
Molecular Dynamics Simulations of the Interactions between a Hydrolyzed Polyacrylamide with the Face and Edge Surfaces of Molybdenite



Abstract
:1. Introduction
2. Methodology
2.1. COSMO-RS Calculation
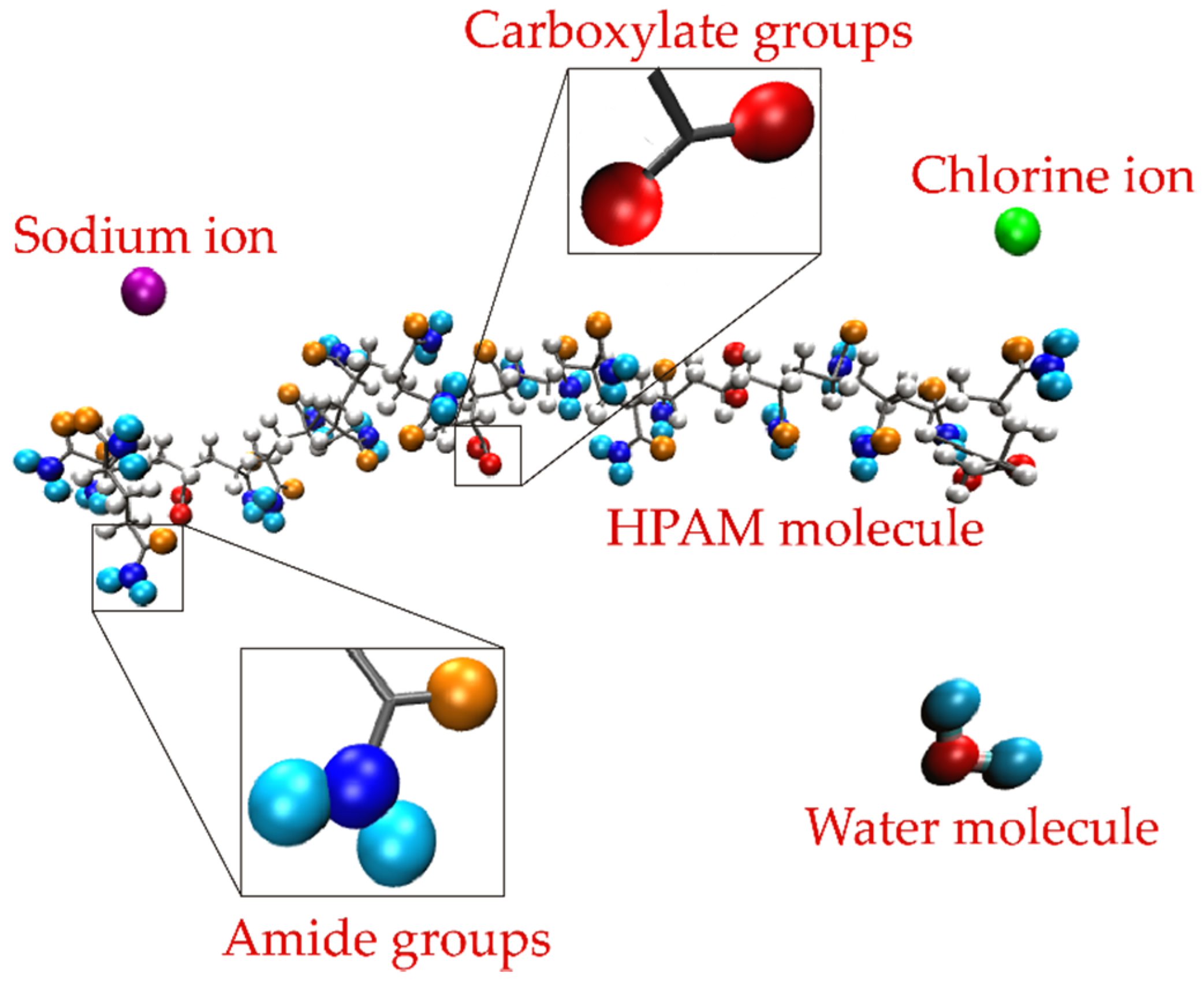
2.2. Molecular Dynamics Simulation
3. Results and Discussion
3.1. Analysis of Sigma Profile
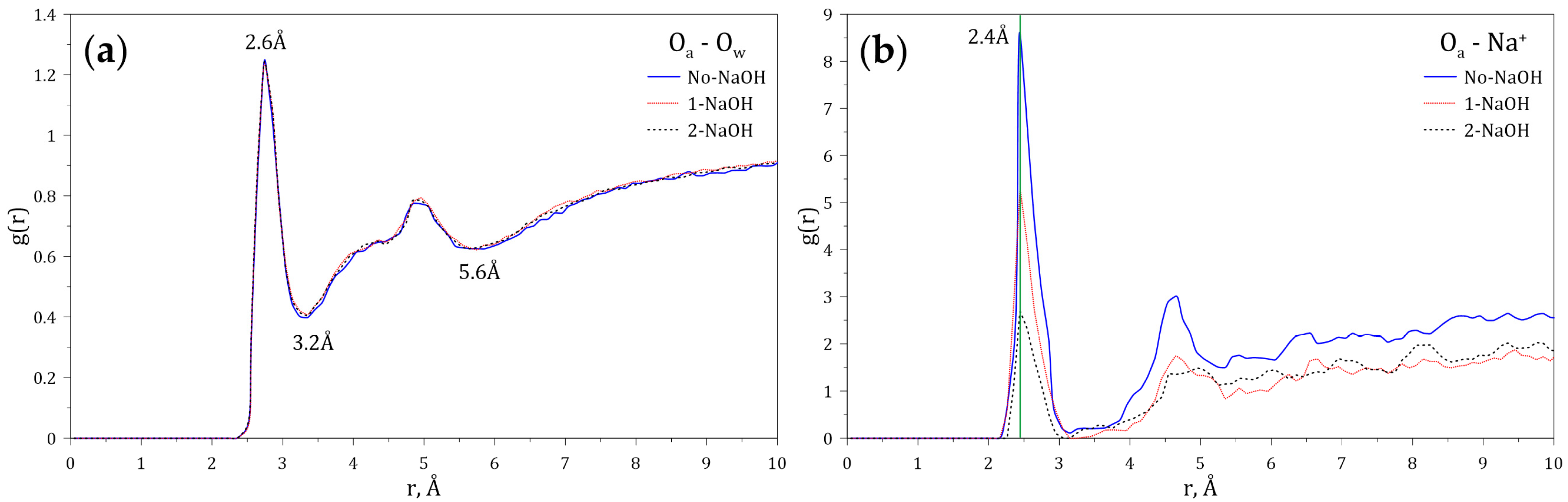
3.2. Interactions between HPAM and Aqueous Medium
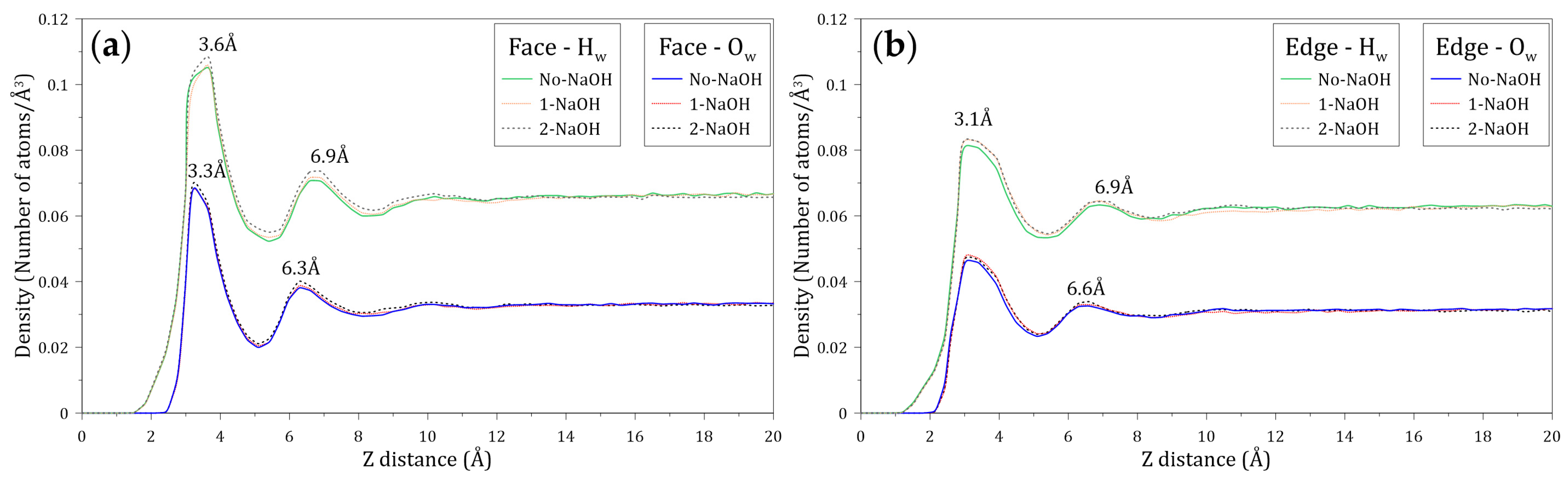
3.3. Interaction of Water with the Face and Edge Surfaces of Molybdenite
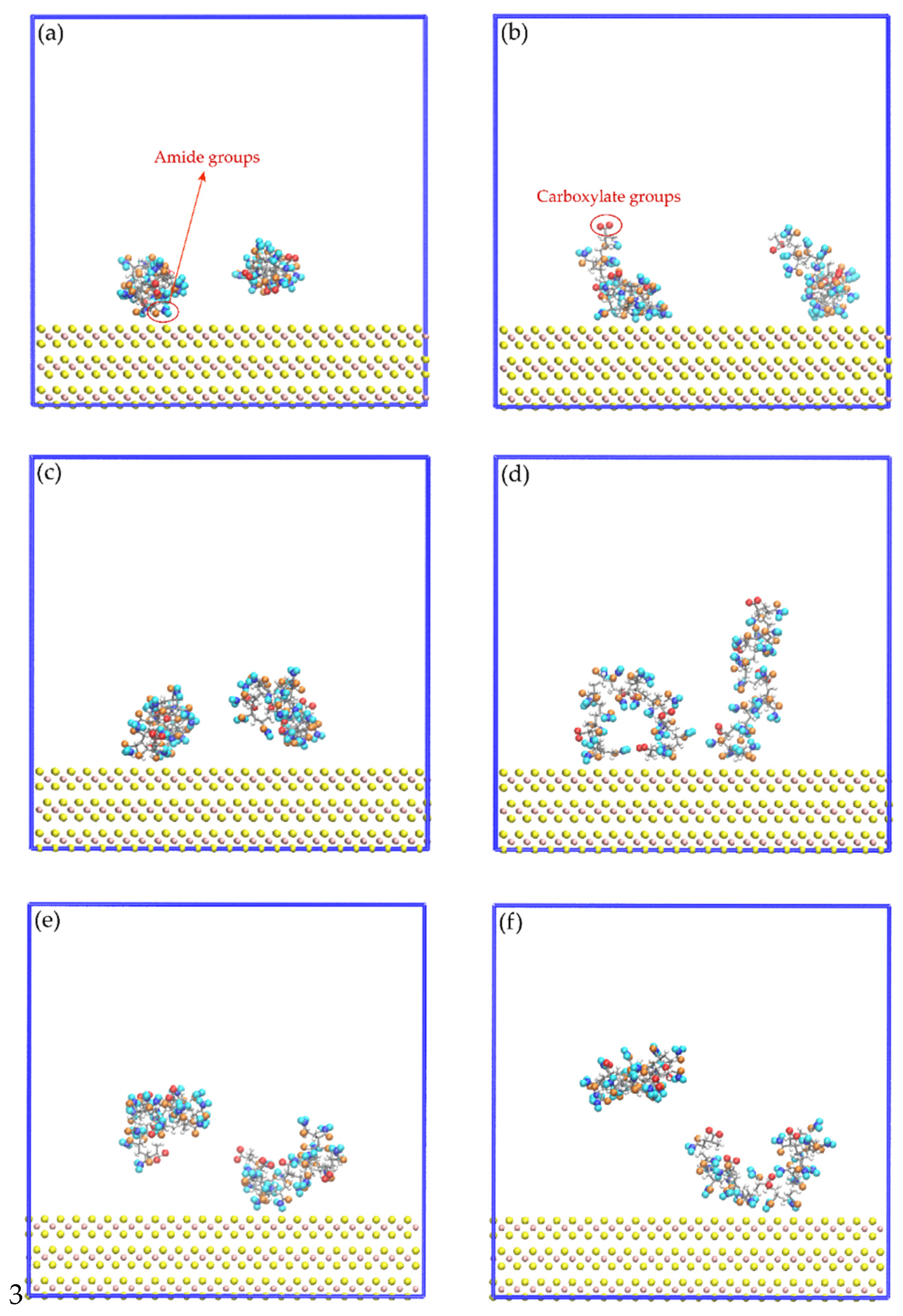
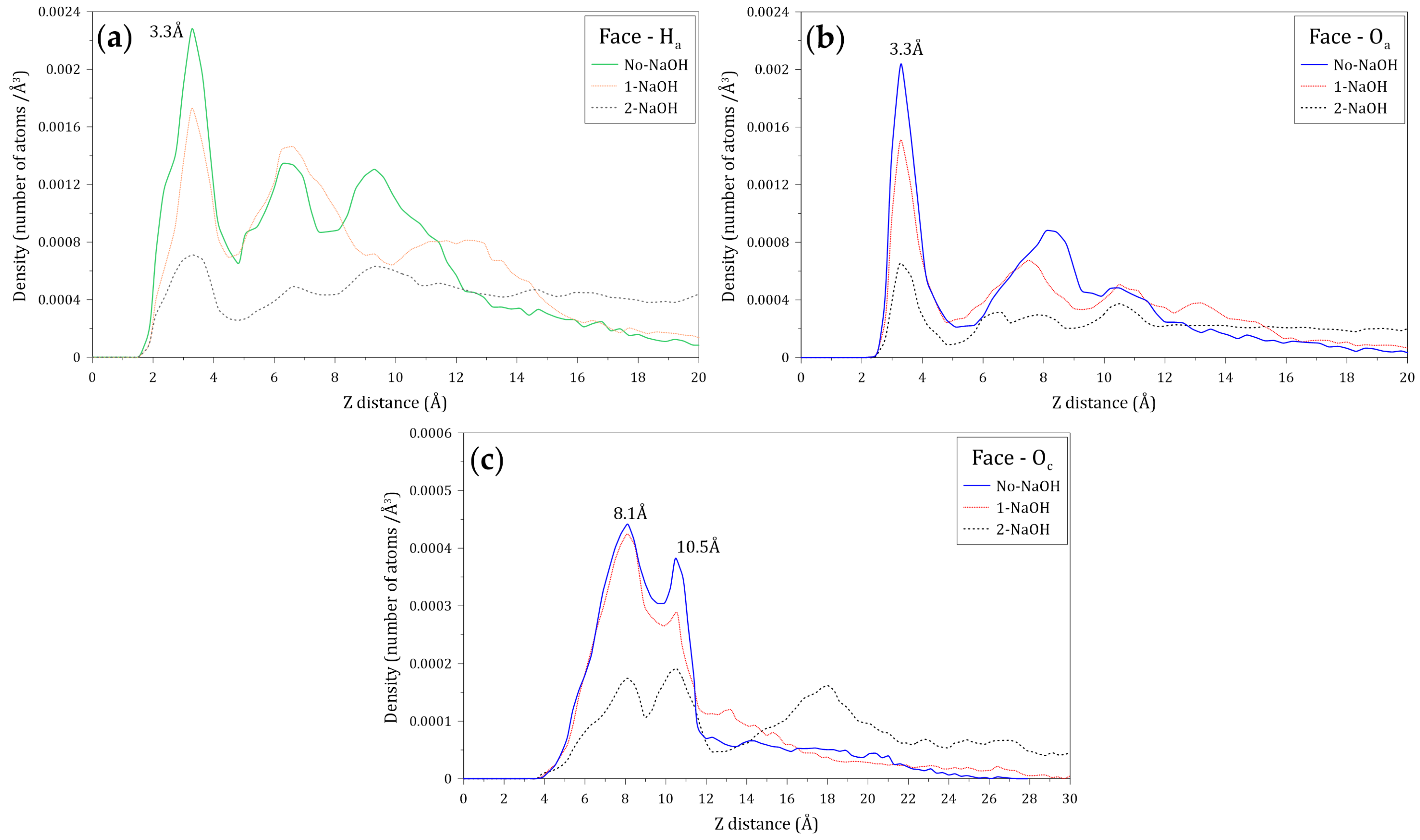
3.4. Interactions of the Amide and Carboxylate Groups of the HPAM with the Molybdenite Face
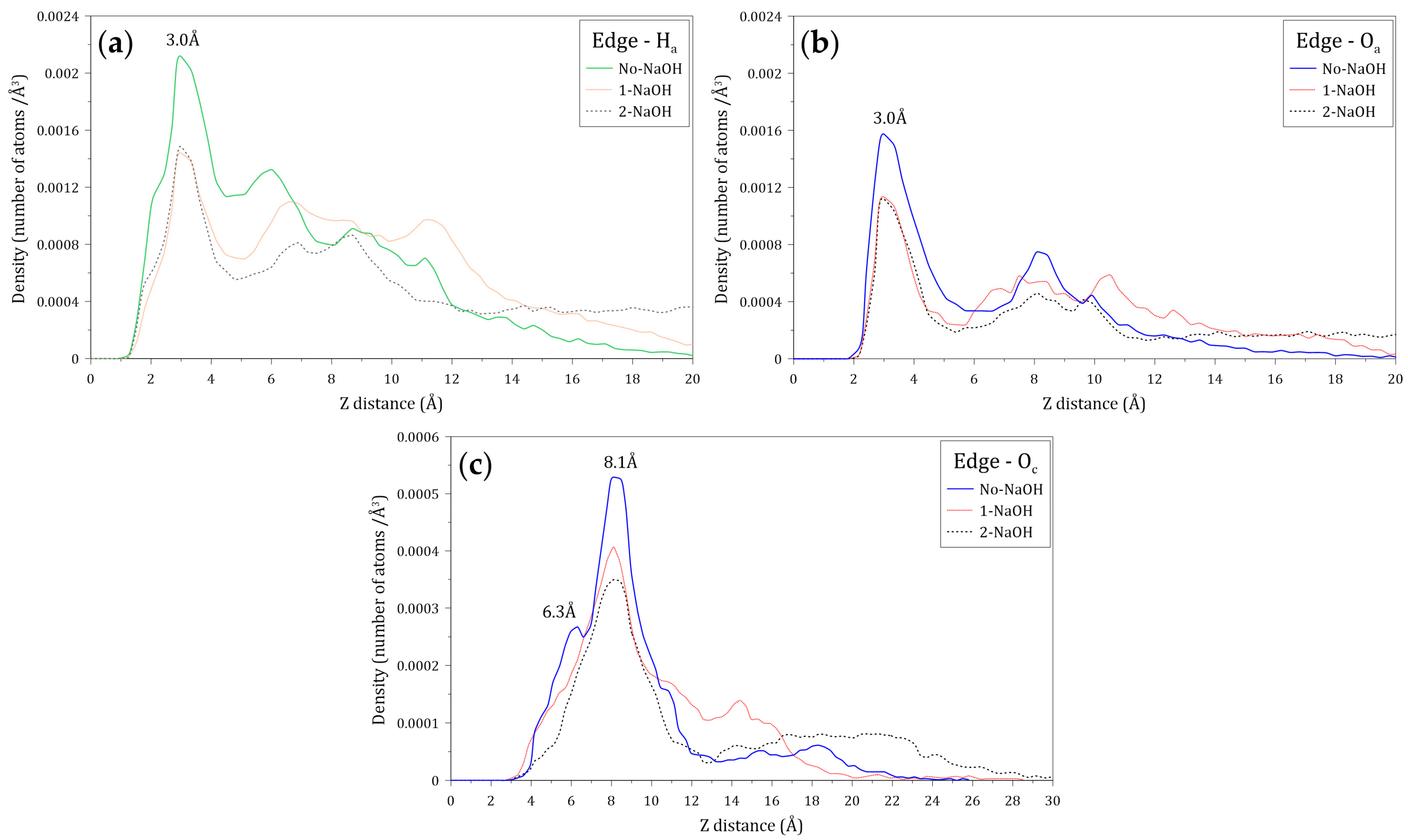
3.5. Interactions of the Amide and Carboxylate Groups of the HPAM with the Molybdenite Edge
4. Conclusions
- The results obtained from molecular dynamics simulations show that the interactions between the HPAM and molybdenite are mainly explained by the interactions of the amide group with the faces and edges of molybdenite.
- Molecular dynamics simulations show that the HPAM molecule rearranges, the amide group moves towards the molybdenite face or edge, and the carboxylate group moves away from the mineral.
- The results obtained in the simulations show that the interactions of the HPAM with the molybdenite edge are slightly stronger than the interaction of this molecule with the mineral face.
- Simulations indicate that the presence of the sodium and hydroxide ions reduces the concentration of HPAM around the face and edge surfaces. HPAM adsorption on molybdenite is expected to be affected at high pH and in salty water.
- The density profiles of the oxygen (Ow) and hydrogen (Hw) atoms of the water molecules located along the Z axis normal to the face and edge surfaces of molybdenite indicate that the water molecules tend to be closer to the edge, which confirms the hydrophilicity character of this mineral surface observed at a macroscopic level.
- The conclusions obtained through molecular dynamic simulations are in line with the results obtained in previous studies completed at a macroscopic level, which indicated that HPAMs adsorb on molybdenite particles and reduce their hydrophobicity.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kinnunen, P.; Obenaus-Emler, J.; Raatikaine, J.; Guignot, S.; Guimerà, J.; Ciroth, A.; Heiskanen, K. Review of closed water loops with ore sorting and tailings valorisation for a more sustainable mining industry. J. Clean. Prod. 2021, 278, 123237. [Google Scholar] [CrossRef]
- Oyarzún, J.; Oyarzún, R. Sustainable development threats, inter-sector conflicts and environmental policy re-quirements in the arid, mining rich, Northern Chile territory. Sustain. Dev. 2011, 19, 263–274. [Google Scholar] [CrossRef]
- Yepsen, R.; Gutierrez, L.; Laskowski, J. Flotation behavior of enargite in the process of flotation using seawater. Miner. Eng. 2019, 142, 105897. [Google Scholar] [CrossRef]
- Ramirez, A.; Gutierrez, L.; Laskowski, J. Use of “oily bubbles” and dispersants in flotation of molybdenite in fresh and seawater. Miner. Eng. 2020, 148, 106197. [Google Scholar] [CrossRef]
- Alvarez, A.; Gutierrez, L.; Laskowski, J. Use of polyethylene oxide to improve flotation of fine molybdenite. Miner. Eng. 2018, 127, 232–237. [Google Scholar] [CrossRef]
- Castro, S.; Laskowski, J.S. Depressing effect of flocculants on molybdenite flotation. Miner. Eng. 2015, 74, 13–19. [Google Scholar] [CrossRef]
- Echeverry, L.; Estrada, D.; Toledo, P.G.; Gutierrez, L. The depressing effect of an anionic polyacrylamide on molyb-denite flotation and the importance of polymer anionicity. Colloids Surf. A Physicochem. Eng. Asp. 2021, 629, 127506. [Google Scholar] [CrossRef]
- Arinaitwe, E.; Pawlik, M. A method for measuring the degree of anionicity of polyacrylamide-based flocculants. Int. J. Miner. Process. 2009, 91, 50–54. [Google Scholar] [CrossRef]
- Quezada, G.R.; Saavedra, J.H.; Rozas, R.E.; Toledo, P.G. Molecular dynamics simulations of the conformation and diffusion of partially hydrolyzed polyacrylamide in highly saline solutions. Chem. Eng. Sci. 2020, 214, 115366. [Google Scholar] [CrossRef]
- Lu, Z.; Ralston, J.; Liu, Q. Face or Edge? Control of Molybdenite Surface Interactions with Divalent Cations. J. Phys. Chem. C 2020, 124, 172–381. [Google Scholar] [CrossRef]
- Chander, S.; Fuerstenau, D.W. On the natural floatability of molybdenite. Trans. Soc. Min. Eng. AIME 1972, 252, 62–68. [Google Scholar]
- Chen, Y.; Chen, X.; Peng, Y. The effect of sodium hydrosulfide on molybdenite flotation in seawater and diluted seawater. Miner. Eng. 2020, 158, 106589. [Google Scholar] [CrossRef]
- Gutierrez, L.; Pawlik, M. Influence of humic acids on oil sand processing. Part I: Detection and quantification of humic acids in oil sand ores and their effect on bitumen wettability. Int. J. Miner. Process. 2014, 126, 117–125. [Google Scholar] [CrossRef]
- Liu, G.; Zhong, H.; Hu, Y.; Zhao, S.; Xia, L. The role of cationic polyacrylamide in the reverse flotation of diasporic bauxite. Miner. Eng. 2007, 20, 1191–1199. [Google Scholar] [CrossRef]
- Wang, K.; Wang, L.; Cao, M.; Liu, Q. Xanthation-modified polyacrylamide and spectroscopic investigation of its ad-sorption onto mineral surfaces. Miner. Eng. 2012, 39, 1–8. [Google Scholar] [CrossRef]
- Jin, J.; Miller, J.D.; Dang, L.X. Molecular dynamics simulation and analysis of interfacial water at selected sulfide mineral surfaces under anaerobic conditions. Int. J. Miner. Process. 2014, 128, 55–67. [Google Scholar] [CrossRef]
- Zhang, L.; Luan, B.; Zhou, R. Parameterization of Molybdenum Disulfide Interacting with Water Using the Free Energy Perturbation Method. J. Phys. Chem. 2019, 123, 7243–7252. [Google Scholar] [CrossRef]
- Chen, P.; Yao, L.; Liu, Y.; Luo, J.; Zhou, G.; Jiang, B. Experimental and theoretical study of dilute polyacrylamide so-lutions: Effect of salt concentration. J. Mol. Model. 2012, 18, 3153–3160. [Google Scholar] [CrossRef]
- Ojeda, R.M.; Llovell, F. Soft-SAFT Transferable Molecular Models for the Description of Gas Solubility in Eutectic Ammonium Salt-Based Solvents. J. Chem. Eng. 2018, 63, 2599–2612. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T.; Lenthe, E.V.; Louwen, J.N. An implementation of the conductor-like screening model of solvation within the amsterdam density functional package—Part II. COSMO for real solvents. Can. J. Chem. 2009, 87, 790–797. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calcu-lations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Mullins, E.; Oldland, R.; Liu, Y.A.; Wang, S.; Sandler, A.A.; Chen, C.C.; Zwolak, M.; Seavey, K. Sigma-profile database for using COSMO-based thermodynamic methods. Ind. Eng. Chem. 2006, 45, 4389–4415. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hoover, W.G. Constant-pressure equations of motion. Phys. Rev. A 1986, 34, 2499–2500. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.; Straatsma, T.P. The Missing Term in Effective Pair Potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar]
- Pengfei, L.; Kenneth, M.; Merz, J. Taking into account the ion-induced dipole interaction in the nonbonded model of ions. J. Chem. Theory Comput. 2014, 10, 289–297. [Google Scholar]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF: A full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [PubMed] [Green Version]
- Graulis, S.; Chateigner, D.; Downs, R.T.; Yokochi, A.F.T.; Quirós, M.; Lutterotti, L.; Manakova, E.; Butkus, J.; Moeck, P.; Bail, A.L. Crystallography Open Database—An open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42, 726–729. [Google Scholar]
- Phadagi, R.; Singh, S.; Hashemi, H.; Kaya, S.; Venkatesu, P.; Ramjugernath, D.; Ebenso, E.E.; Bahadur, I. Understanding the role of Dimethylformamide as co-solvents in the dissolution of cellulose in ionic liquids: Experimental and theoretical approach. J. Mol. Liq. 2021, 328, 115392. [Google Scholar]
- Vishnoi, A.; Banerjee, T.; Ghosh, P.; Musharaf Ali, S.; Shenoy, K.T. Theoretical prediction of distribution coefficients of Sr2+ in nuclear waste/ionic liquid phases using COSMO-RS model. Sep. Purif. Technol. 2014, 133, 138–148. [Google Scholar]
- Qin, Y.; Chen, X.; Wang, L.; Wei, X.; Nong, W.; Wei, X.; Liang, J. Experimental Determination and Computational Prediction of Dehydroabietic Acid Solubility in (−)-α-Pinene + (−)-β-Caryophyllene + P-Cymene System. Molecules 2022, 27, 1220. [Google Scholar]
- Yuan, R.; Li, Y.; Li, C.; Fang, H.; Wang, W. Study about how the metal cationic ions affect the properties of partially hydrolyzed hydrophobically modified polyacrylamide (HMHPAM) in aqueous solution. Colloids Surf. A Physicochem. Eng. Asp. 2013, 434, 16–24. [Google Scholar]
- Payaka, A.; Tongraar, A.; Rode, B.M. QM/MM Dynamics of CH3COO−Water Hydrogen Bonds in Aqueous Solution. J. Phys. Chem. 2010, 114, 10443–10453. [Google Scholar]
- Abraham, F.F. The interfacial density profile of a Lennard-Jones fluid in contact with a (100) Lennard-Jones wall and its relationship to idealized fluid/wall systems: A Monte Carlo simulation. J. Chem. Phys. 1978, 68, 3713–3716. [Google Scholar]
- Yu, C.J.; Richter, A.G.; Datta, A.; Durbin, M.K.; Dutta, P. Observation of molecular layering in thin liquid films using X-ray reflectivity. Phys. Rev. Lett. 1999, 82, 2326–2329. [Google Scholar]
- Song, S.; Zhang, X.; Yang, B.; Lopez-Mendoza, A. Flotation of molybdenite fines as hydrophobic agglomerates. Sep. Purif. Technol. 2012, 98, 451–455. [Google Scholar]
- Castro, S.; Lopez-Valdivieso, A.; Laskowski, J.S. Review of the flotation of molybdenite. Part I: Surface properties and floatability. Int. J. Miner. Process. 2016, 148, 48–58. [Google Scholar]
- He, T.; Li, H.; Jin, J.; Peng, Y.; Wang, Y.; Wan, H. Improving fine molybdenite flotation using a combination of aliphatic hydrocarbon oil and polycyclic aromatic hydrocarbon. Results Phys. 2019, 12, 1050–1055. [Google Scholar]
- Dang, L.X.; Montgomery Pettitt, B. A theoretical study of like ion pairs in solution. J. Phys. Chem. 1990, 94, 4303–4308. [Google Scholar]
- Lee, S.H. Molecular dynamics simulation of ion mobility. 2. alkali metal and halide ions using the spc/e model for water at 25 °C. J. Phys Chem. 1996, 100, 1420–1425. [Google Scholar]
- Lynden-Bell, R.M.; Rasaiah, J.C. From hydrophobic to hydrophilic behaviour: A simulation study of solvation entropy and free energy of simple solutes. J. Chem. Phys. 1997, 107, 1981. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Atom | ɛ (Kcal/mol) | rm (Å) | Mulliken Charge (q) |
|---|---|---|---|---|
| Molybdenite (MoS2) | Mo | 0.056 | 3.056 | 0.458 |
| S | 0.274 | 4.035 | −0.229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Echeverry-Vargas, L.; Estrada, D.; Gutierrez, L. Molecular Dynamics Simulations of the Interactions between a Hydrolyzed Polyacrylamide with the Face and Edge Surfaces of Molybdenite. Polymers 2022, 14, 3680. https://doi.org/10.3390/polym14173680
Echeverry-Vargas L, Estrada D, Gutierrez L. Molecular Dynamics Simulations of the Interactions between a Hydrolyzed Polyacrylamide with the Face and Edge Surfaces of Molybdenite. Polymers. 2022; 14(17):3680. https://doi.org/10.3390/polym14173680
Chicago/Turabian StyleEcheverry-Vargas, Luver, Darwin Estrada, and Leopoldo Gutierrez. 2022. "Molecular Dynamics Simulations of the Interactions between a Hydrolyzed Polyacrylamide with the Face and Edge Surfaces of Molybdenite" Polymers 14, no. 17: 3680. https://doi.org/10.3390/polym14173680
APA StyleEcheverry-Vargas, L., Estrada, D., & Gutierrez, L. (2022). Molecular Dynamics Simulations of the Interactions between a Hydrolyzed Polyacrylamide with the Face and Edge Surfaces of Molybdenite. Polymers, 14(17), 3680. https://doi.org/10.3390/polym14173680