
Experimental and Theoretical Study of Cyclic Amine Catalysed Urethane Formation

 , , and
, , and 
Abstract
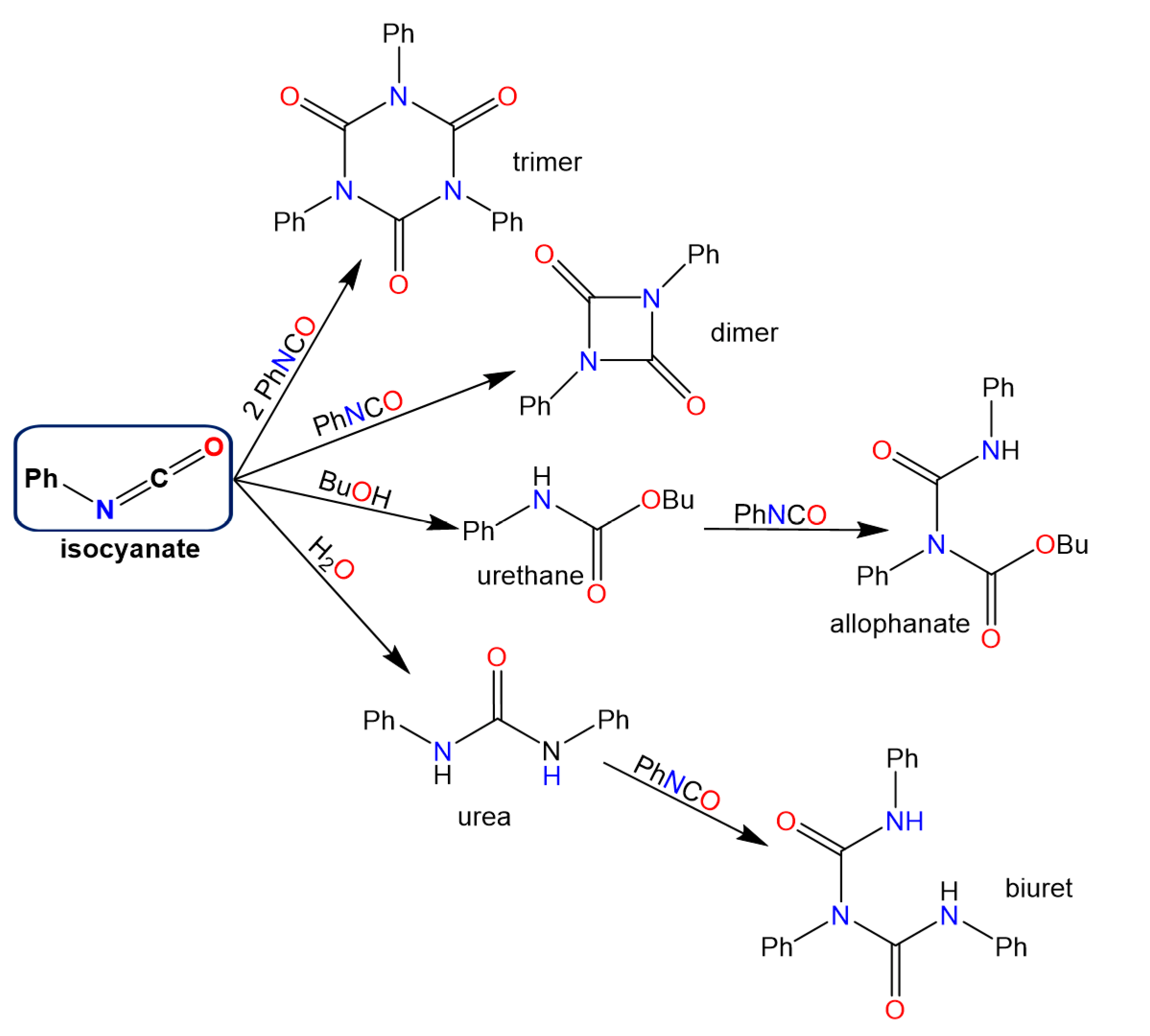
:1. Introduction
2. Methods
2.1. Materials and Methods
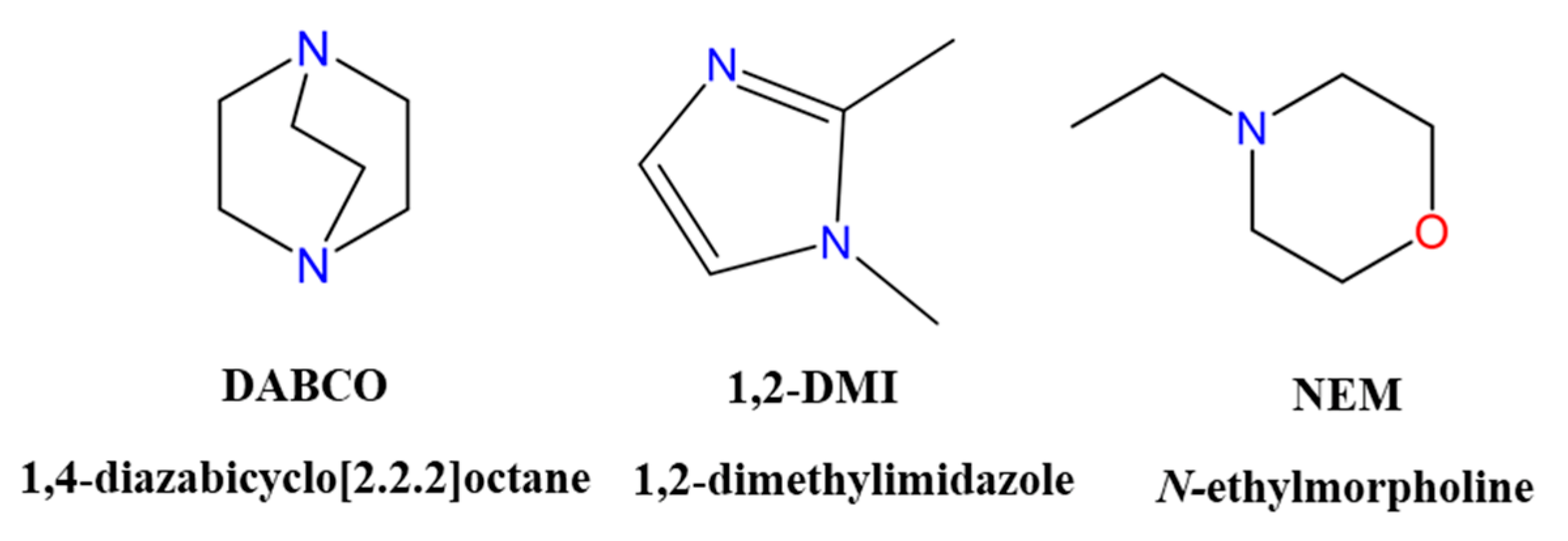
Materials
2.2. Kinetic Experiments
2.3. Analysis Method
2.4. Theoretical Method
3. Results and Discussion
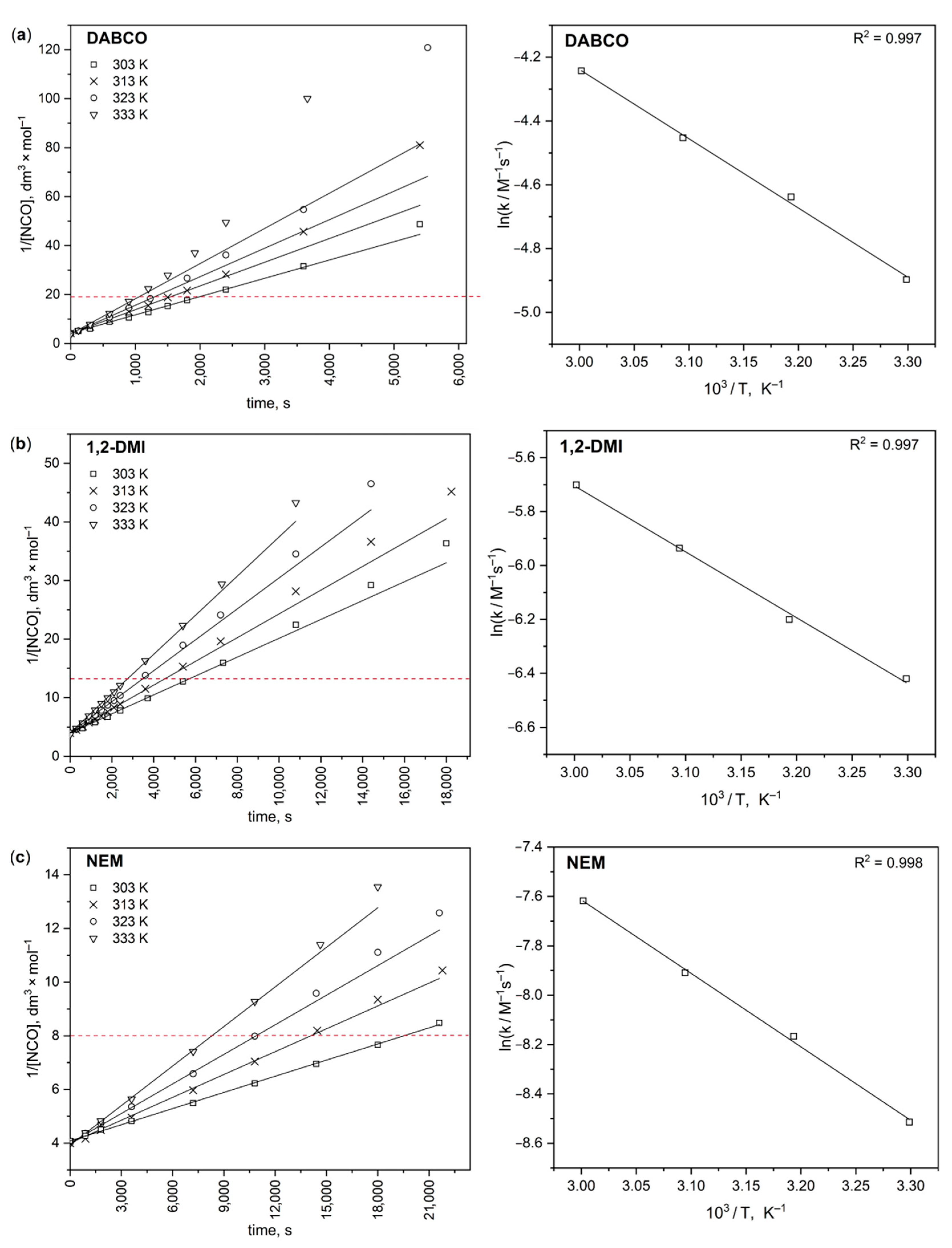
3.1. Results of the Kinetic Experiments
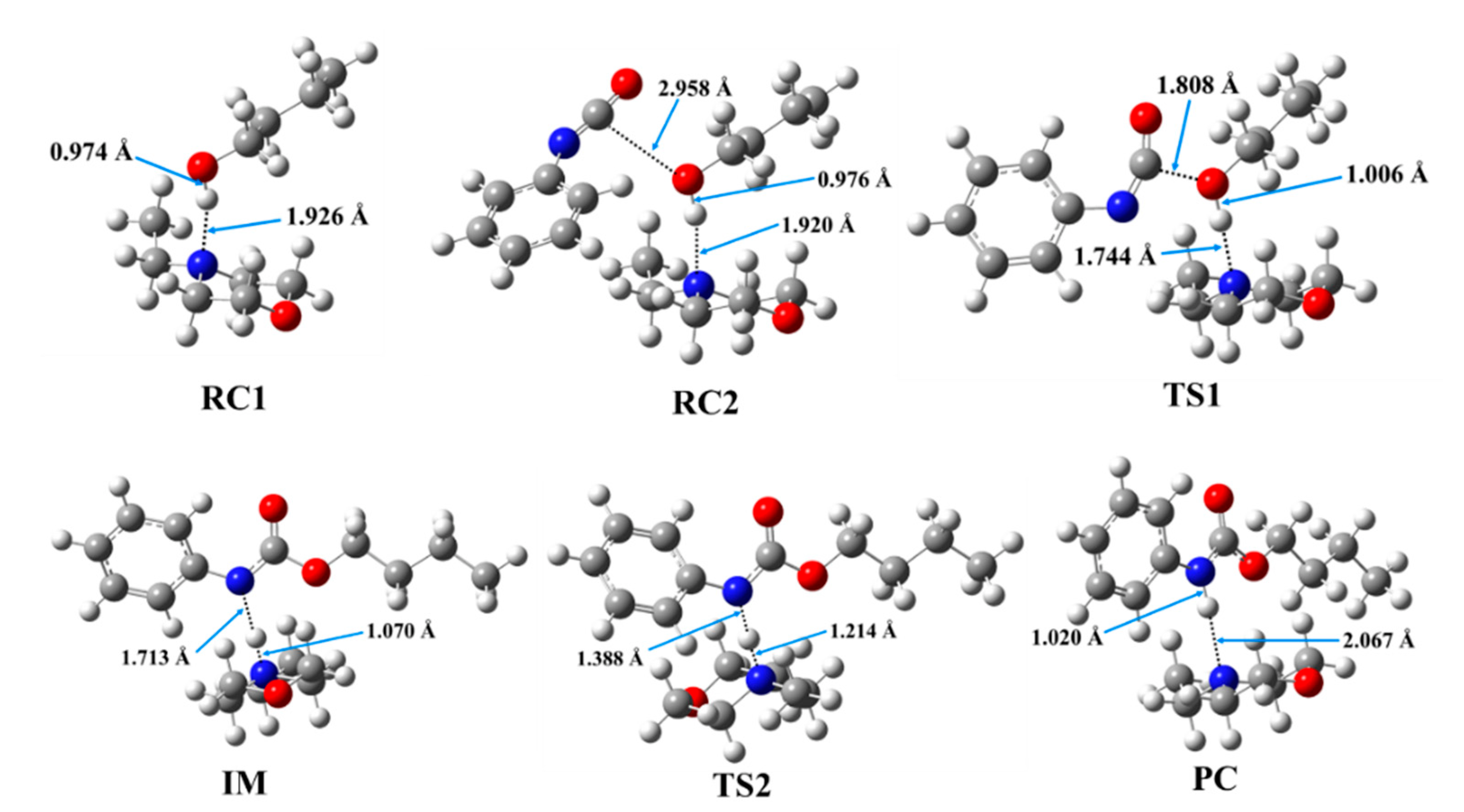
3.2. Results of the Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Avar, G.; Meier-Westhues, U.; Casselmann, H.; Achten, D. Polyurethanes; Elsevier: Amsterdam, The Netherlands, 2012; Volume 10, ISBN 9780080878621. [Google Scholar]
- Yanping, Y. The Development of Polyurethane. Mater. Sci. Mater. Rev. 2018, 1, 507. [Google Scholar] [CrossRef]
- Xu, Q.; Lin, J.; Jiang, G. Synthesis, Characterization and Properties of Soybean Oil-Based Polyurethane. Polymers 2022, 14, 2201. [Google Scholar] [CrossRef] [PubMed]
- Samaniego-aguilar, K.; Arrillaga, A.; Anakabe, J.; Gamez-perez, J.; Cabedo, L. In Service Performance of Toughened PHBV/TPU Blends Obtained by Reactive Extrusion for Injected Parts. Polymers 2022, 14, 2337. [Google Scholar] [CrossRef] [PubMed]
- Szycher, M. Szycher’S Handbook of Polyurethanes, 2nd ed.; Taylor & Francis Group: Abingdon, UK, 2013; ISBN 9781439863138. [Google Scholar]
- Sonnenschein, M.F. Polyurethanes Science, Technology, Markets, and Trends, 1st ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2015; ISBN 9789896540821. [Google Scholar]
- Muuronen, M.; Deglmann, P.; Tomović, Ž. Design Principles for Rational Polyurethane Catalyst Development. J. Org. Chem. 2019, 84, 8202–8209. [Google Scholar] [CrossRef]
- Eling, B.; Tomović, Ž.; Schädler, V. Current and Future Trends in Polyurethanes: An Industrial Perspective. Macromol. Chem. Phys. 2020, 221, 2000114. [Google Scholar] [CrossRef]
- Suzuki, T.; Tokumoto, K.; Takahashi, Y.; Kiso, H.; Van Maris, R.; Tucker, J. Zero Emission Polyurethane Catalyst. TOSOH Res. Technol. Rev. 2013, 57, 13–21. [Google Scholar]
- Van Maris, R.; Tamano, Y.; Yoshimura, H.; Gay, K.M. Polyurethane Catalysis by Tertiary Amines. J. Cell. Plast. 2005, 41, 305–322. [Google Scholar] [CrossRef]
- Waleed, H.Q.; Csécsi, M.; Hadjadj, R.; Thangaraj, R.; Pecsmány, D.; Owen, M.; Szőri, M.; Fejes, Z.; Viskolcz, B.; Fiser, B. Computational Study of Catalytic Urethane Formation. Polymers 2022, 14, 8. [Google Scholar] [CrossRef]
- Waleed, H.Q.; Csécsi, M.; Hadjadj, R.; Thangaraj, R.; Owen, M.; Szőri, M.; Fejes, Z.; Viskolcz, B.; Fiser, B. The Catalytic Effect of DBU on Urethane Formation—A Computational Study. Mater. Sci. Eng. 2012, 46, 70–77. [Google Scholar] [CrossRef]
- Chaffanjon, P.; Grisgby, R.A., Jr.; Rister, E.L., Jr.; Zimmerman, R.L. Use of real-time FTIR to characterize kinetics of amine catalysts and to develop new grades for various polyurethane applications, including low emission catalysts. J. Cell. Plast. 2003, 39, 187–210. [Google Scholar] [CrossRef]
- Malwitz, N.; Wong, S.W.; Frisch, K.C.; Manis, P.A. Amine Catalysis of Polyurethane Foams. J. Cell. Plast. 1987, 23, 461–502. [Google Scholar] [CrossRef]
- Du, W.; Zhang, G.; Wang, S.; Tan, L.; Chen, H. Cure Kinetics of an Optical Polythiourethane with Amine Catalyst by IR Analysis. Int. J. Polym. Sci. 2019, 2019, 8194379. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, M. DFT Analysis of Catalytic Urethanation. Bull. Chem. Soc. Jpn. 2011, 84, 933–935. [Google Scholar] [CrossRef]
- Wen, Z. DFT Study of the Catalytic Mechanism for Urethane Formation in the Presence of Basic Catalyst 1,4-Diazabicyclo[2.2.2]Octane. Commun. Comput. Chem. 2014, 2, 22–35. [Google Scholar] [CrossRef]
- Williams, D.B.G.; Lawton, M. Drying of Organic Solvents: Quantitative Evaluation of the Efficiency of Several Desiccants. J. Org. Chem. 2010, 75, 8351–8354. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Szori, M.; Abou-Abdo, T.; Fittschen, C.; Csizmadia, I.G.; Viskolcz, B. Allylic Hydrogen Abstraction II. H-Abstraction from 1,4 Type Polyalkenes as a Model for Free Radical Trapping by Polyunsaturated Fatty Acids (PUFAs). Phys. Chem. Chem. Phys. 2007, 9, 1931–1940. [Google Scholar] [CrossRef]
- Izsák, R.; Szori, M.; Knowles, P.J.; Viskolcz, B. High Accuracy Ab Initio Calculations on Reactions of OH with 1-Alkenes. The Case of Propene. J. Chem. Theory Comput. 2009, 5, 2313–2321. [Google Scholar] [CrossRef]
- Wang, X.; HU, W.; Gui, D.; Chi, X.; Wang, M.; Tian, D.; Liu, J.; Ma, X.; Pang, A. DFT Study of the Proton Transfer in the Urethane Formation between 2,4-Diisocyanatotoluene and Methanol. Bull. Chem. Soc. Jpn. 2013, 86, 255–265. [Google Scholar] [CrossRef]
- Kolboe, S. Proton Affinity Calculations with High Level Methods. J. Chem. Theory Comput. 2014, 10, 3123–3128. [Google Scholar] [CrossRef]
- Ephraim, S.; Woodward, A.E.; Mesrobian, R.B. Kinetic Studies of the Reaction of Phenyl Isocyanate with Alcohols in Various Solvents. J. Am. Chem. Soc. 1958, 80, 1326–1328. [Google Scholar] [CrossRef]
- Inoue, S.I.; Nagai, Y.; Okamoto, H. Amine-Manganese Complex as an Efficient Catalyst for Polyurethane Syntheses. Polym. J. 2002, 34, 298–301. [Google Scholar] [CrossRef] [Green Version]
- Farkas, A.; Flynn, K.G. The Catalytic Effects of 1,4-Diaza [2.2.2] Bicycloöctane for Isocyanate Reactions. J. Am. Chem. Soc. 1960, 82, 642–645. [Google Scholar] [CrossRef]
- El Ghobary, H.; Muller, L. Process for Preparing Polyurethane Foam 1. EU. Patent No. 1 018 525 A1, 12 July 2000. [Google Scholar]
- Muha, K.; Hulme, S.P.; Harakal, M.E. Low Odor Amine Catalysts for Polyurethane Flexible Slabstock Foams Based on Polyester Polyols. U.S. Patent No 5,591,780, 20 September 1997. [Google Scholar]
- Silva, A.L.; Bordado, J.C. Recent Developments in Polyurethane Catalysis: Catalytic Mechanisms Review. Catal. Rev. Sci. Eng. 2004, 46, 31–51. [Google Scholar] [CrossRef]
- Cheikh, W. Data House of Polyurethane Combined Theoretical and Experimental Methods. Doctoral Thesis, University of Miskolc, Miskolc, Hungary, 2021. [Google Scholar]
- Hunter, E.P.L.; Lias, S.G. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data 1998, 27, 413–656. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature, °C | DABCO | 1,2-DMI | NEM |
|---|---|---|---|
| k × 104, M−1 s−1 | k × 104, M−1 s−1 | k × 104, M−1 s−1 | |
| 30 | 74.7 ± 0.6 | 16.3 ± 0.1 | 2.01 ± 0.01 |
| 40 | 96.8 ± 0.8 | 20.3 ± 0.2 | 2.84 ± 0.03 |
| 50 | 116.4 ± 0.8 | 26.4 ± 0.3 | 3.68 ± 0.03 |
| 60 | 143.6 ± 3.1 | 33.4 ± 0.4 | 4.92 ± 0.06 |
| Ea, kJ mol−1 | 18.1 ± 0.7 | 20.3 ± 0.8 | 24.8 ± 0.8 |
| A, M−1 s−1 | 9.7 ± 2.4 | 5.1 ± 1.5 | 3.7 ± 1.1 |
| ΔE0 (kJ/mol) | ||||||||
|---|---|---|---|---|---|---|---|---|
| R | RC1 | RC2 | TS1 | IM | TS2 | PC | P | |
| Cat.-free | 0.0 | - | −11.2 * | 119.1 | - | - | - | −92.6 |
| DABCO | 0.0 | −26 | −46.6 | −0.9 | −96.2 | −107.6 | −124.7 | −92.6 |
| 1,2-DMI | 0.0 | −21.8 | −33.5 | 7.2 | −78.4 | −86.9 | −119.9 | −92.6 |
| NEM | 0.0 | −28.7 | −49.1 | −0.2 | −95.9 | −106 | −132.2 | −92.6 |
| ΔH (kJ/mol) | ||||||||
| R | RC1 | RC2 | TS1 | IM | TS2 | PC | P | |
| Cat.-free | 0.0 | - | −8.9 * | 116.5 | - | - | - | −94.8 |
| DABCO | 0.0 | −24.5 | −41.4 | −0.8 | −96.9 | −109.2 | −124.6 | −94.8 |
| 1,2-DMI | 0.0 | −20.6 | −28.6 | 7.2 | −78.8 | −88.6 | −119.6 | −94.8 |
| NEM | 0.0 | −27.3 | −44.4 | −0.9 | −97.2 | −107.9 | −132.2 | −94.8 |
| ΔG (kJ/mol) | ||||||||
| R | RC1 | RC2 | TS1 | IM | TS2 | PC | P | |
| Cat.-free | 0.0 | - | 28.9 * | 170 | - | - | - | −41.5 |
| DABCO | 0.0 | 14.5 | 28 | 91.5 | 2.2 | −6.9 | −28.8 | −41.5 |
| 1,2-DMI | 0.0 | 19.5 | 46.7 | 103.1 | 19.7 | 14.9 | −24.6 | −41.5 |
| NEM | 0.0 | 13.3 | 34.6 | 100.7 | 9.1 | −0.9 | −33.9 | −41.5 |
| Catalysts | PAcalc (kJ/mol) | PAexp (kJ/mol) [31] |
|---|---|---|
| DABCO | 983.9 | 963.4 |
| 1,2-DMI | 1002.9 | 984.7 |
| NEM | 973.2 | - |
| Catalyst | RC1 | RC2 | TS1 | IM | TS2 | PC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N-H * | O-H | N-H * | O-H | C-O | N-H * | O-H | C-O | N-H * | N-H ** | N-H * | N-H ** | N-H * | N-H ** | |
| DABCO | 1.856 | 0.976 | 1.835 | 0.979 | 3.073 | 1.661 | 1.012 | 1.853 | 1.074 | 1.668 | 1.207 | 1.367 | 1.998 | 1.023 |
| 1,2-DMI | 1.878 | 0.973 | 1.898 | 0.972 | 3.059 | 1.704 | 1.001 | 1.832 | 1.082 | 1.610 | 1.150 | 1.450 | 1.972 | 1.020 |
| NEM | 1.926 | 0.974 | 1.920 | 0.976 | 2.958 | 1.744 | 1.006 | 1.808 | 1.070 | 1.713 | 1.214 | 1.388 | 2.067 | 1.020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waleed, H.Q.; Pecsmány, D.; Csécsi, M.; Farkas, L.; Viskolcz, B.; Fejes, Z.; Fiser, B. Experimental and Theoretical Study of Cyclic Amine Catalysed Urethane Formation. Polymers 2022, 14, 2859. https://doi.org/10.3390/polym14142859
Waleed HQ, Pecsmány D, Csécsi M, Farkas L, Viskolcz B, Fejes Z, Fiser B. Experimental and Theoretical Study of Cyclic Amine Catalysed Urethane Formation. Polymers. 2022; 14(14):2859. https://doi.org/10.3390/polym14142859
Chicago/Turabian StyleWaleed, Hadeer Q., Dániel Pecsmány, Marcell Csécsi, László Farkas, Béla Viskolcz, Zsolt Fejes, and Béla Fiser. 2022. "Experimental and Theoretical Study of Cyclic Amine Catalysed Urethane Formation" Polymers 14, no. 14: 2859. https://doi.org/10.3390/polym14142859
APA StyleWaleed, H. Q., Pecsmány, D., Csécsi, M., Farkas, L., Viskolcz, B., Fejes, Z., & Fiser, B. (2022). Experimental and Theoretical Study of Cyclic Amine Catalysed Urethane Formation. Polymers, 14(14), 2859. https://doi.org/10.3390/polym14142859