (MPa) | YM b (MPa) | Elongation at Break (%) | Tensile Test Conditions | WVP c × 1020 (g m h−1 kPa−1 m−2) | Reference |
|---|---|---|---|---|---|---|---|
| αs1—Casein Films | -- | 0.004 | -- | 38 | Film size 20 × 50 mm. Loading Rate—50 mm/min | -- | [62] |
| αs1—Casein Films | Transglutaminase (Enzyme) | 0.01 | -- | 75 | -- | ||
| α, β and κ—Casein Films f | -- | 52 ± 0.20 | 1107 ± 11 | 8 ± 2 | Loading rate—20 mm/min d | -- | [63] |
| α, β and κ—Casein Films g | -- | 49 ± 3 | 1391 ± 48 | 6 ± 2 | -- | ||
| α, β and κ—Casein Films h | 10% Glycerol | 36 ± 0.40 | 693 ± 38 | 25 ± 9 | -- | ||
| α, β and κ—Casein Films i | 10% Glycerol | 21 ± 0.30 | 497 ± 40 | 17 ± 1 | -- | ||
| α, β and κ—Casein Films j | 10% Glycerol | 33 ± 2 | 765 ± 109 | 15 ± 7 | -- | ||
| α, β and κ—Casein Films k | 10% Glycerol | 48 ± 2 | 1004 ± 40 | 8 ± 3 | -- | ||
| Whey Protein Films | 40% Sorbitol | 18 | 650 | 5 | Loading rate—100 mm/min d | -- | [64] |
| Whey Protein Films | 15% Glycerol | 29 | 1100 | 4 | -- | ||
| Fish Gelatin Films l | 10 wt.% glycerol | 36.52 ± 2.98 | 1.79 ± 0.54 | Sample size 4.75 × 22.25 mm e | 5.33 ± 0.16 | [56] | |
| Fish Gelatin Films m | 10 wt.% glycerol | 43.02 ± 0.52 | 2.31 ± 0.33 | 5.47 ± 0.10 | |||
| Fish Gelatin Films n | 10 wt.% glycerol | 52.36 ± 3.16 | 2.88 ± 0.68 | 6.52 ± 0.16 |
| Natural Polymer | Plasticizer/Crosslinker | TS a (MPa) | YM b (MPa) | Elongation at Break (%) | Tensile Test Conditions c | WVP d (g m h−1 kPa−1 m−2) | Reference |
|---|---|---|---|---|---|---|---|
| Glutenin-rich Wheat Gluten Film | 20% Glycerol | 5 | -- | 100 | Sample Size: 2.54 × 10 cm. Loading rate: 508 mm/min | 6.94 × 104 | [73] |
| Gliadin-rich Wheat Gluten Film | 20% Glycerol | 15 | -- | 350 | -- | 1.11 × 105 | |
| SPI Film | 50% Glycerin | 2.80 ± 0.30 | -- | 165.70 ± 15 | Film size: 2.54 × 15 cm | 9.66 × 10−9 | [74] |
| Zein Film | -- | 6.70 ± 0.37 | 409.86 ± 7.62 | 1.96 ± 0.18 | Film size: 40 × 10 × 0.47 ± 0.12 mm | 1.69 × 10−4 | [75] |
| Zein Film | 10% Tributyl Citrate | 17.80 ± 4.26 | 556.29 ± 29.42 | 4.53 ± 0.54 | 1.64 × 10−4 |
| S. No | Biomass Resource | Pretreatment Method | Main Treatment Method | LCI Resources | Life Cycle Impact Assessment | Reference |
|---|---|---|---|---|---|---|
| CNF Manufacturing Process | ||||||
| 1 | Bleached kraft wood sulfate pulp | Tempo oxidation | Sonication | Literature data/SimaPro USLCI/Ecoinvent | Method: Cumulative energy demand (CED) and Eco-Indicator 99 (EI99) | [124] |
| 2 | Bleached kraft wood sulfate pulp | Tempo oxidation | Homogenization | Literature data/SimaPro USLCI/Ecoinvent | Method: CED and EI99 | [124] |
| 3 | Bleached kraft wood sulfate pulp | Chloroacetic acid etherification | Sonication | Literature data/SimaPro USLCI/Ecoinvent | Method: CED and EI99 | [124] |
| 4 | Bleached kraft wood sulfate pulp | Chloroacetic acid etherification | Homogenization | Literature data/SimaPro USLCI/Ecoinvent | Method: CED and EI99 | [124] |
| 5 | Bleached & unbleached sulfate pulp | Enzymatic | Micro fluidizer | Process research institute, Literature & Ecoinvent database | Method: CED and ReCiPe | [125] |
| 6 | Bleached & unbleached sulfate pulp | Carboxy methylation pretreatment | Micro fluidizer | Process research institute, Literature & Ecoinvent database | Method: CED and ReCiPe | [125] |
| 7 | Bleached & unbleached sulfate pulp | No pretreatment | Homogenization | Process research institute, Literature & Ecoinvent database | Method: CED and ReCiPe | [125] |
| 8 | Carrot waste | Enzymatic | Homogenization | Laboratory scale process data & Ecoinvent | Method: CED and ReCiPe | [126] |
| CNC Manufacturing Process | ||||||
| 1 | Unripe coconut fiber | Chopping/Washing/Bleaching | Acid Hydrolysis/Dialysis | Laboratory scale process data & Ecoinvent | Method: ReCiPe | [127] |
| 2 | Cotton fiber | Chopping | Acid Hydrolysis/Dialysis | Laboratory scale process data & Ecoinvent | Method: ReCiPe | [127] |
| 3 | Bleached kraft pulp | -- | Acid Hydrolysis | US LCI and US Ecoinvent | Method: TRACI | [128] |
| Natural Polymers | Global Warming [kg CO2 eq.] | Acidification [mol. H+ eq.] | Eutrophication [mol. N eq.] | Ozone Depletion [kg CFC-11 eq.] | Water Depletion [m3] |
|---|---|---|---|---|---|
| Chitosan (Shrimp shell) | 12.2 | 0.684 | 2.82 | 7.05 × 10−6 | −0.236 |
| Chitosan (Crab shell) | 77.1 | −0.261 | 3.12 | 1.23 × 10−5 | 5.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponnusamy, P.G.; Mani, S. Material and Environmental Properties of Natural Polymers and Their Composites for Packaging Applications—A Review. Polymers 2022, 14, 4033. https://doi.org/10.3390/polym14194033
Ponnusamy PG, Mani S. Material and Environmental Properties of Natural Polymers and Their Composites for Packaging Applications—A Review. Polymers. 2022; 14(19):4033. https://doi.org/10.3390/polym14194033
Chicago/Turabian StylePonnusamy, Prabaharan Graceraj, and Sudhagar Mani. 2022. "Material and Environmental Properties of Natural Polymers and Their Composites for Packaging Applications—A Review" Polymers 14, no. 19: 4033. https://doi.org/10.3390/polym14194033
APA StylePonnusamy, P. G., & Mani, S. (2022). Material and Environmental Properties of Natural Polymers and Their Composites for Packaging Applications—A Review. Polymers, 14(19), 4033. https://doi.org/10.3390/polym14194033