3.1. Characterization of the Original Lignins Using FTIR
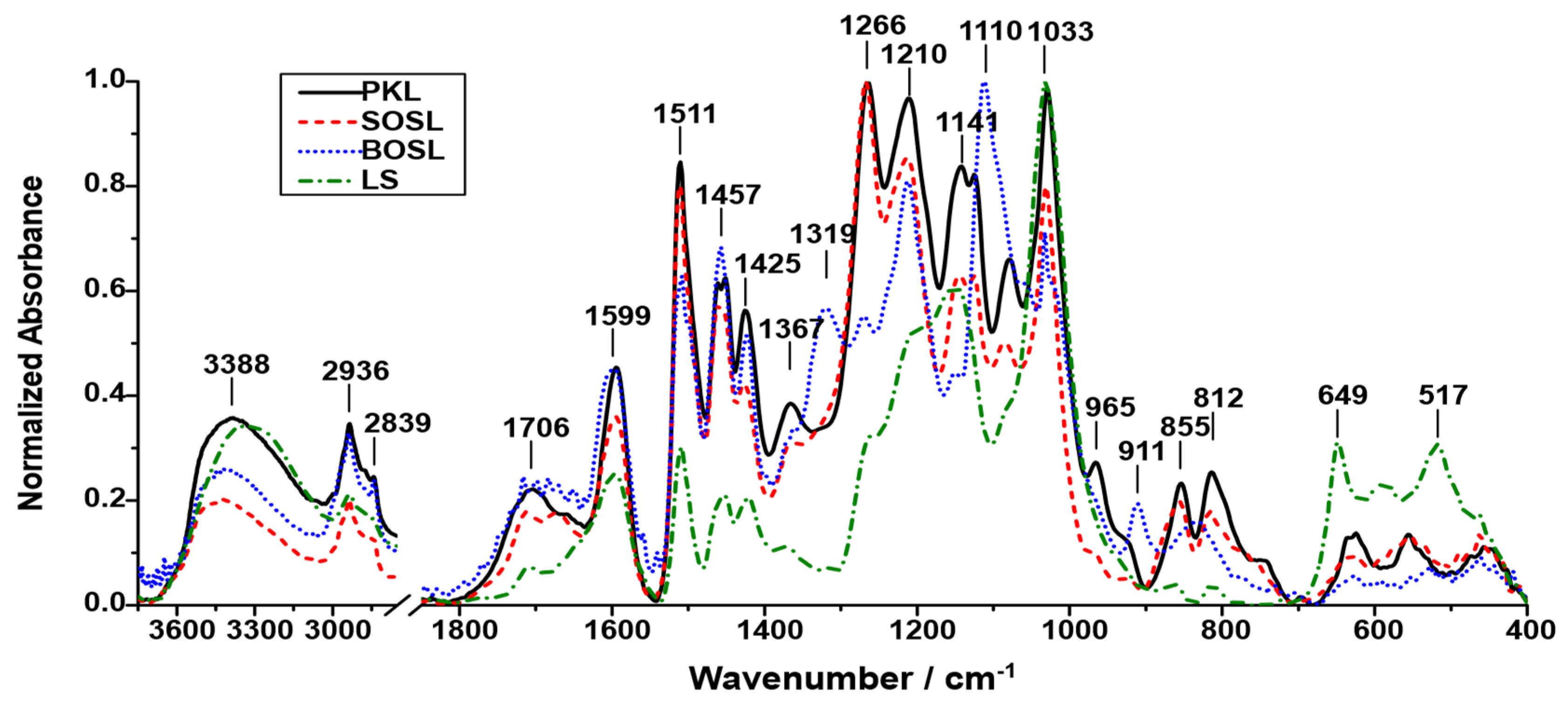
To obtain information regarding the chemical composition of the four lignins, they were analyzed using FTIR (
Figure 2). The signals detected for the lignins occurred at similar wavenumbers; however, the relative intensity varied depending on the lignin type.
The proposed assignments of the signals are listed in
Table 1.
The FTIR results provide insight into the chemical composition of the lignin; however, they must be examined with care. Due to the high complexity of the compound and the resulting high variety of functional groups, the signals overlapped, and a true baseline could not be determined. Thus, the real intensity of the signals could not be determined, and the results should only be considered for observing tendencies and not precise quantities. Despite this, some conclusions may be obtained from the FTIR spectra.
While the positions of the signals were roughly similar among the different lignins, the intensity varied, indicating that the abundance of the different functional groups producing the signals depends on the lignin source. The LS was the lignin that differed the most from the other lignins. In the spectrum recorded for LS, the highest intensity signal was 1033 cm−1. It could originate from aromatic in-plane deformation of G-type lignin or C=O stretching in lignin. As the LS was extracted from softwood, it is likely that G-type building blocks were the most abundant ones, making this the most probable signal source. Other pronounced signals for the LS were at 1141, 1511, and 1599 cm−1, originating from C-H in-plane deformation or phenolic OH, aromatic skeletal vibrations (G- > S-type lignin), and aromatic skeletal vibrations (S- > G-type lignin), respectively. Additionally, the broad signal at 3388 cm−1 originating from O-H stretching had a high intensity. This can be explained by the hydrophilicity of LS, which led to more water molecules being enclosed by the lignin. The signal at 649 cm−1 represents the sulfonate groups, which are characteristic of LS. The signals originating from the other functional groups were less pronounced than for the other lignins.
The BOSL is the only hardwood lignin examined in this study. Hence, signals originating from functional groups of S-type lignin have higher intensities compared to the other softwood lignins, which can be seen, for example, by the most pronounced signal at 1110 cm−1, originating from the C-H in-plane deformation of S-type lignin. However, there were also signals indicating a high number of G-type lignin structures such as the signal at 1210 cm−1 (C-C and C-O stretching in G-type lignin) or at 1511 cm−1 (aromatic skeletal vibrations G- > S-type lignin).
Compared to the spectrum of BOSL, the spectrum of SOSL had less significant signals at 1110 and 1457 cm−1. Strong signals were observed at wavenumbers of 1266, 1210, 1511, and 1033 cm−1. The signals at 1210 and 1511 cm−1, as well as one of two potential assignments of the signal at 1033 cm−1, were more typical for G-type lignin, which was to be expected considering the softwood origin of the SOSL. The highest signal at 1266 cm−1 was produced by C=O stretching vibrations in the lignin structures.
In the spectrum of the PKL, most of the signals with high intensity were similar to those of the SOSL. The main differences were that for the PKL, almost all signals had a higher intensity relative to the highest peak than for the SOSL. Considering that the spectrum depicts the normalized absorbance, the reason for this is that the abundance of the different functional groups was more uniform than in the SOSL, resulting in not one clear maximum signal upon normalization but various more similar ones.
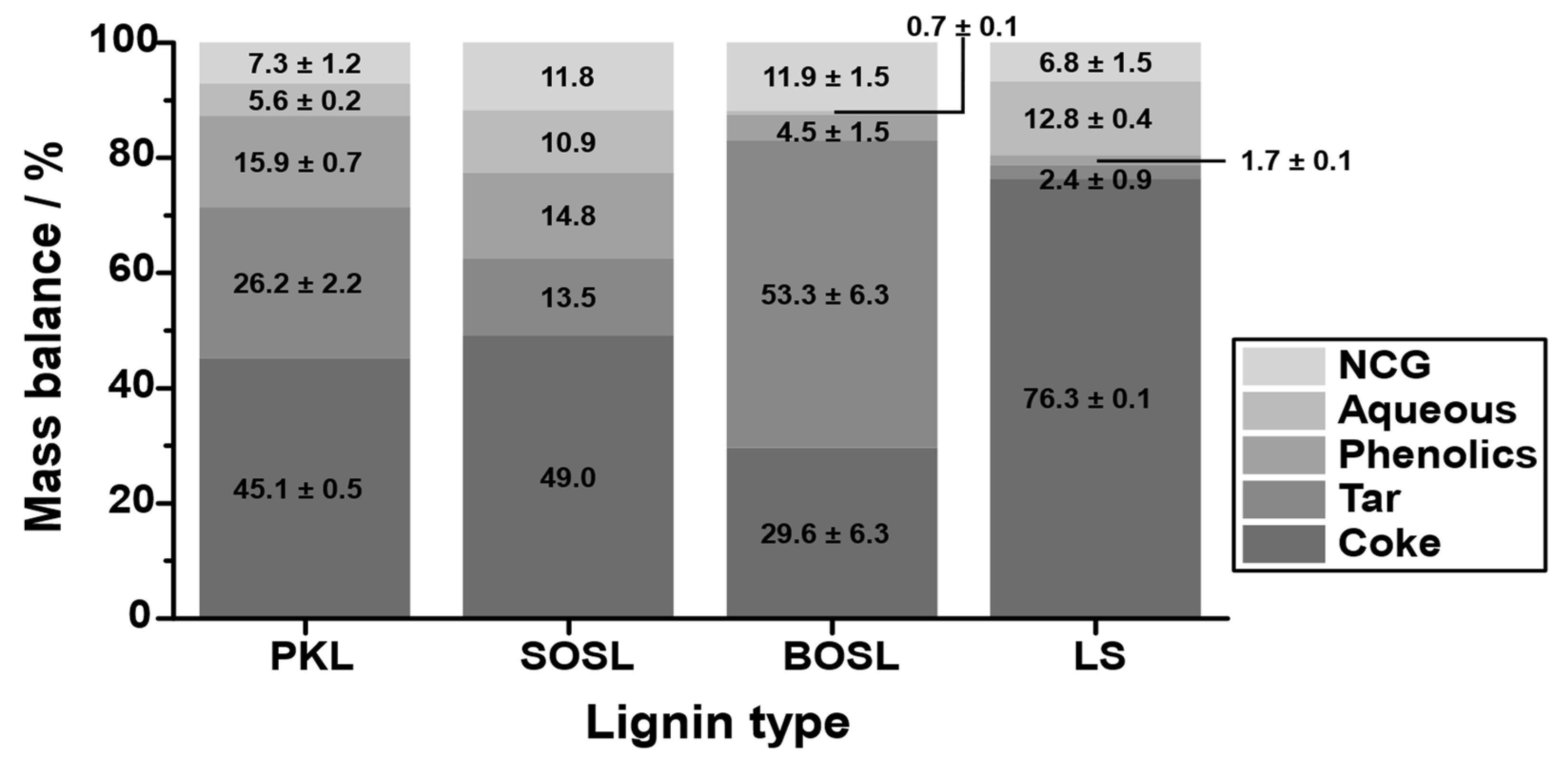
3.4. Influence of the Lignin Origin on the Products
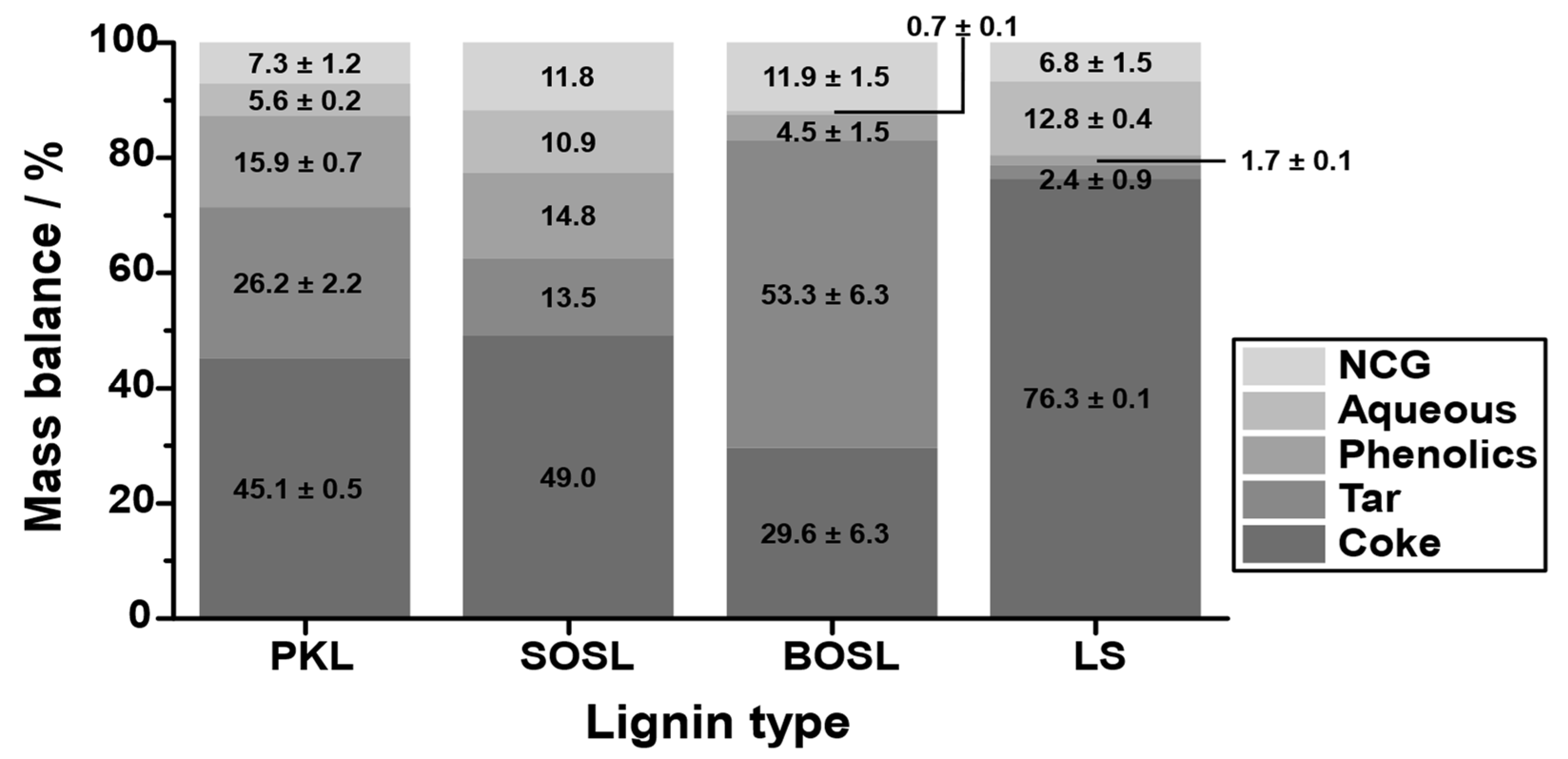
The products were separated into an aqueous phase, a light organic (phenolic) phase, a noncondensable gas (NCG) phase, a heavy organic (tar-like) phase, and coke. The quantity of the different fractions varied significantly among the different lignins (
Figure 5). While the coke yield of the PKL and the SOSL was similar at 45.1–49.0% and that of the BOSL was lower (29.6%), much more coke was obtained during the pyrolysis of the LS (76.3%). This higher coke yield was mainly compensated for by lower phenolics and tar production—together only approximately 4%. These yields were by far the lowest among the investigated technical lignins. Only 1.7% phenolics were obtained during the pyrolysis of LS. The phenolic fraction of the BOSL was slightly larger at 4.5%; however, it was still much lower than those of the PKL and SOSL, which were at 15.9% and 14.8%, respectively. The aqueous fraction of the LS was the largest together with the SOSL (12.8% and 10.9%), while the BOSL pyrolysis produced almost no aqueous products (0.7%).
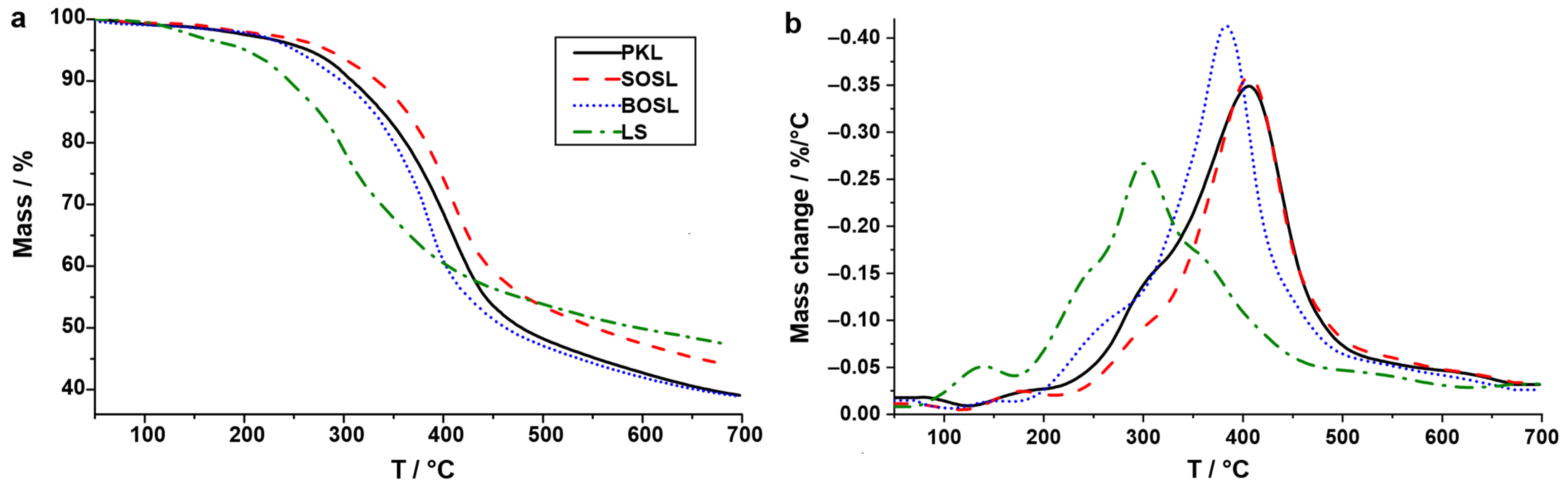
The decomposition yields differed significantly from those obtained by TGA, especially considering the low temperature applied. This underlines the differences in the heating mechanism and their influence on the cleavage mechanism [
10].
Comparing the three softwood lignins, an important factor on the mass balance could be the molecular mass. Kraft and organosolv lignins usually have a significantly lower molecular mass than lignosulfonates [
4]. For smaller molecules, fewer bonds have to be cleaved to obtain phenolic chemicals that can be obtained as condensates. This could explain the high amount of coke for the LS as well as the higher phenolic yields for the BOSL, SOSL, and PKL. However, the phenolic yield of the BOSL was much lower than that of the PKL and SOSL. This indicates that the molecular mass was only one of many influences on the obtained products. Due to the hardwood origin of the BOSL, the structure of it was different to the SOSL, which was probably the reason for the different mass balances. In general, due to the lower reactivity of the S-type lignin, the natural hardwood lignins were more linear than the softwood lignins and contained fewer C-C bonds [
6]. However, the characteristics of hardwood and softwood lignins after organosolv treatment depend on the treatment conditions and can vary significantly [
28]. Thus, it is difficult to generally compare softwood and hardwood organosolv lignins. Considering the obtained results, the main difference between the organosolv lignins was that for the BOSL, a much larger tar fraction was obtained instead of the phenolics and aqueous phases.
The aqueous or water-soluble fraction was obtained in different yields depending on the lignin type. To determine the remaining organic molecules in the aqueous fraction, the fractions were analyzed using GC-FID. However, the detected quantity of carbon-containing molecules was negligible for all lignins. Comparing the aqueous yields with the mass loss up to 200 °C measured with TGA indicated that, especially for the LS and SOSL, the aqueous fraction was, to a large degree, not just bound water but originated from chemical cleavage. In the TGA, only approximately 2.0% or 3.7% (SOSL and LS, respectively) of mass loss by water at lower temperatures was detected, while the aqueous fraction in these cases comprised more than 10% of the mass balance. From the lignins that were used in this research, only the LS was water soluble. It has more hydrophilic groups, and it could be that despite the drying step before pyrolysis, bound water was still present and later on found in the aqueous fraction. Additional mass in the aqueous phase probably originated from the cleavage of hydroxy groups or other water-soluble functional groups. A reason for the higher yield in the aqueous phase for LS could be that fewer monomers were cleaved and obtained as condensates. Thus, more functional groups were still present in the reactor at higher temperatures, allowing further cleavage of small molecules, which are soluble in water. Varying yields among the remaining lignins were probably due to the chemical structure of the lignins.
There are several factors that can influence the NCG yield. Because the gases were not collected in the experimental setup used herein, direct analysis was not possible. Due to the vacuum pump system, portions of water were sucked into the pumping system and afterwards condensed only at the exit. This water was also considered as part of the NCG fraction. Further analysis of the chemical composition of the gaseous fraction was not carried out; however, a previous study found that most of the gases obtained during lignin microwave-assisted pyrolysis at lower temperatures were CO
2, CO, CH
4, and H
2, with CO
2 being, by far, the main product [
16].
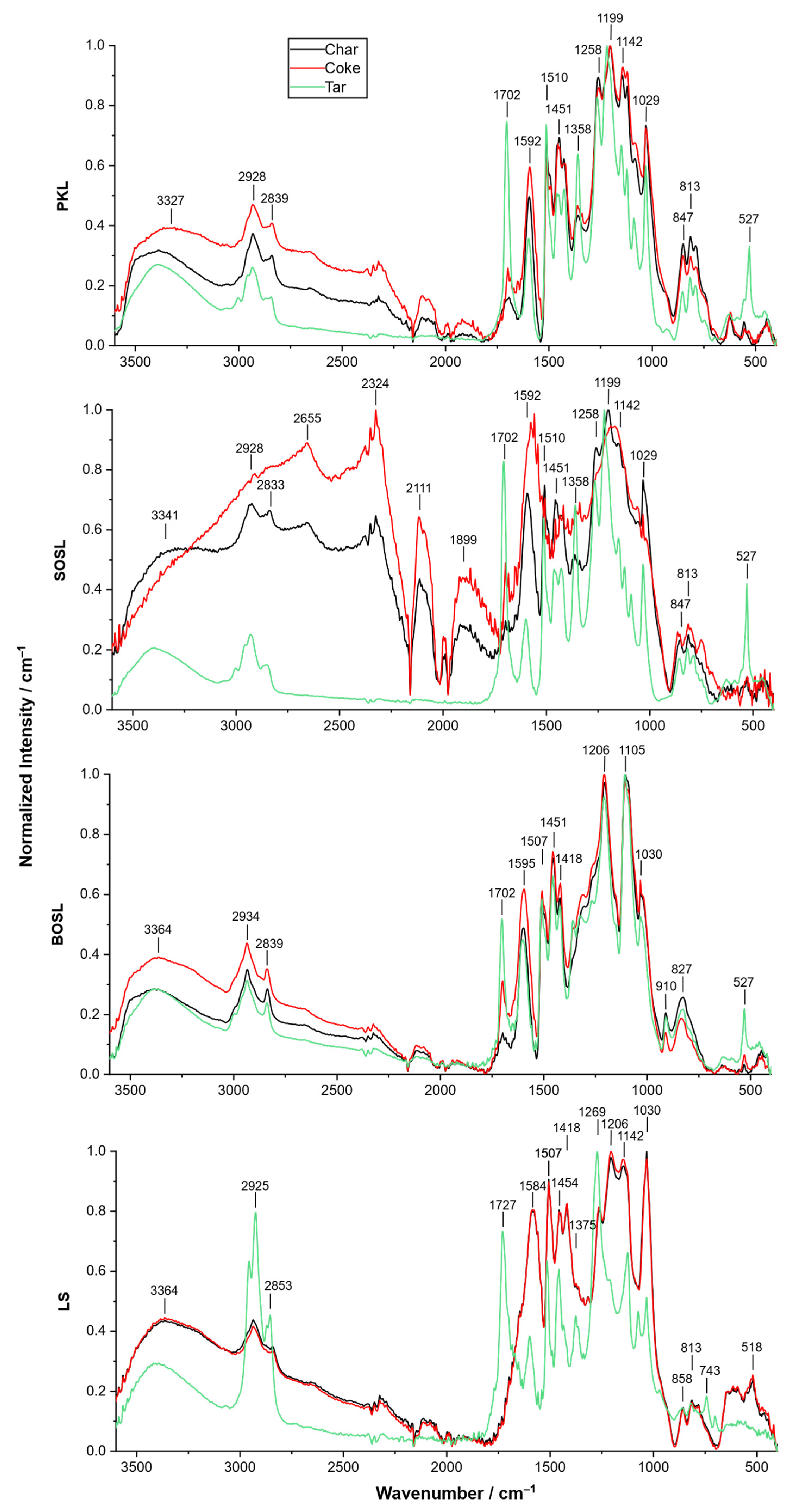
As mentioned above, pyrolysis of the BOSL produced significantly larger amounts of tar compared to the other lignin types. To explain these results, the chemical nature of the chars and cokes have to be discussed in further detail. For this, the FTIR spectra of the char, coke, and tar of the different lignins were recorded (
Figure 6). The considerations regarding the results obtained with FTIR concerning the signal overlap due to the high number of different functional groups described earlier was also valid for these results.
The spectrum of the SOSL coke differed from the spectra of the coke of the other lignins in a way that different signals were pronounced. Especially, those between 2650 and 1590 cm−1 had a high normalized intensity.
In all spectra of the cokes and chars, signals occurred at similar wavenumbers, indicating that the chemical structures of the different chars and cokes resembled each other. However, the intensity of the signals varied, outlining that the functional groups were differently abundant due to the differences in their chemical structures. Only for the LS, were the spectra of the char and the coke almost identical. For all the lignins, the normalized signals at higher wavenumbers of 3600–1800 cm−1 were more pronounced for the cokes than for the chars. Similarly, the intensities at 1580–1600 cm−1 were comparably higher for the cokes.
Meanwhile, comparing the spectra of the chars and cokes to those of the tars indicated more differences between the structures. For the PKL, SOSL, and BOSL, the normalized signals between 3600 and 1800 cm−1 were more pronounced for the cokes and chars than for the tars. This was not the case for the tar of LS, where strong signals occurred at 2925 and 2853 cm−1. However, the other signals at higher wavelengths were also less pronounced for the tar of LS compared to the coke and char. All of the tars had a pronounced signal at approximately 1702 cm−1, possibly originating from C=O groups. The tars of the PKL, SOSL, and BOSL produced an additional strong signal at approximately 527 cm−1, which could not be assigned.
Proposed assignments of the signals are listed in
Table 2.
The char and coke of LS had almost identical spectra. This can be explained by the observation that only small amounts of tar could be extracted from the char. Thus, the extraction did not change the chemical composition of the char significantly, and the materials were almost identical. The small amount of tar that was extracted, however, had a highly different structure than the tar and coke. It contained a higher proportional content of CH3 or CH2 (2940–1830 cm−1) as well as C=O groups (1727 cm−1). In general, the ratio of aromatic and functional groups was higher than OH groups, which were cleaved by the pyrolysis as indicated by the lower signal at 3364 cm−1.
For the PKL and BOSL, the normalized spectra of the cokes had a higher intensity at higher wavenumbers than those of the chars, indicating that the ratio of functional groups in the cokes after acetone extraction changed. The signals at lower wavenumbers mainly originated from the aromatic structures in lignin, such as guaiacol structures, and were more pronounced for the chars. The signals at higher wavenumbers, which originated from hydroxy groups or water and unspecific methyl or methylene rests had a higher normalized intensity in the cokes than in the chars. Thus, mainly aromatic groups were extracted from the char, leading to a lower relative intensity in the coke. This is in line with the spectra obtained from the tars, which had a higher relative intensity at lower wavelengths, an indication of a high number of aromatic groups in the tars. The high viscosity of the tars provides additional information on the extracted compounds. The monomeric aromatic compounds won as bio-oil are often liquids with lower viscosity. Thus, the extracted tar contained aromatic structures that were not cleaved completely to the monomers but remained as oligomers or small polymers, explaining the higher viscosity. These molecules had boiling points that were too high for evaporation during the pyrolysis process but they could be extracted by acetone.
The spectra of the char and coke of the SOSL were different compared to each other, as high-intensity signals for the coke were found at 2650, 2321, 2115, and 1909 cm−1. The signals at wavenumbers of 1650 and 1909 cm−1 could not be assigned; in the literature on FTIR of lignin, they were not described as typically occurring signals. The overall signal intensity of the coke of the SOSL was lower than for the other measurements, indicating a large decrease in functional groups measured by FTIR. This can be derived from the highly normalized intensity of atmospheric carbon monoxide and carbon dioxide (2115 and 2321 cm−1, respectively) compared to the signals of the material itself. Hence, artifacts and false positives could be a larger factor, and further conclusions from the FTIR of the tar cannot be drawn. As for the other lignins, the spectrum of the tar indicates a high amount of functional and aromatic groups in the extracted product.
As mentioned above, the tar yield of the BOSL was much higher than that of the other lignins. The reason for this could be that unlike the SOSL and PKL, no porous larger network was formed during the pyrolysis of the BOSL. Thus, the final pyrolysis products had lower molecular sizes and were more soluble in acetone. The reason for the lower number of newly formed bonds could be the lower reactivity of the S-type lignin, which is more common in hardwood.
The low yield of phenolic condensates (6.0%) leads to the conclusion that a large part of the remaining oligomers could not be further cleaved under the given parameters.
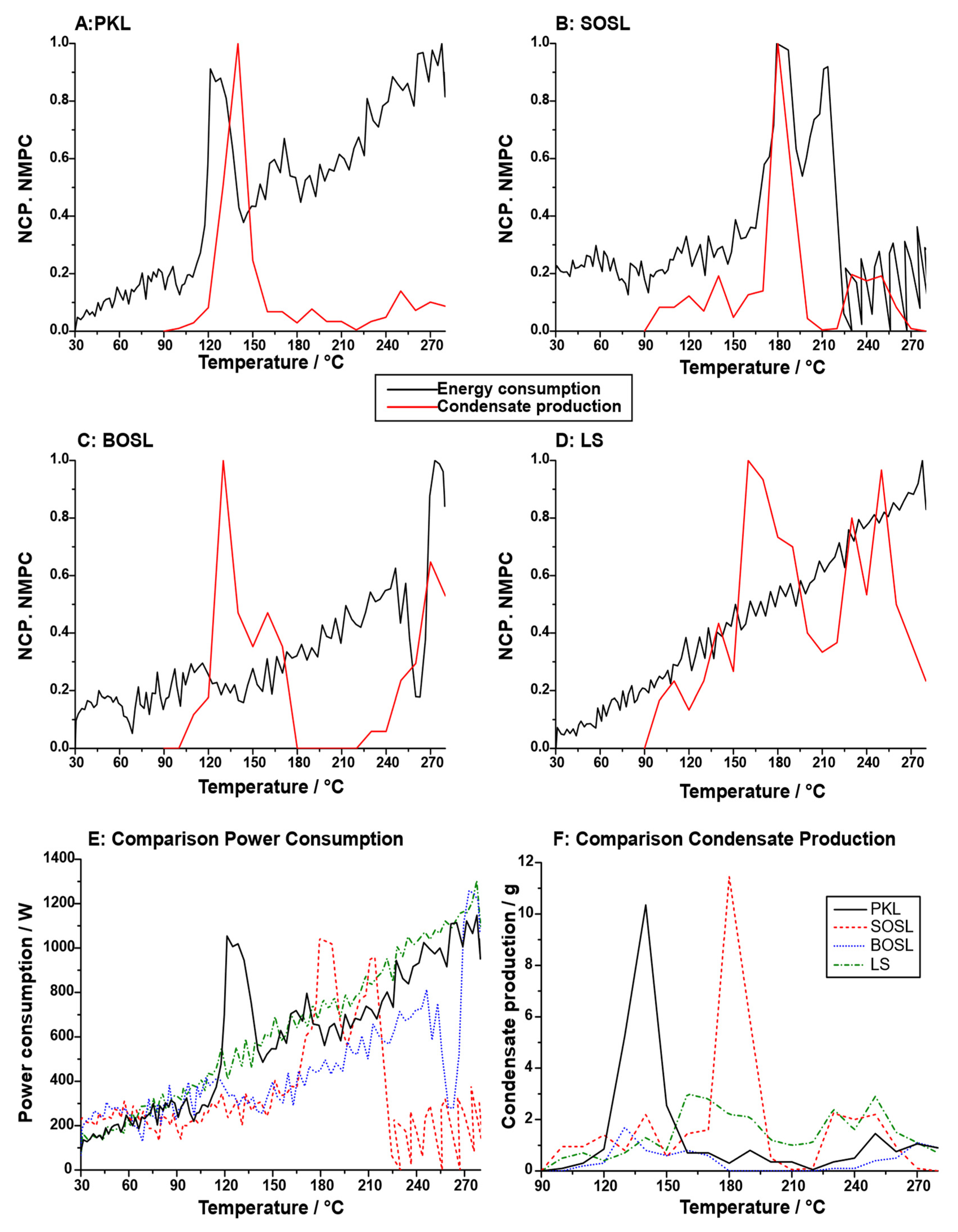
For potential applications and further insight into the pyrolysis process, not only the overall mass balances but also the temperature at which the condensates were obtained and the energy needed to pyrolyze the lignin should be considered. To obtain an estimate of this, the normalized condensate production rate and the microwave power consumption were determined (
Figure 7A–D). The normalized condensate production rate varied significantly between the different technical lignins. While for both the PKL and SOSL, a clear maximum could be determined (PKL: 140 °C; SOSL: 180 °C), both the BOSL and LS condensate production was high at two different temperatures (BOSL: 130 °C and 270 °C; LS: 160 °C and 250 °C). The overall microwave energy consumption was lower for the OSLs than for the LS and PKL (
Figure 7E). The reason for this is that at high temperatures, for both the OSLs, the microwave power consumption decreases significantly, as exothermic reactions take place.
The high phenolics yields make SOSL and the PKL the most interesting lignins for applications in which phenolics are the desired products, which is the aim of most lignin cleavage processes. Of these two, the PKL could be considered more suitable, because the condensates can be obtained at a lower temperature, reducing the energy and time needed for production. However, the exothermic reaction that occurs at higher temperatures during the cleavage of both the SOSL and BOSL could reduce the energy needed for cleaving these lignin types. This is especially the case for the SOSL. Using this exothermic reaction could be a way to decrease the energy consumption of the process; however, potential corrosion of equipment due to the high temperature and high pressure has to be considered. The average energy consumption during the pyrolysis process of the PKL was calculated to be 0.53 kWh. Due to the exothermic reactions, those of the SOSL and BOSL were 0.31 and 0.37 kWh, respectively. This underlines the potential of using the exothermic reaction step to produce bio-oil at lower energy consumption. The energy consumption of the LS was the highest, on average at 0.56 kWh despite the overall low cleavage reactions.
As mentioned above, both the PKL and SOSL had a temperature at which a high fraction of the condensates was produced (
Figure 7F). Thus, the bonds that were cleaved for condensate release were most probably of a similar nature. Microwave-assisted pyrolysis of lignin occurs in three degradation stages: first, the initial stage, during which the moisture is released from the lignin; afterwards, the second stage of fast degradation occurs, during which the
β-O-4 bonds as well as other ether bonds are cleave, while side chains are also released and small oxygenated molecules, such as furans, acids, and ketones are formed; during the third stage, carbon–carbon bond breaking is the dominant reaction [
16]. Considering the low temperatures of the cleavage, the main signal herein was expected to originate primarily from the second stage.
While for both the SOSL and the PKL a clear maximum condensate production at a given temperature was observed, the BOSL and the LS had several local maxima. During the pyrolysis of the BOSL, almost no aqueous phase was produced. Hence, the two local maxima of condensate production most probably both originated from phenolics production. This indicates that unlike the PKL and the SOSL, there were either two separate cleavage steps of one starting structure or that there were two different chemical compounds in the starting material. However, the overall condensate production was very low, and the normalization led to the over-exaggeration of the peaks as is visible in
Figure 7F. Despite the very low condensate production, there was a decrease in the microwave power consumption at higher temperatures of the BOSL pyrolysis, indicating an exothermic process.
The condensate production rate of the LS has to be considered separately from the other lignin types. Since almost no phenolic fraction was produced, the LS was the only lignin with aqueous products as the main cleavage products. Hence, different cleavage reactions have to also be expected. The condensate production rate indicated that the cleavage products were produced without a clear maximum but at several temperatures with similar rates. Thus, it is likely that the condensates originated from various cleavage reactions, for example, cleavage of sulfonate groups and hydroxy groups.
It has to be mentioned that the temperature was measured with an external IR thermoelement. Thus, the temperature was measured on the surface of the sample, resulting in potential inaccuracies. Microwave heating results in a higher temperature in the middle of a sample [
10]. Thus, the true temperature in the sample is expected to be higher than measured and described. In addition, due to the differences in microwave acceptance and sample position, local hot spots can occur. This effect can be further strengthened by heterogeneity due to the formation of char [
31]. This may lead to significantly higher temperatures at hot spots than in the overall material, increasing cleavages at these spots.
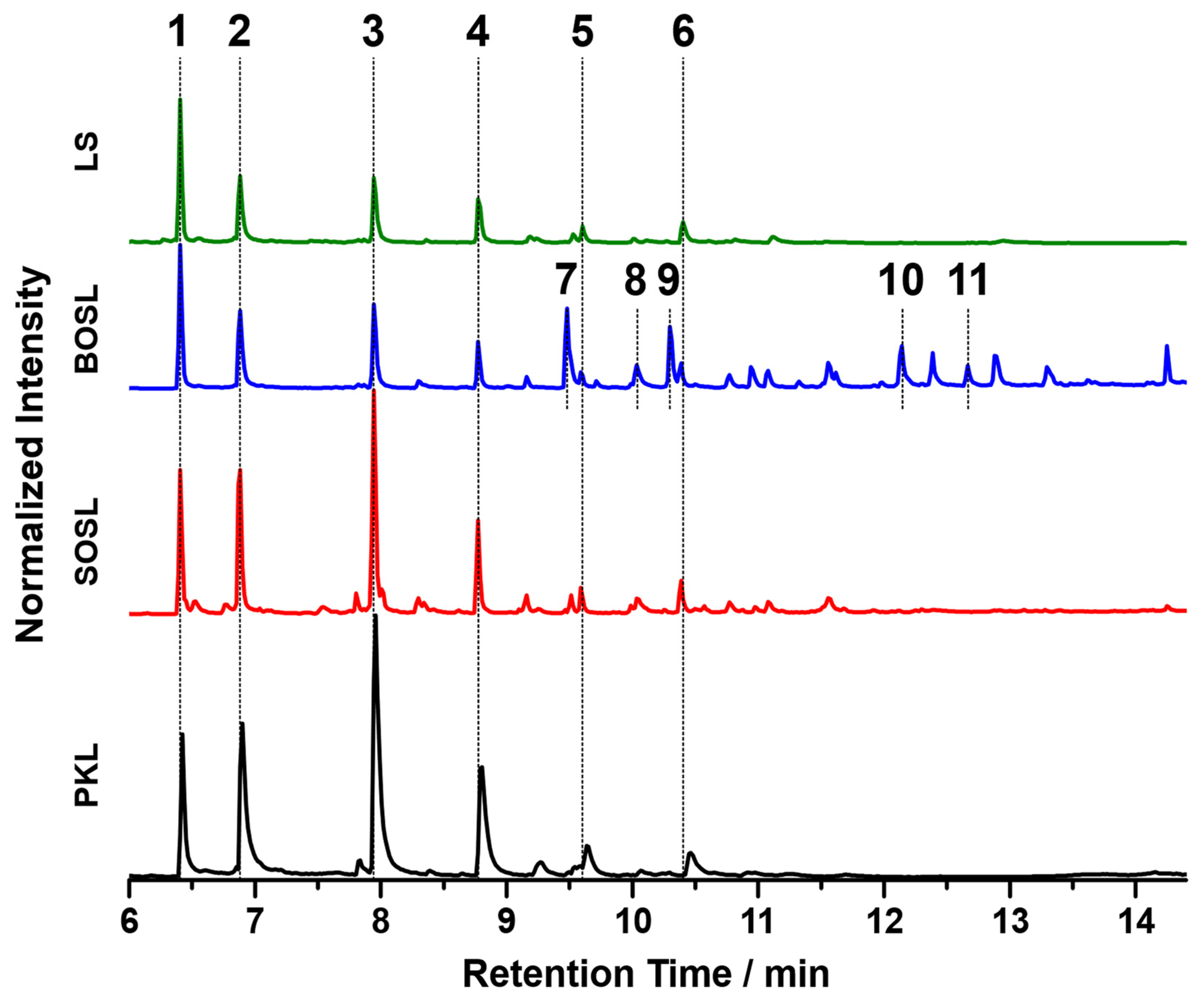
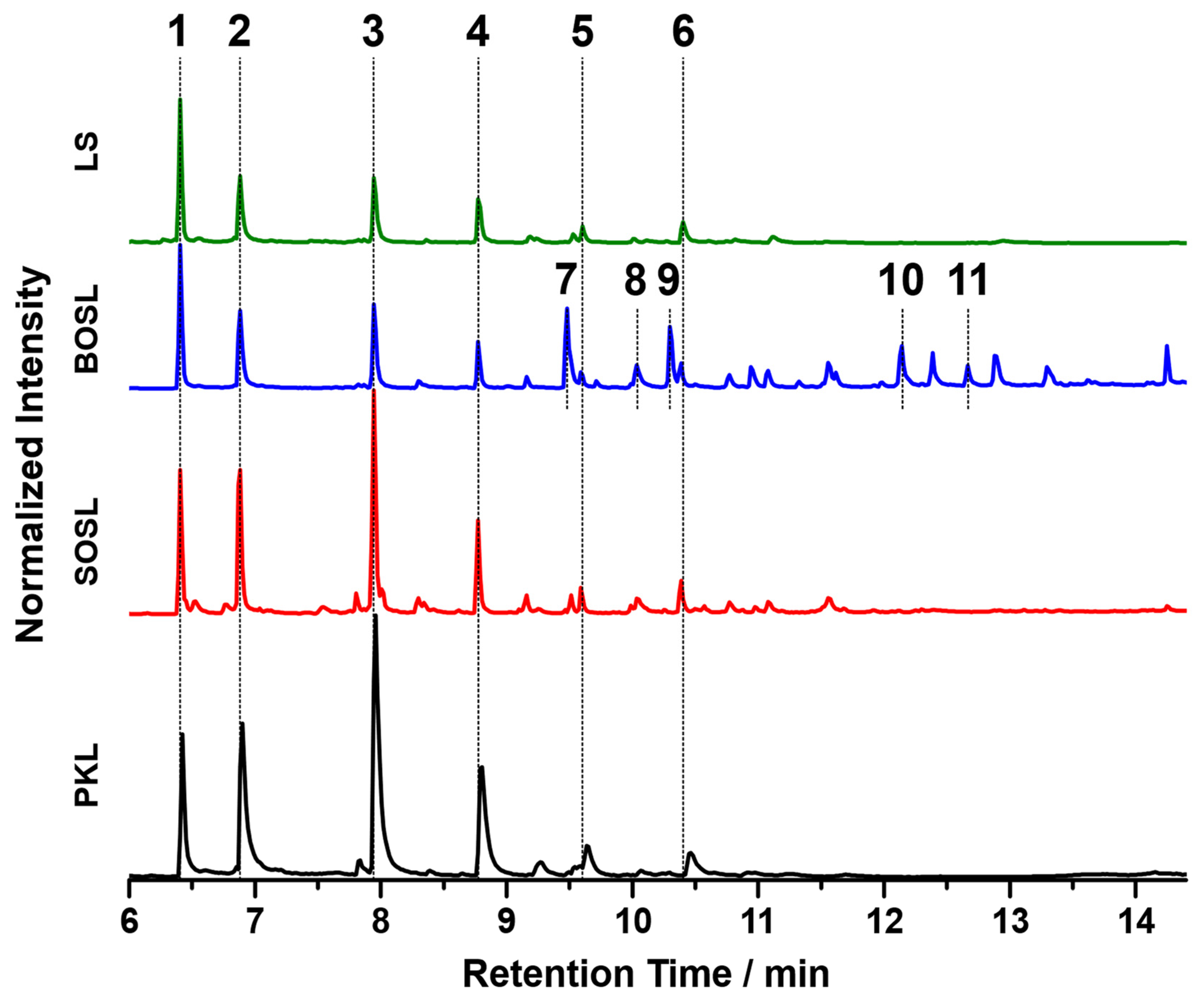
For comparison of the organic cleavage products, the chromatograms of the phenolic fractions of the four lignins are depicted in
Figure 8. The tentative assignments of signals by GC-MS are listed in
Table 3. For a better comparison, the chromatograms were normalized to the signal of the internal standard iodobenzene (signal 1 in
Figure 8). The three bio-oils from the PKL, SOSL, and LS, all originating from softwood lignins, had very similar products. While the bio-oil of the BOSL originating from hardwood lignin also contained these products, and its chromatogram had significantly more different signals with similar high intensities. The five main products of the softwood lignins according to the proposed assignments were 2-methoxyphenol (signal 2; guaiacol), 4-methylguaiacol (signal 3), 4-ethylguaiacol (signal 4), 4-propylguaiacol (signal 5), and 4-prop-1-enylguaiacol (signal 6). The signal intensity varied significantly depending on the lignin source. As such, the signals of the PKL and SOSL, compared to the internal standard, had a much higher intensity than those of the LS. The results indicate that the pyrolysis of softwood lignins produces clear main products, while the other side products were only obtained in small quantities. The pyrolysis of hardwood lignin yielded a more distributed bio-oil composition of various compounds. Some of the additional compounds detected for the bio-oil of the BOSL were proposed as 2,6-dimethoxy-phenol (signal 7), syringaldehyde (signal 10; 4-hydroxy-3,5-dimethoxy-benzaldehyde), and acetosyringone (signal 11; 4′-hydroxy-3′,5′-dimethoxy-acetophenone). Additionally, for both the organosolv lignins, vanillin (signal 8; 4-hydroxy-3-methoxybenzaldehyde), one of the few products that was commercially won from lignin, was detected [
32].
The proposed assignments of the signals visible in the chromatograms in
Figure 8 are listed in
Table 3. The signals having a high intensity only in the chromatogram of the bio-oil of the BOSL are marked with a (B) in the “Number in
Figure 8” column.
The main reason for the more diverse composition of the bio-oil of the BOSL was the composition of the hardwood lignins. While softwood lignin is often mainly built from G-type precursors, the additional S-type precursors in hardwood upon cleavage produce compounds such as syringaldehyde or 2,6-dimethoxyphenol. These chemicals have both ortho-positions occupied with methoxy groups, as is also the case in the precursor of S-type lignin: sinapyl alcohol [
3].
The chromatograms of the three softwood lignins indicated that the method applied for cleaving the lignins had a high selectivity towards certain bonds in the lignin structure, leading to the production of a few compounds with a high yield. This could make the used method interesting for applications aimed at obtaining these specific cleavage products.
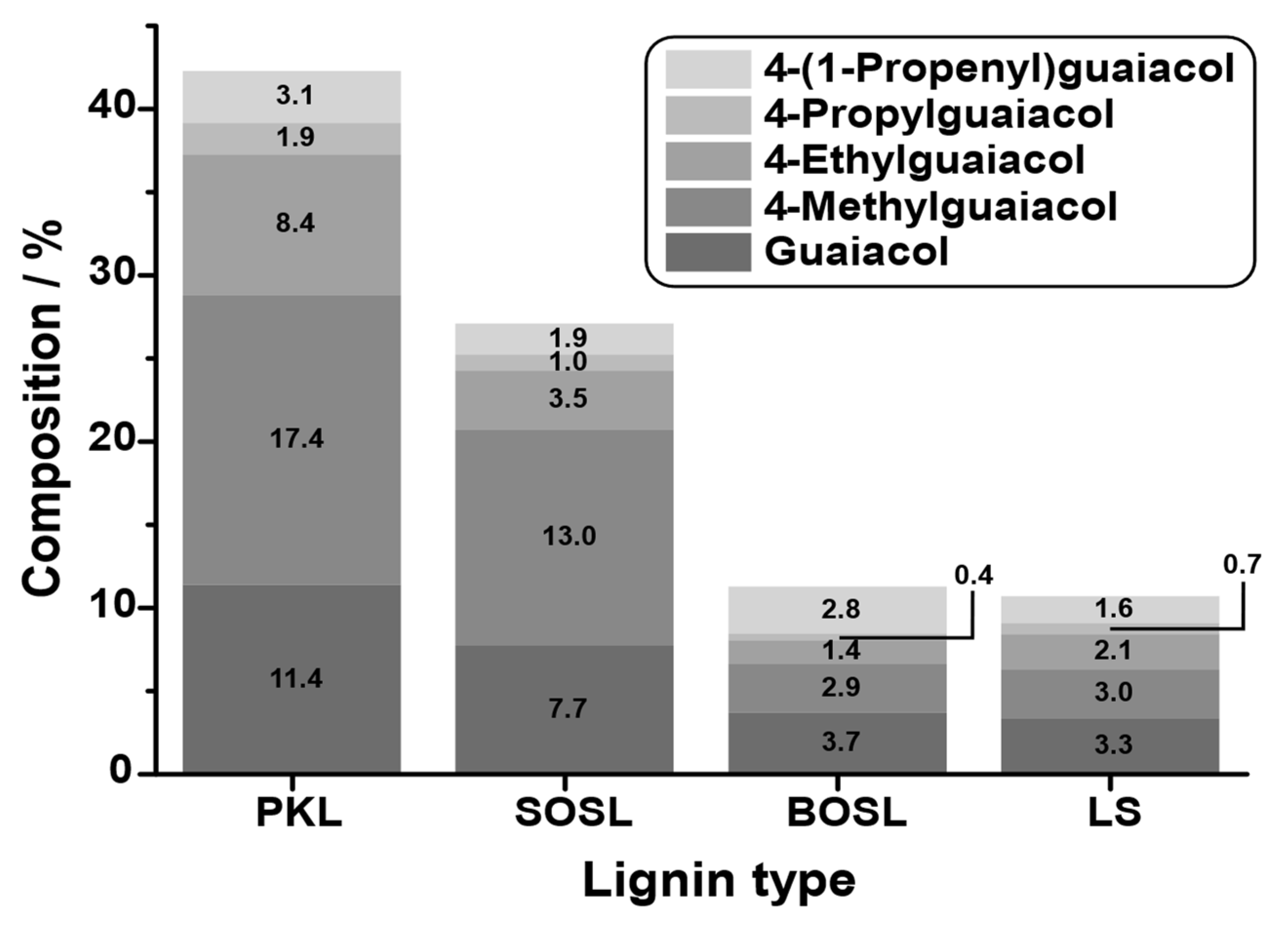
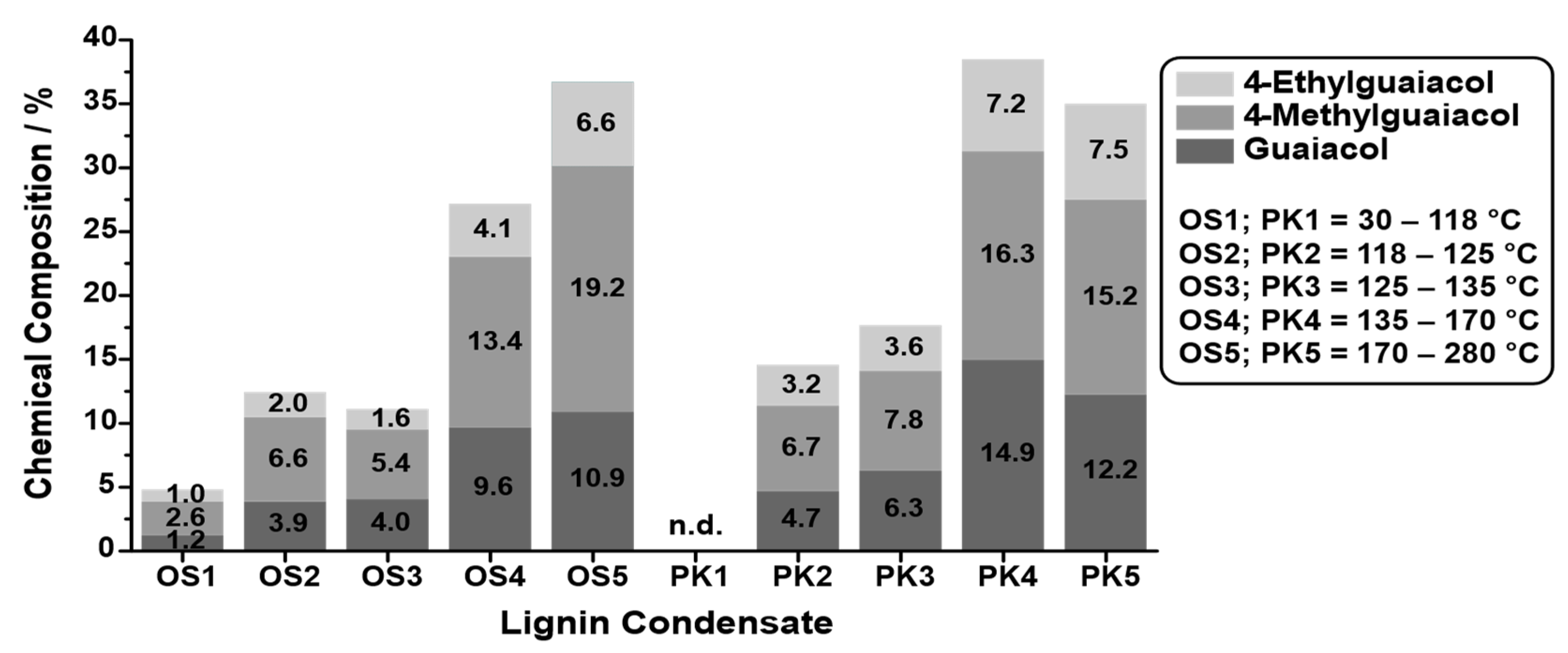
To compare the quantities of small aromatic compounds in the bio-oil, the five main products that could be detected in all lignins, namely, guaiacol, 4-methylguaiacol, 4-ethylguaiacol, 4-propylguaiacol, and 4-(1-propenyl)guaiacol, were selected, and their quantity was determined by GC-FID. The resolution and peak width of the signals obtained by GC-FID were less broad and more symmetrical than those obtained by GC-MS (compare
Figure A1 in
Appendix A). This minimizes the effects of the overlap, leading to a more precise quantification. The results are depicted in
Figure 9. The quantity of the main products varied significantly depending on the lignin source. While for the PKL, the five selected main products accounted for approximately 42% of the phenolic fraction composition, this value decreased for the SOSL, BOSL, and LS in that order, namely, being approximately 28%, 12%, and 11%, respectively.
The results indicate that a higher phenolic fraction yield also results in a higher percentage of monomers in the bio-oil. Thus, it is likely that the main difference in the yields originated from more monomers being cleaved overall. Due to the low yield of the selected monomers, especially in the BOSL and LS bio-oils, many additional compounds have to be part of the bio-oil’s composition. In the case of the BOSL, some of these compounds were already specified in the GC-MS; however, the low yield of the quantified molecules indicates that even more side products must be present in all bio-oils. These undetectable compounds could have a very low boiling point or a boiling point higher than the temperature to which the GC was heated (250 °C). However, after the liquid–liquid separation, the bio-oil was purified from the separation agent by evaporation. At this point, other also easily evaporated compounds were separated from the bio-oil. Thus, compounds with a low boiling point can be excluded, and the remaining compounds in the bio-oil must be compounds with a high boiling point. These compounds could be lignin oligomers, which are carried into the condensation system by other evaporating compounds, by the vacuum, or by the increasing pressure in the reactor due to the gas formation. The loss of oligomers in the reactor due to the fact of any of these reasons could additionally decrease the monomeric cleavage product yield of the reaction, as they are not cleaved further once carried into the condensation system.
Considering the selection of the cleavage products for quantification, the obtained yield of monomeric cleavage products for the BOSL was underestimated with this method. The true monomeric yield for the BOSL is expected to be closer to that of the SOSL due to the additional S-type lignin originating monomers.



 and
and 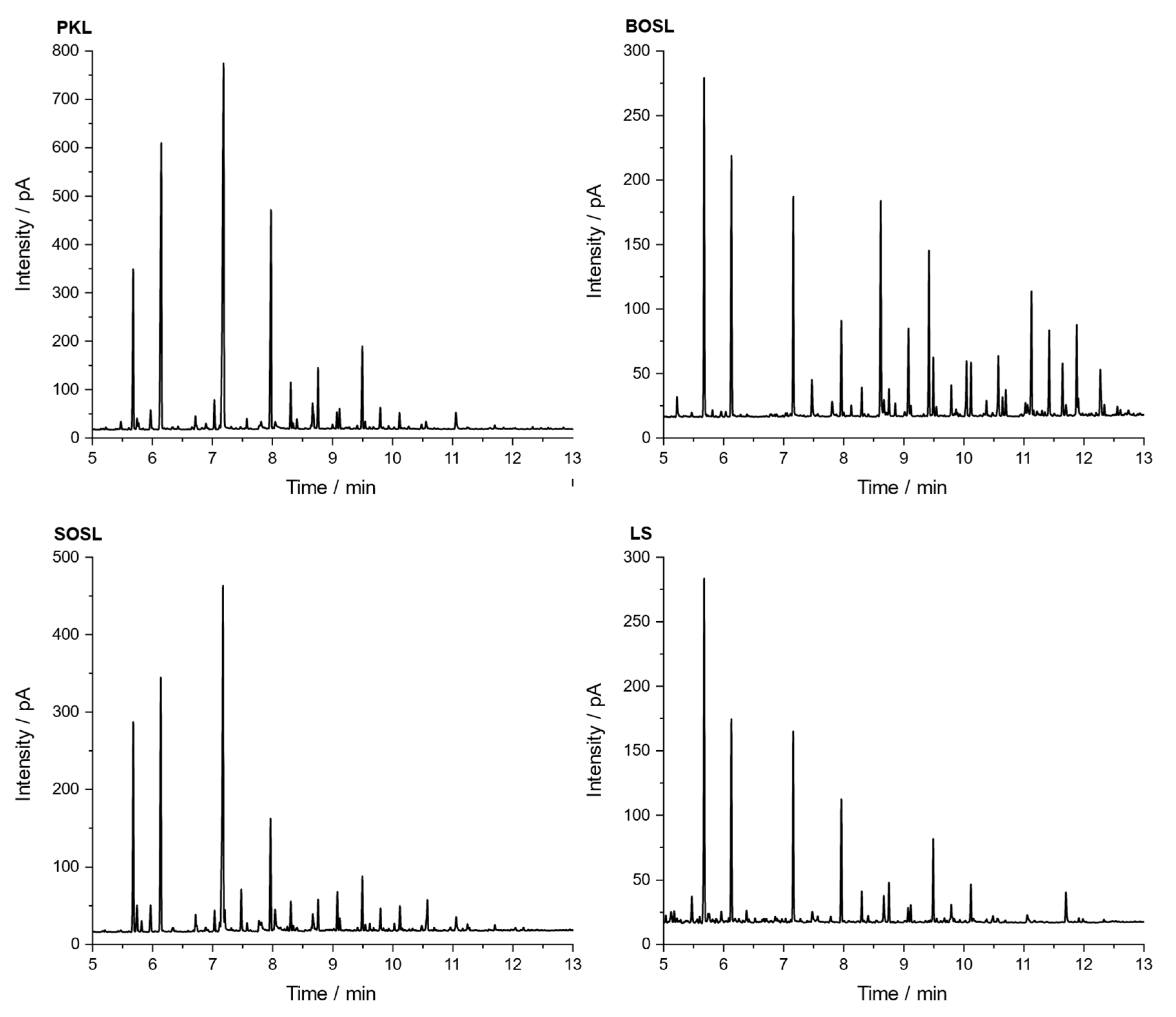










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}