
Synthesis of New Type Polymers by Quasi-Living Atom Transfer Radical Polymerization

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Properties of Used Materials
2.2. Cleaning of Used Materials
2.3. Polymers Synthesis
2.3.1. Synthesis of 1,1,1,1-tetrakis [2′-bromo-2′-methylpropionyloxymethyl] Methane Initiator
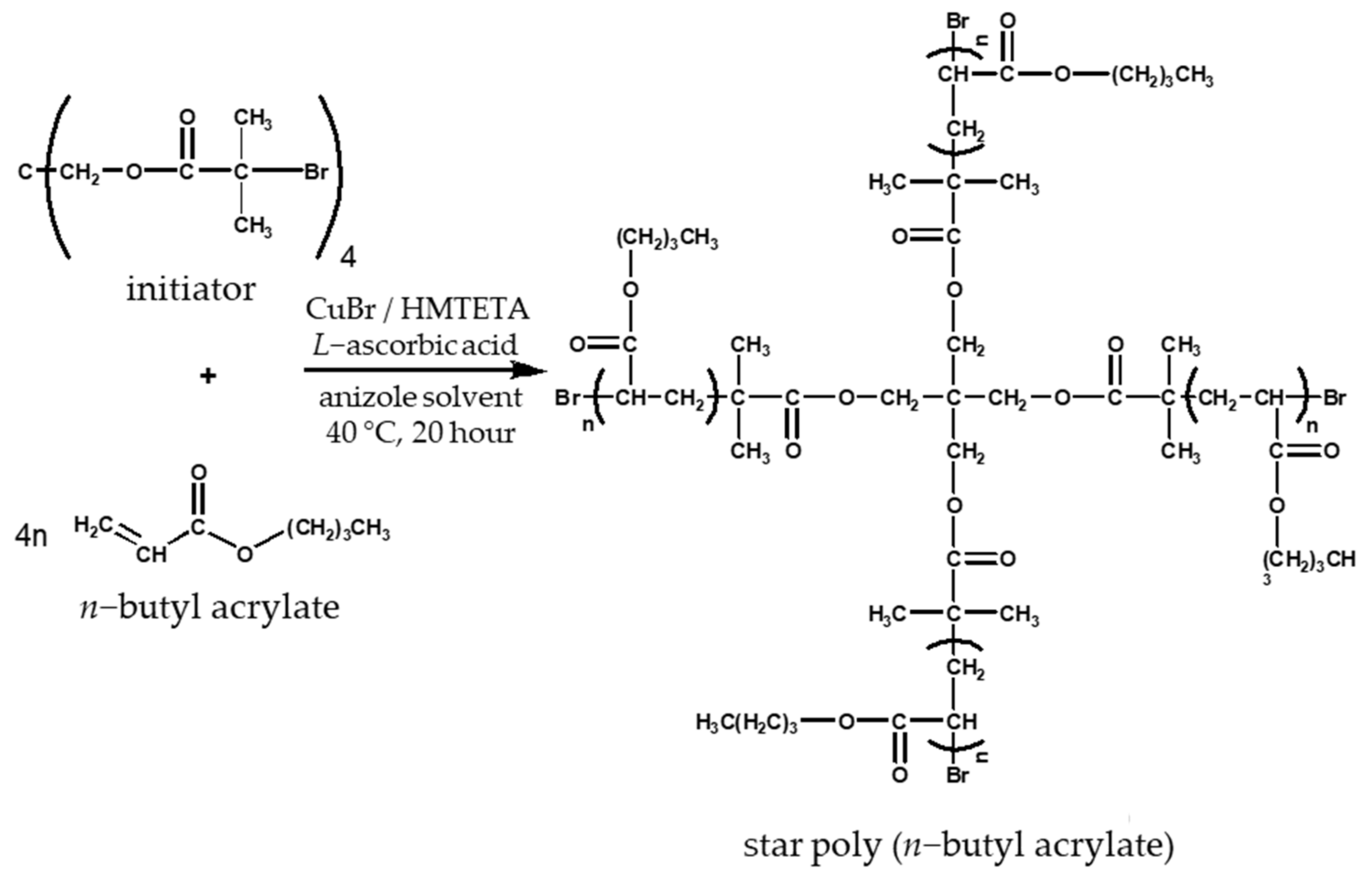
2.3.2. Synthesis of Poly (n-butyl acrylate) Star Polymers by Quasi-Living Radical Polymerization
2.4. Modification of the Chain End of Star Polymers by a Substitution Reaction
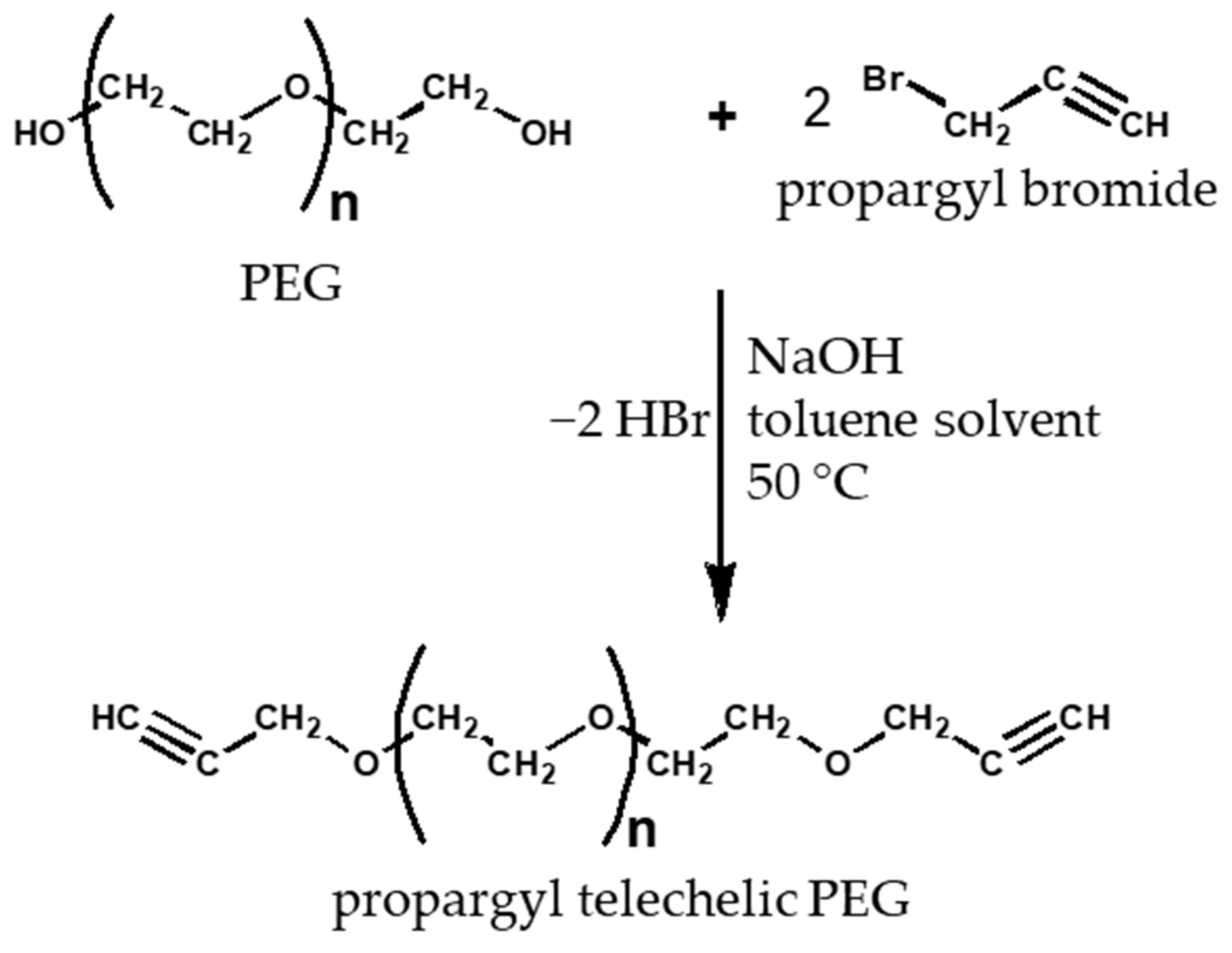
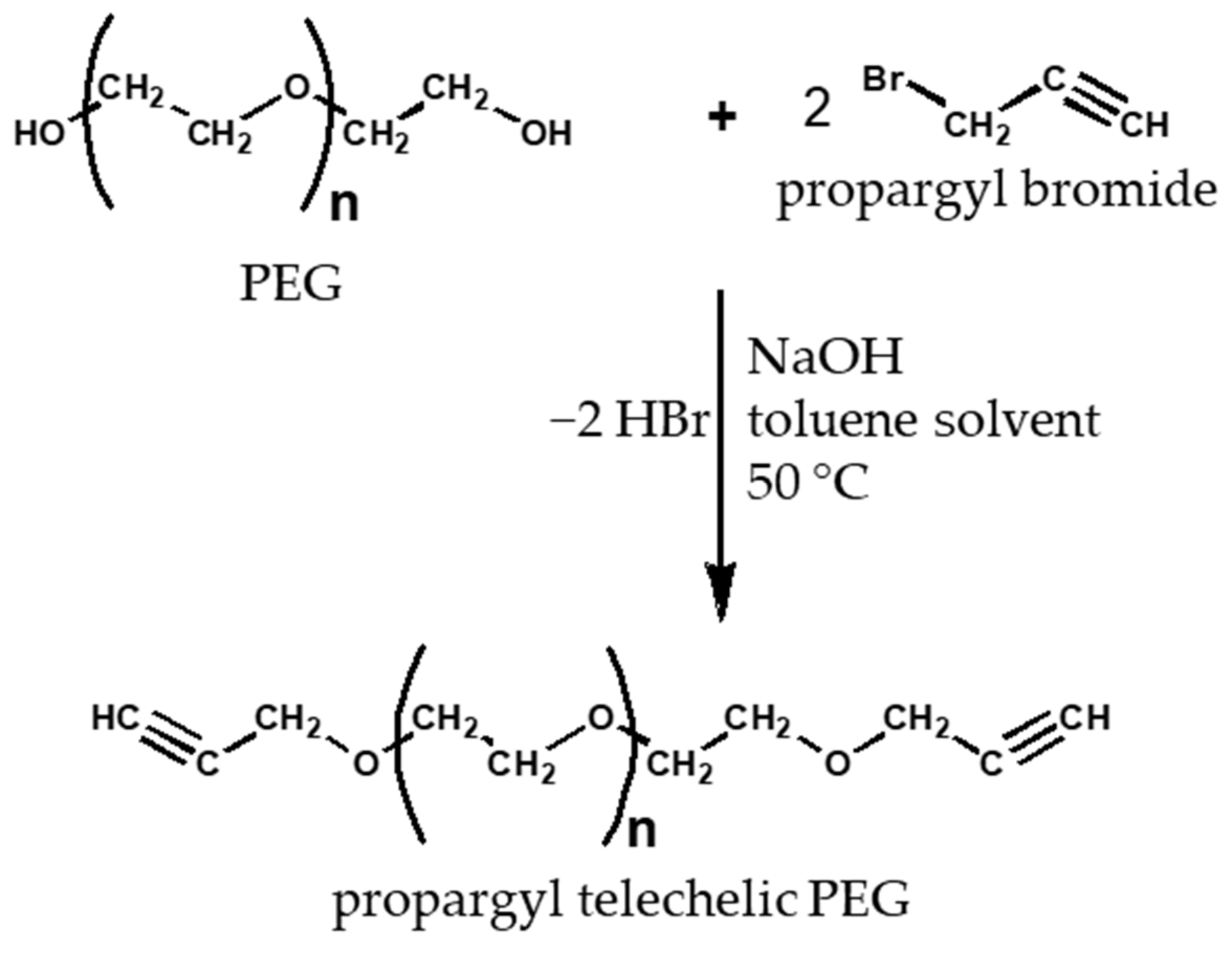
2.5. Production of Propargyl Telechelic Polyethylene Glycol
2.6. Synthesis of a New Type of Amphiphilic Conetworks
2.7. Method of Analysis
2.7.1. Gel Permeation Chromatography
2.7.2. Nuclear Magnetic Resonance
2.7.3. Mass Spectroscopy
2.7.4. Differential Scanning Calorimetry
2.7.5. Thermogravimetry
2.7.6. Elemental Analysis
2.7.7. Determination of Swelling Degree
3. Results
3.1. Synthesis of 1,1,1,1-tetrakis [2′-bromo-2′-methylpropionyloxymethyl] Methane Initiator
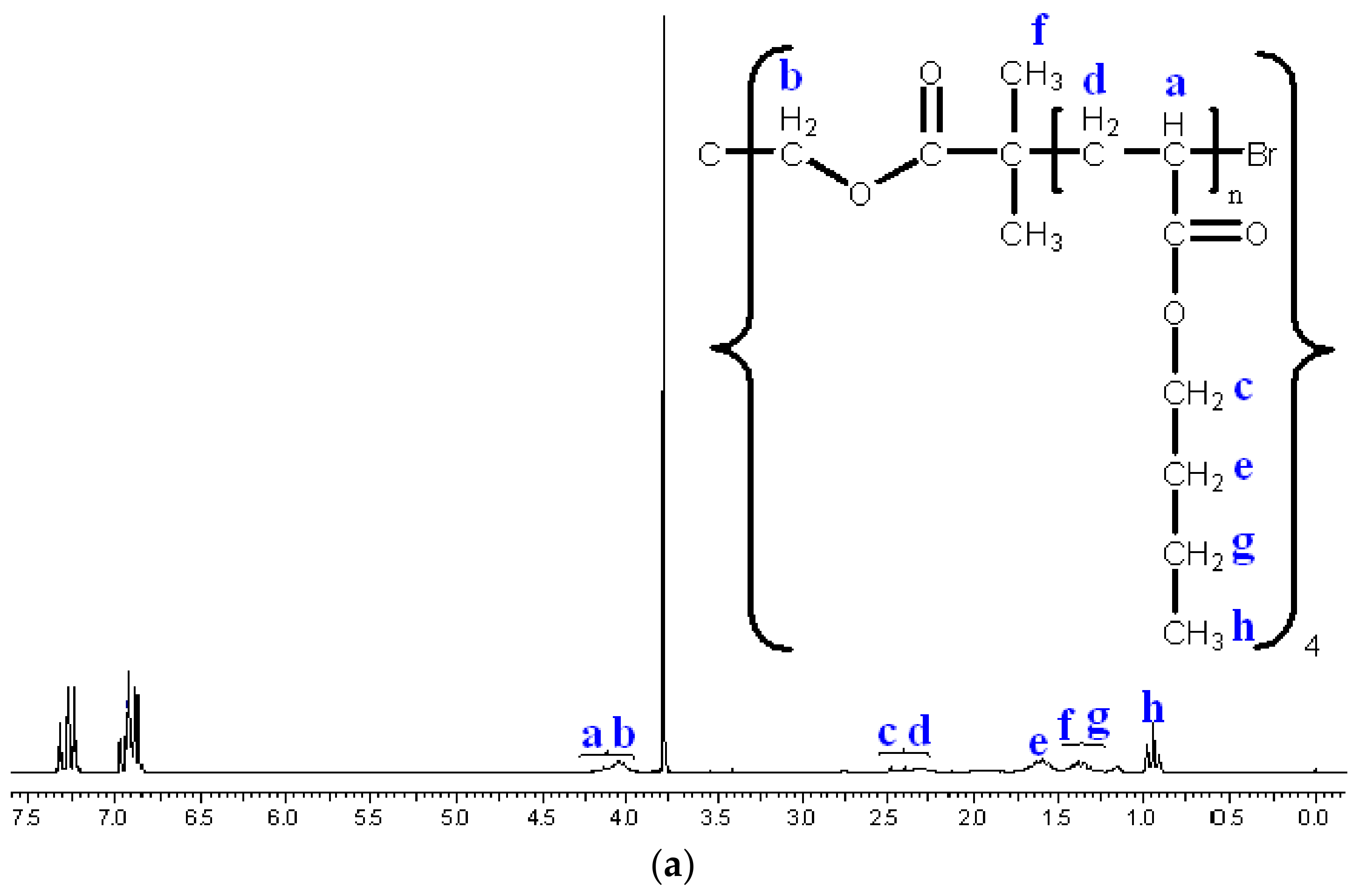
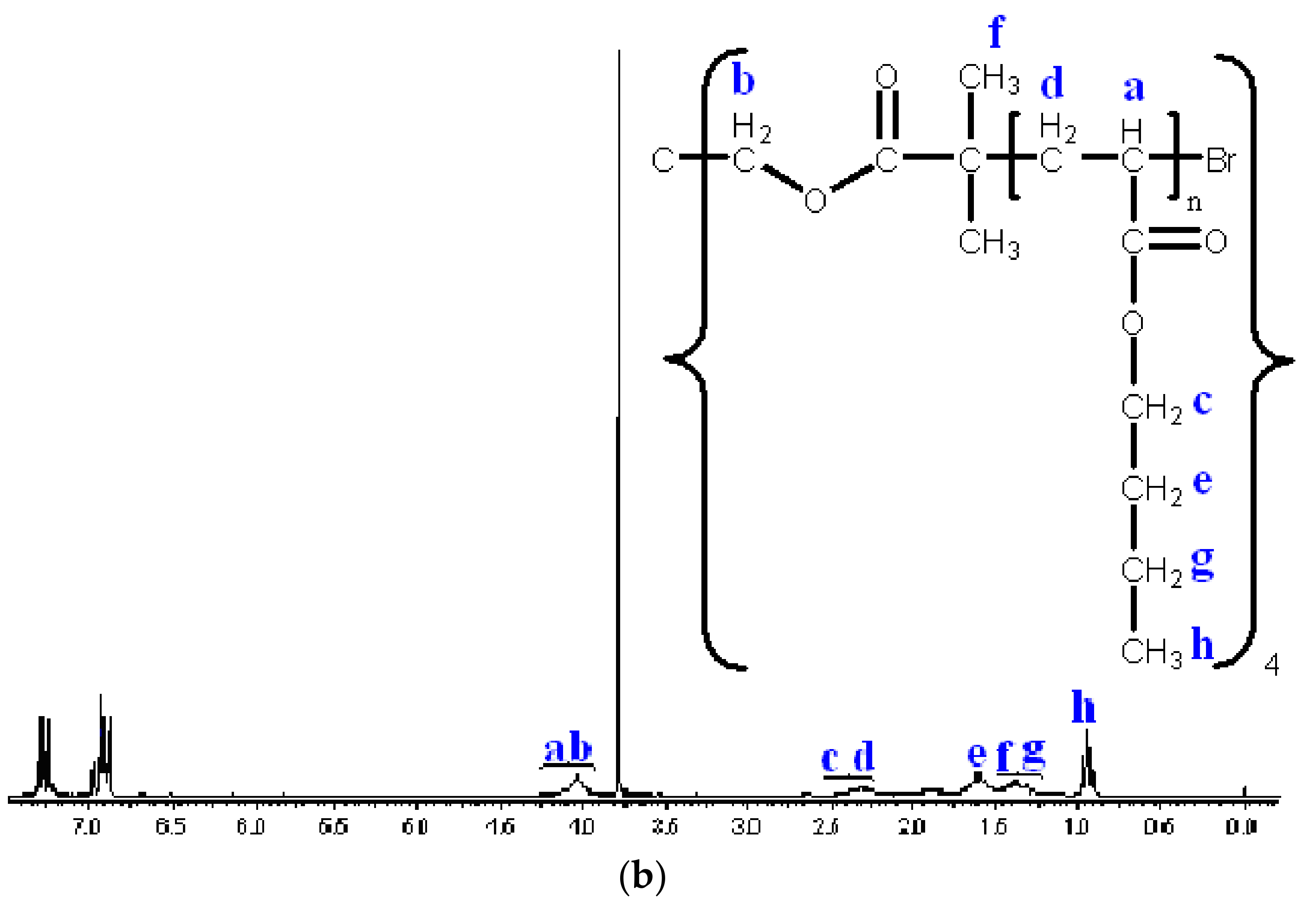
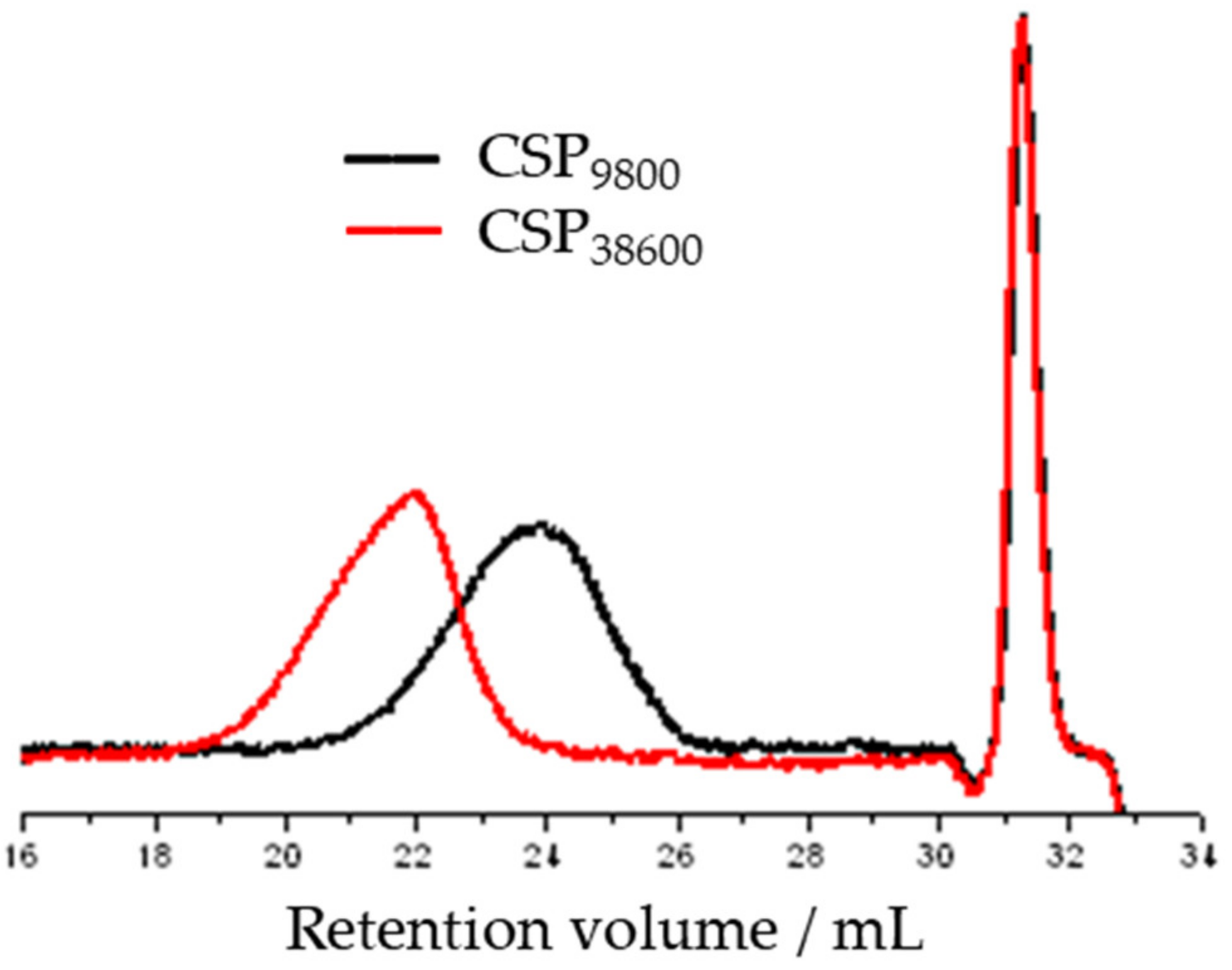
3.2. Synthesis of Star n-butyll acrylate Polymers by Atomic Radical Polymerization
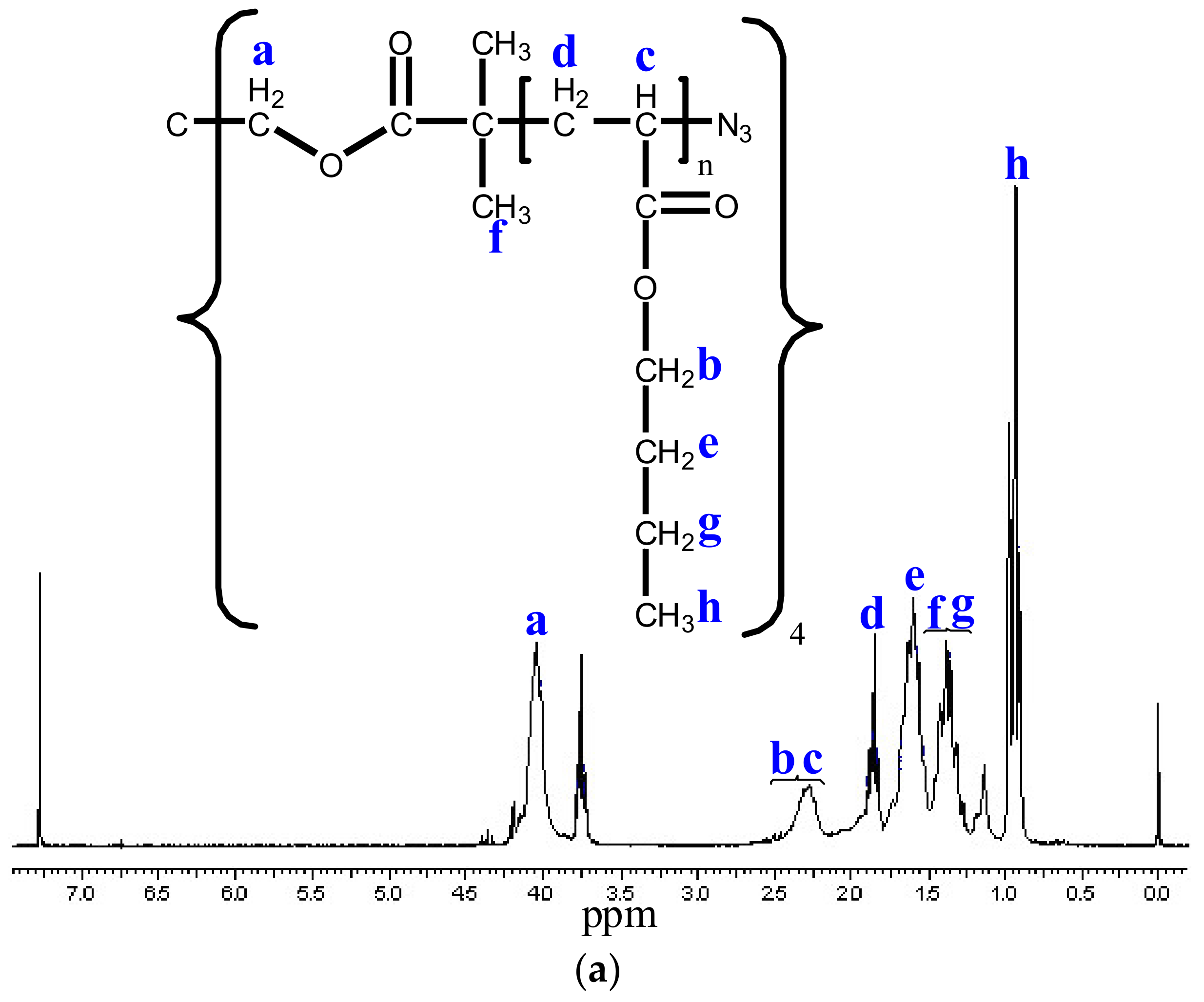
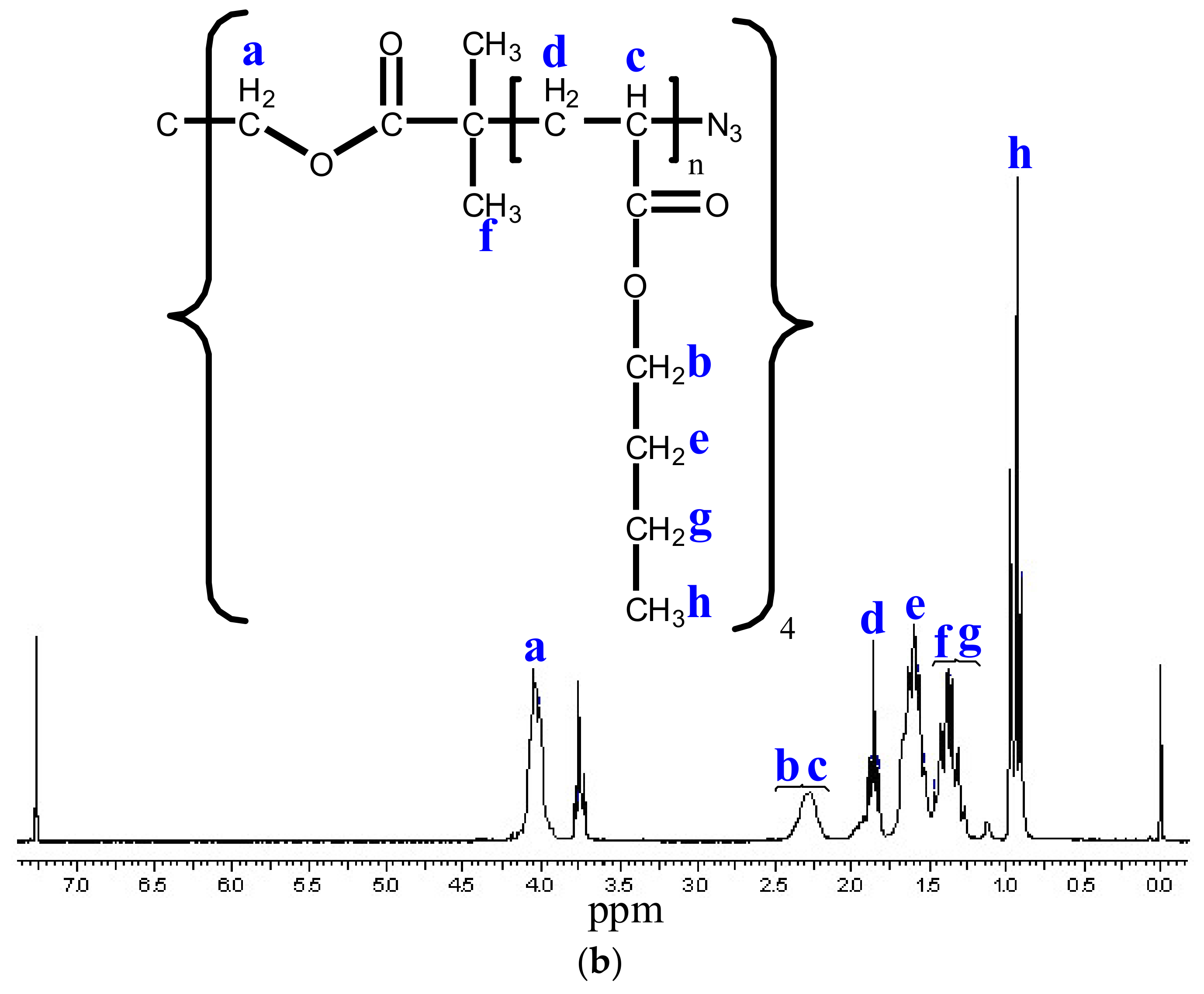
3.3. Modification of the Chain End of Star Polymers by a Substitution Reaction
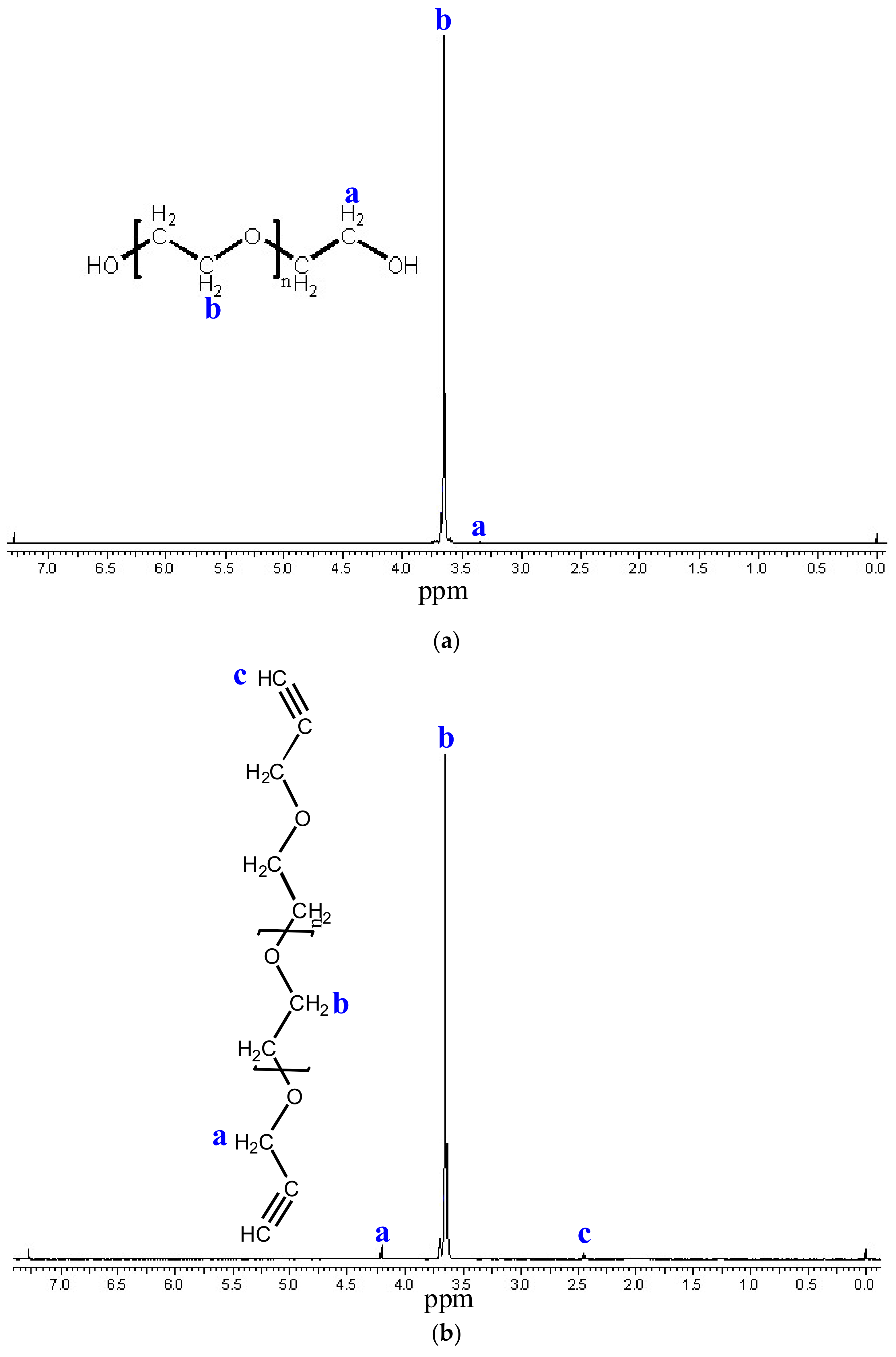
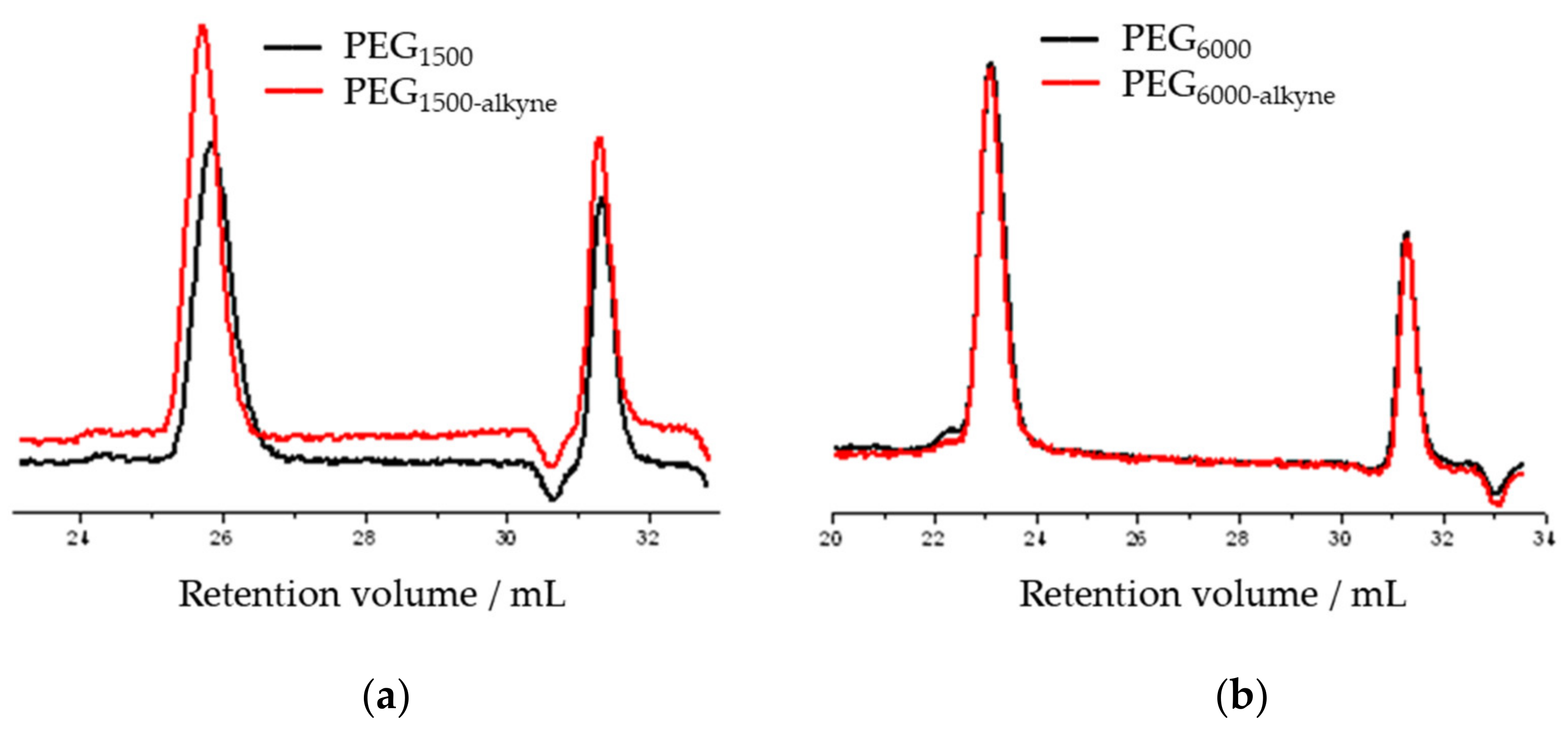
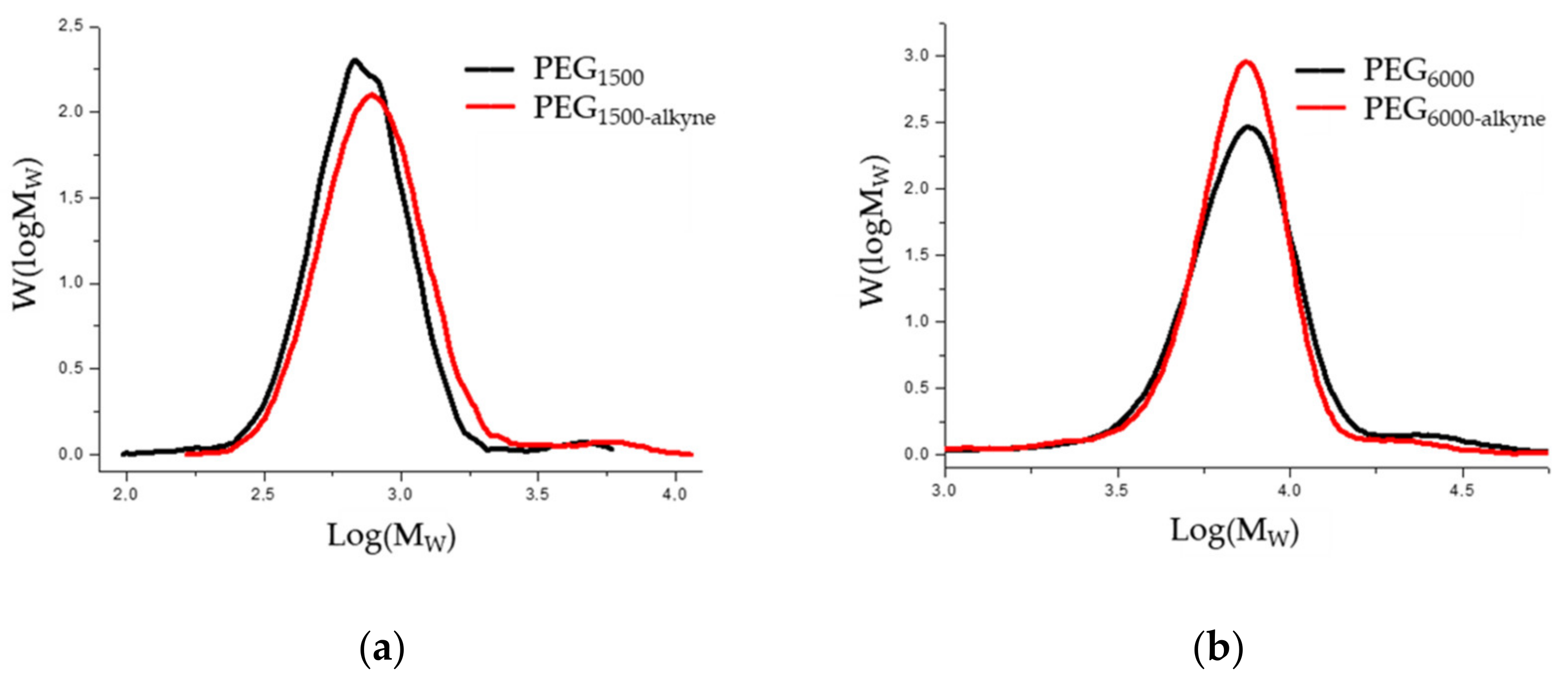
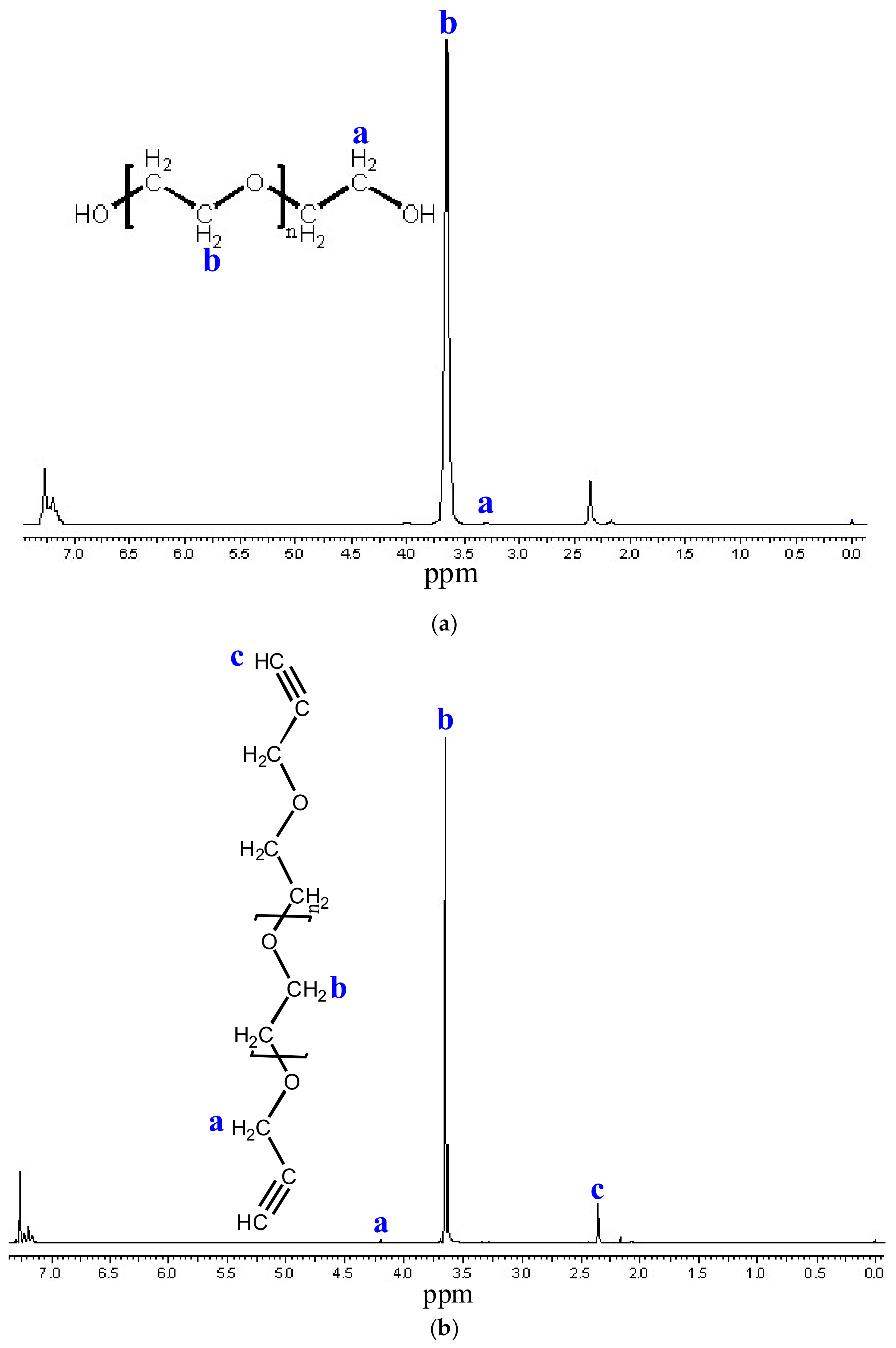
3.4. Functionalization of Poly (Ethylene Glycol)
3.5. Synthesis of a New Type of Amphiphilic Conetworks
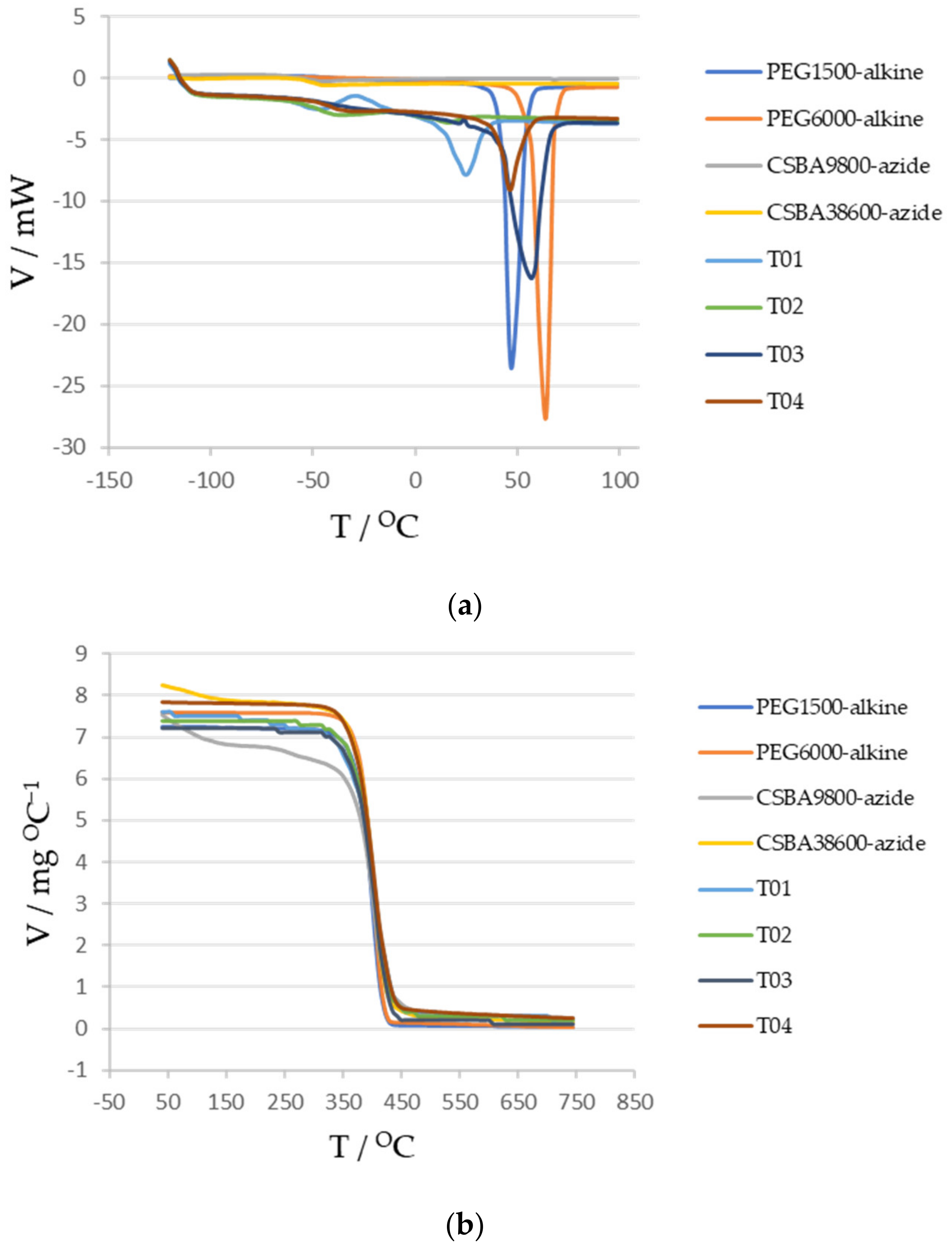
3.6. Differential Scanning Calorimetry and Thermogravimetry
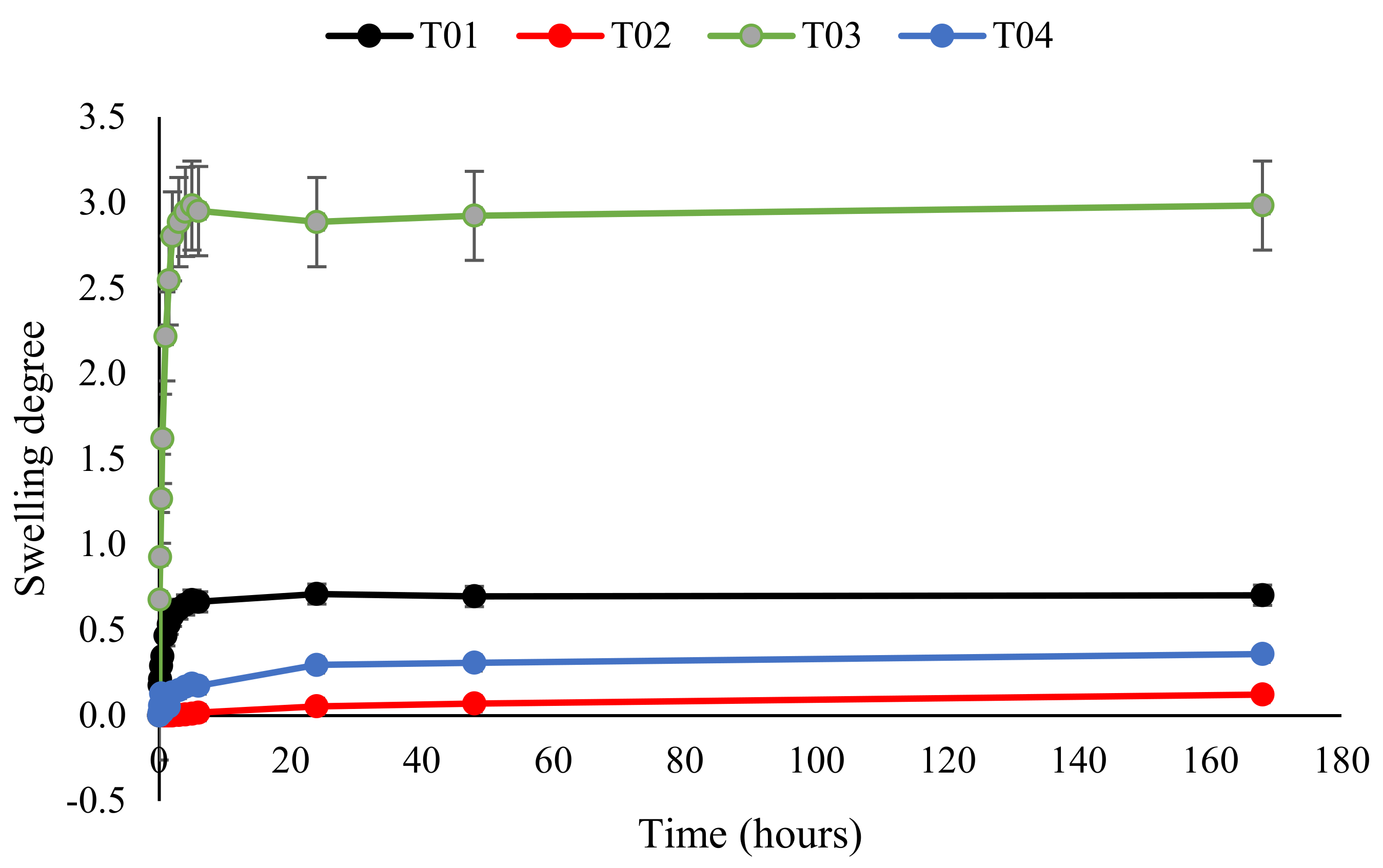
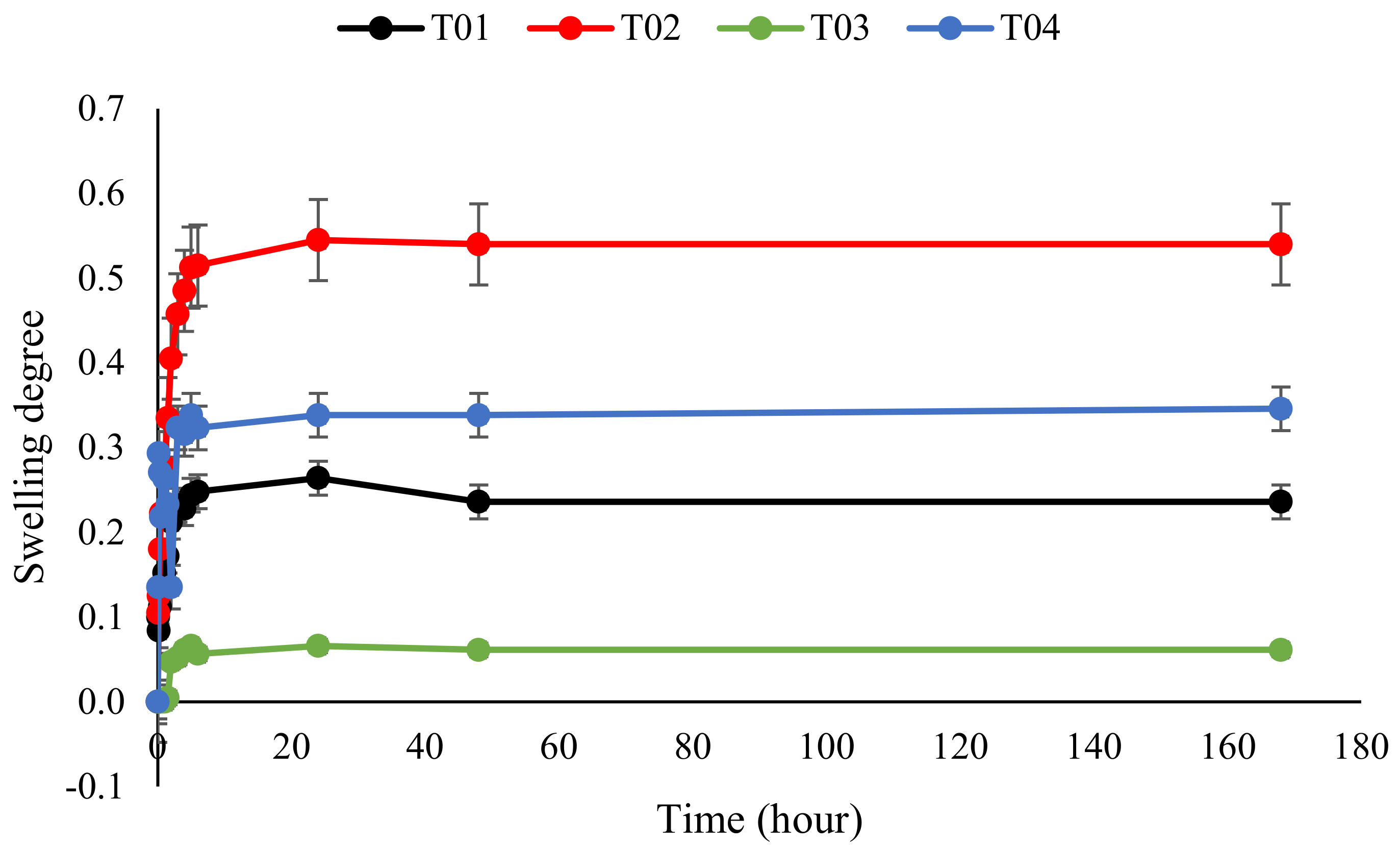
3.7. Swelling of Polymer Conetworks
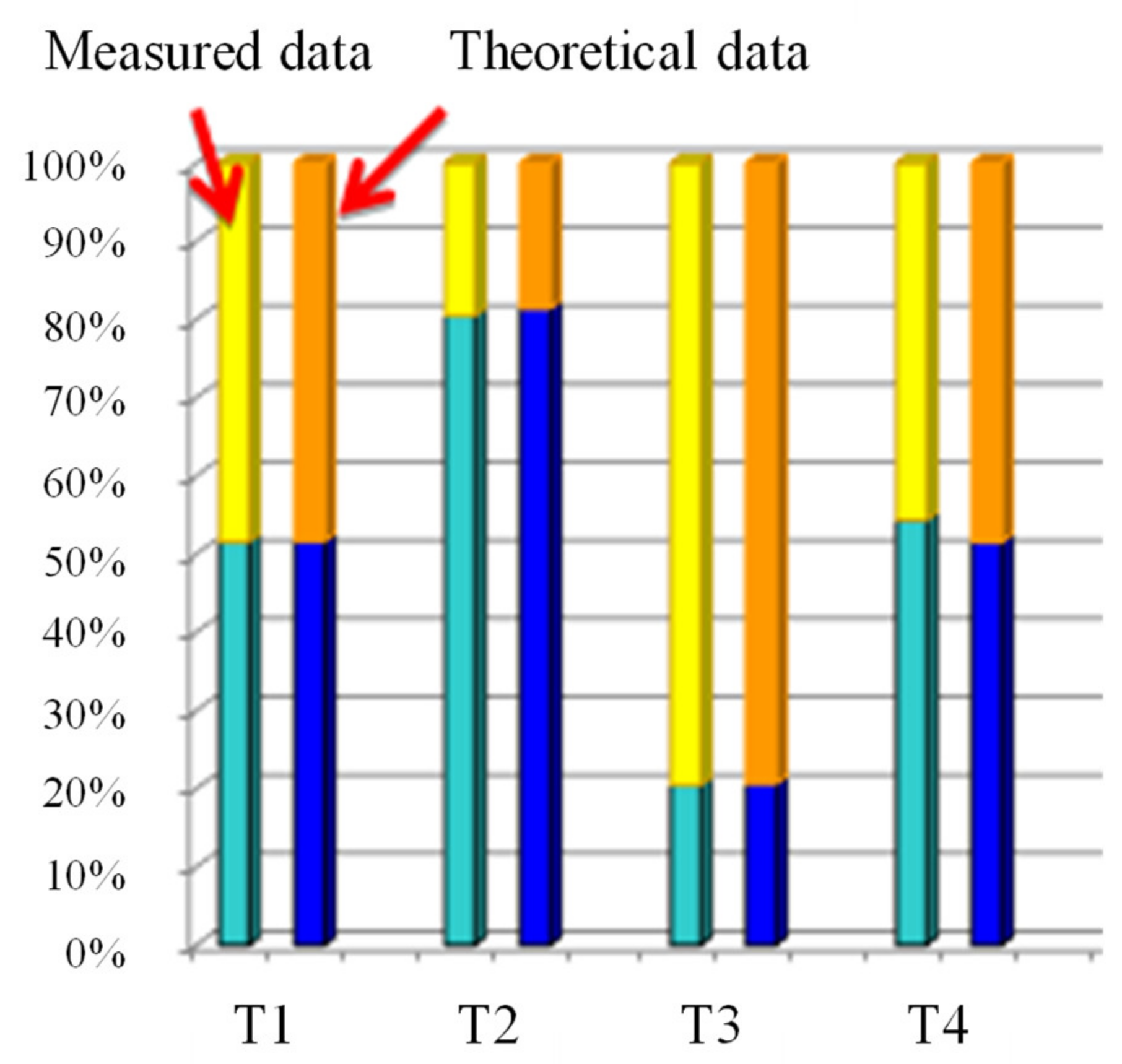
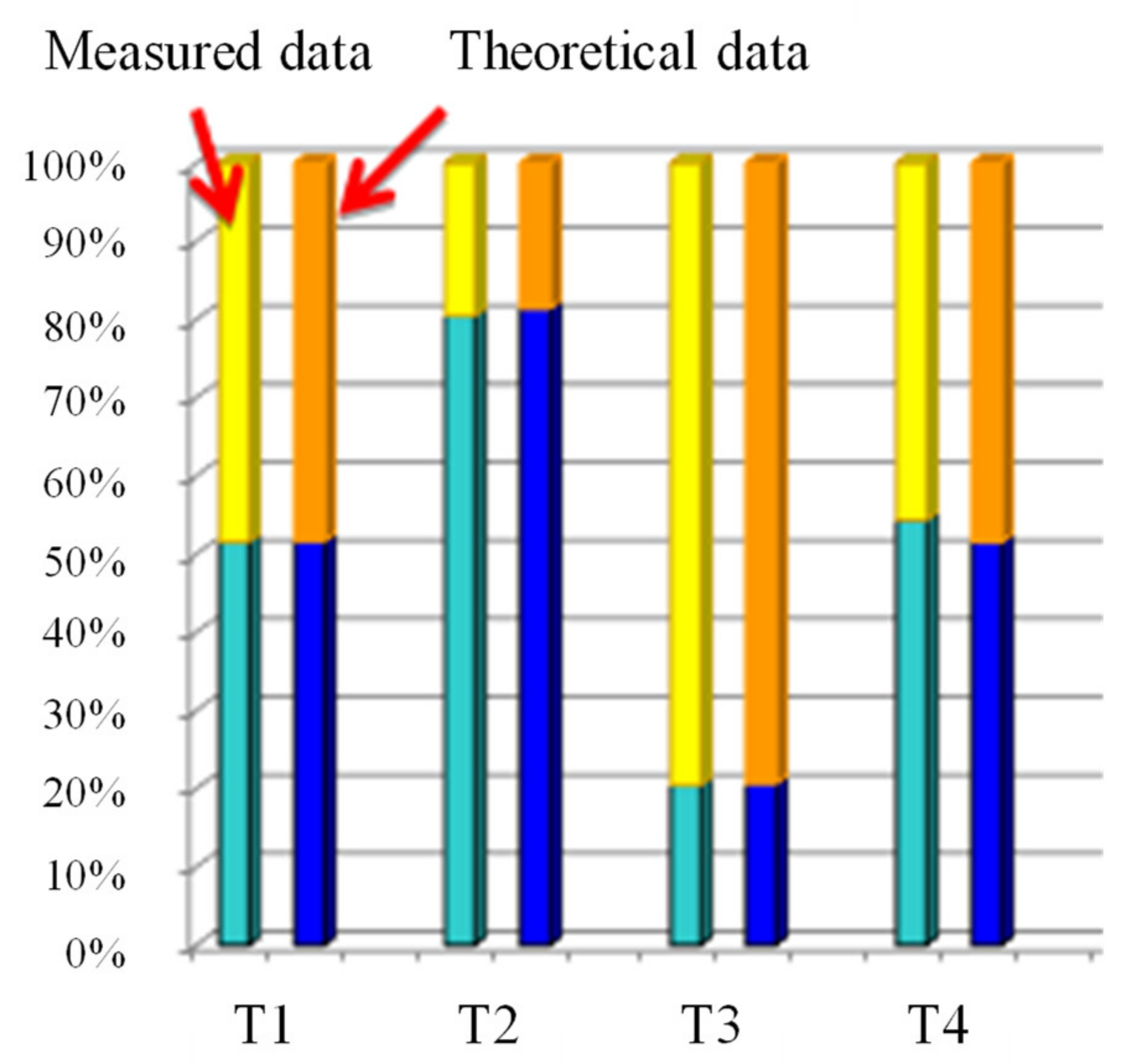
3.8. Elemental Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baekeland, L.H. Method of Making Insoluble Products of Phenol and Formaldehyde. U.S. Patent 942,699, 7 December 1909. [Google Scholar]
- Hermann Staudinger Foundation of Polymer Science—Landmark. Available online: https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/staudingerpolymerscience.html (accessed on 23 February 2022).
- The Nobel Prize in Chemistry 1953. Available online: https://www.nobelprize.org/prizes/chemistry/1953/staudinger/biographical/ (accessed on 23 February 2022).
- Chen, D.; Kennedy, J.P.; Allen, A.J. Amphiphilic Networks. I. Network Synthesis by Copolymerization of Methacryloyl-Capped Polyisobutylene with 2-(Dimethylamino) Ethyl Methacrylate and Characterization of The Networks. J. Macromol. Sci. Part A Chem. 1988, 25, 389–401. [Google Scholar] [CrossRef]
- Weber, M.; Stadler, R. Hydrophilic-hydrophobic two-component polymer networks: 1. Synthesis of reactive poly(ethylene oxide) telechelics. Polymer 1988, 29, 1064–1070. [Google Scholar] [CrossRef]
- MWeber, M.; Stadler, R. Hydrophilic-hydrophobic two-component polymer networks: 2. Synthesis and characterization of poly(ethylene oxide)-linked-polybutadiene. Polymer 1988, 29, 1071–1078. [Google Scholar]
- Szwarc, M.; Levy, M.; Milkovich, R. Polymerization initiated by electron transfer to monomer: A new method of formation of block polymers. J. Am. Chem. Soc. 1956, 78, 2656–2657. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Iván, B. Designed Polymers by Carbocationic Macromolecular Engineering: Theory and Practice; Hanse Publishers: München, Germany; New York, NY, USA,, 1992. [Google Scholar]
- Garside, M. Global Plastic Production 1950–2018—Statista. Available online: https://www.statista.com/statistics/282732/global-production-ofplastics-since-1950 (accessed on 12 January 2022).
- Rikkou-Kalourkoti, M.; Patrickios, C.S. Synthesis and Characterization of End-Linked Amphiphilic Copolymer Conetworks Based on a Novel Bifunctional Cleavable Chain Transfer Agent. Macromolecules 2012, 45, 7890–7899. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Bera, A.; Nutan, B.; Jewrajka, S.K. Reactive Compatibilizer Mediated Precise Synthesis and Application of Stimuli Responsive Polysaccharides-Polycaprolactone Amphiphilic Co-Network Gels. Polymer 2016, 99, 470–479. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Jewrajka, S.K. CHAPTER 3: Designing Multi-Component Biodegradable/Biocompatible Amphiphilic Polymer Co-Networks for Biomedical Applications. In Amphiphilic Polymer Co-Networks; RSC Publishing: London, UK, 2020; pp. 47–76. [Google Scholar]
- Chandel, A.K.S.; Kumar, C.U.; Jewrajka, S.K. Effect of Polyethylene Glycol on Properties and Drug Encapsulation–Release Performance of Biodegradable/Cytocompatible Agarose–Polyethylene Glycol–Polycaprolactone Amphiphilic Co-Network Gels. ACS Appl. Mater. Interfaces 2016, 8, 3182–3192. [Google Scholar] [CrossRef]
- Bera, A.; Chandel, A.K.S.; Kumar, C.U.; Jewrajka, S.K. Degradable/cytocompatible and pH responsive amphiphilic conetwork gels based on agarose-graft copolymers and polycaprolactone. J. Mater. Chem. B 2015, 3, 8548–8557. [Google Scholar] [CrossRef]
- Nutan, B.; Chandel, A.K.S.; Jewrajka, S.K. Liquid Prepolymer-Based in Situ Formation of Degradable Poly(Ethylene Glycol)-Linked-Poly(Caprolactone)-Linked-Poly(2-Dimethylaminoethyl)Methacrylate Amphiphilic Conetwork Gels Showing Polarity Driven Gelation and Bioadhesion. ACS Appl. Bio Mater. 2018, 1, 1606–1619. [Google Scholar] [CrossRef]
- Konstantinov, I.A.; Broadbelt, L.J. Chapter 2—A Quantum Mechanical Approach for Accurate Rate Parameters of Free-Radical Polymerization Reactions. In Computational Quantum Chemistry; Soroush, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 17–46. ISBN 978-0-12-815983-5. [Google Scholar]
- Chandel, A.K.S.; Nutan, B.; Raval, I.H.; Jewrajka, S.K. Self-Assembly of Partially Alkylated Dextran-Graft-Poly[(2-Dimethylamino)Ethyl Methacrylate] Copolymer Facilitating Hydrophobic/Hydrophilic Drug Delivery and Improving Conetwork Hydrogel Properties. Biomacromolecules 2018, 19, 1142–1153. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/“living” Radical Polymerization. Atom Transfer Radical Polymerization in the Presence of Transition-Metal Complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Kato, M.; Kamigaito, M.; Sawamoto, M.; Higashimura, T. Polymerization of Methyl Methacrylate with the Carbon Tetrachloride/Dichlorotris- (Triphenylphosphine)Ruthenium(II)/Methylaluminum Bis(2,6-Di-Tert-Butylphenoxide) Initiating System: Possibility of Living Radical Polymerization. Macromolecules 1995, 28, 1721–1723. [Google Scholar] [CrossRef]
- Matyjaszewski, K. (Ed.) Controlled/Living Radical Polymerization: From Synthesis to Materials; American Chemical Society: Washington, DC, USA, 2006; ISBN 978-0-8412-3991-3. [Google Scholar]
- Coessens, V.; Pintauer, T.; Matyjaszewski, K. Functional Polymers by Atom Transfer Radical Polymerization. Prog. Polym. Sci. 2001, 26, 337–377. [Google Scholar] [CrossRef]
- Baek, K.-Y.; Kamigaito, M.; Sawamoto, M. Synthesis of End-Functionalized Poly(Methyl Methacrylate) by Ruthenium-Catalyzed Living Radical Polymerization with Functionalized Initiators. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 1937–1944. [Google Scholar] [CrossRef]
- Baek, K.-Y.; Kamigaito, M.; Sawamoto, M. Star Poly(Methyl Methacrylate) with End-Functionalized Arm Chains by Ruthenium-Catalyzed Living Radical Polymerization. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 1972–1982. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/“Living” Radical Polymerization. Halogen Atom Transfer Radical Polymerization Promoted by a Cu(I)/Cu(II) Redox Process. Macromolecules 1995, 28, 7901–7910. [Google Scholar] [CrossRef]
- Ando, T.; Kato, M.; Kamigaito, M.; Sawamoto, M. Living Radical Polymerization of Methyl Methacrylate with Ruthenium Complex: Formation of Polymers with Controlled Molecular Weights and Very Narrow Distributions. Macromolecules 1996, 29, 1070–1072. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Brown, W.C.; Morelli, B.C.; Tang, W.; Poli, R.; Matyjaszewski, K. Origin of Activity in Cu-, Ru-, and Os-Mediated Radical Polymerization. Macromolecules 2007, 40, 8576–8585. [Google Scholar] [CrossRef]
- Kong, L.-Z.; Pan, C.-Y. Preparation of Dendrimer-like Copolymers Based on Polystyrene and Poly(l-Lactide) and Formation of Hollow Microspheres. Polymer 2008, 49, 200–210. [Google Scholar] [CrossRef]
- Van Camp, W. Novel Routes for the Design of Poly((meth)acrylic acid) Containing Polymer Structures by Controlled Radical Polymerization. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2007. [Google Scholar]
- Matyjaszewski, K.; Nakagawa, Y.; Gaynor, S.G. Synthesis of Well-Defined Azido and Amino End-Functionalized Polystyrene by Atom Transfer Radical Polymerization. Macromol. Rapid Commun. 1997, 18, 1057–1066. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. Polymersome/Silica Capsules by ‘Click’-Chemistry. Macromol. Rapid Commun. 2008, 29, 1097–1103. [Google Scholar] [CrossRef]
- Dimonie, M.; Teodorescu, M. Phase Transfer Catalysis Synthesis of α,ω-Diallylpoly(Ethylene Oxide). Makromol. Chem. Rapid Commun. 1993, 14, 303–307. [Google Scholar] [CrossRef]
- Malkoch, M.; Vestberg, R.; Gupta, N.; Mespouille, L.; Dubois, P.; Mason, A.F.; Hedrick, J.L.; Liao, Q.; Frank, C.W.; Kingsbury, K.; et al. Synthesis of Well-Defined Hydrogel Networks Using Click Chemistry. Chem. Commun. 2006, 26, 2774–2776. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Lewis, D.R.; Díaz, D.D.; Finn, M.G.; Koberstein, J.T.; Turro, N.J. Synthesis of Degradable Model Networks via ATRP and Click Chemistry. J. Am. Chem. Soc. 2006, 128, 6564–6565. [Google Scholar] [CrossRef]


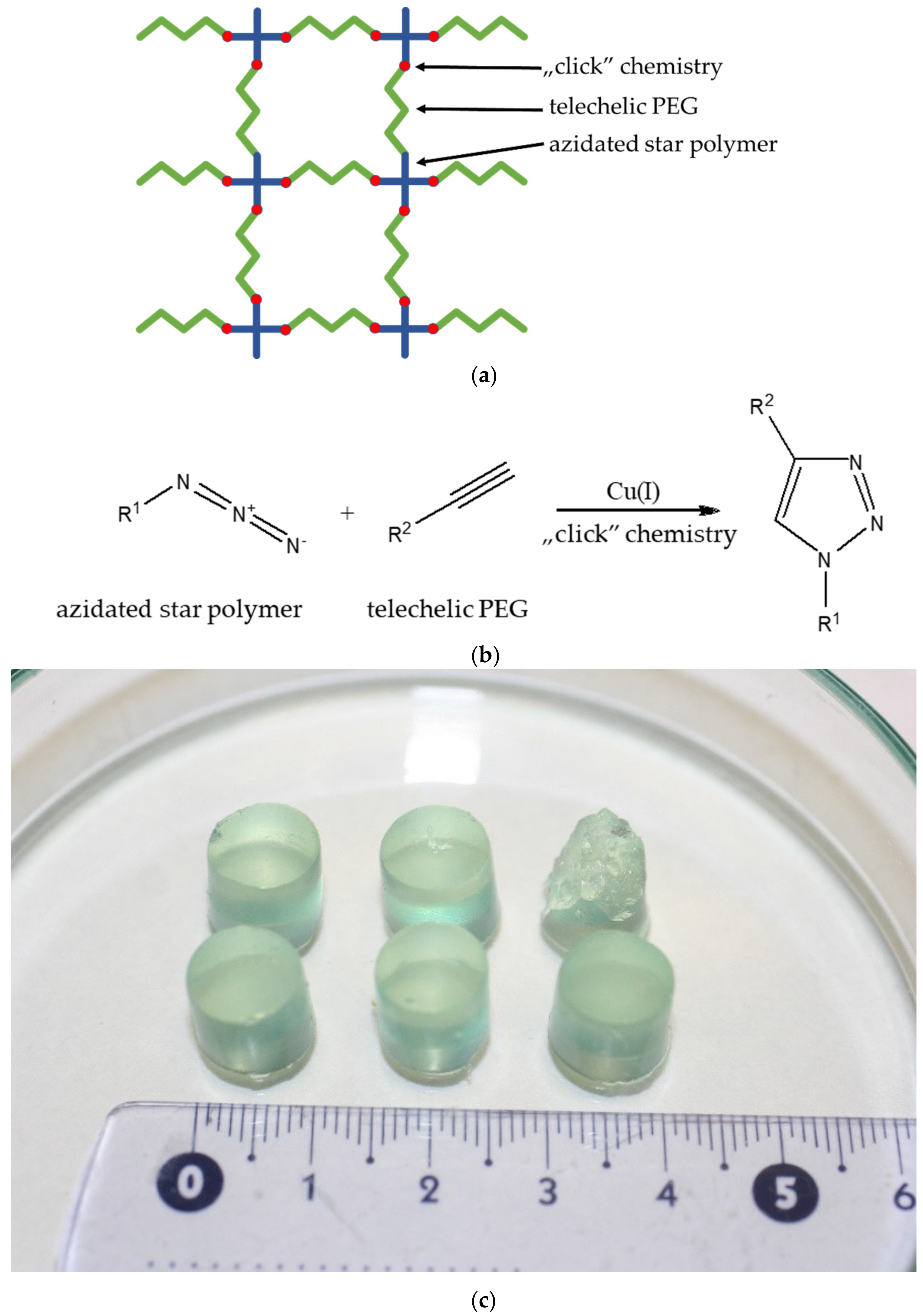
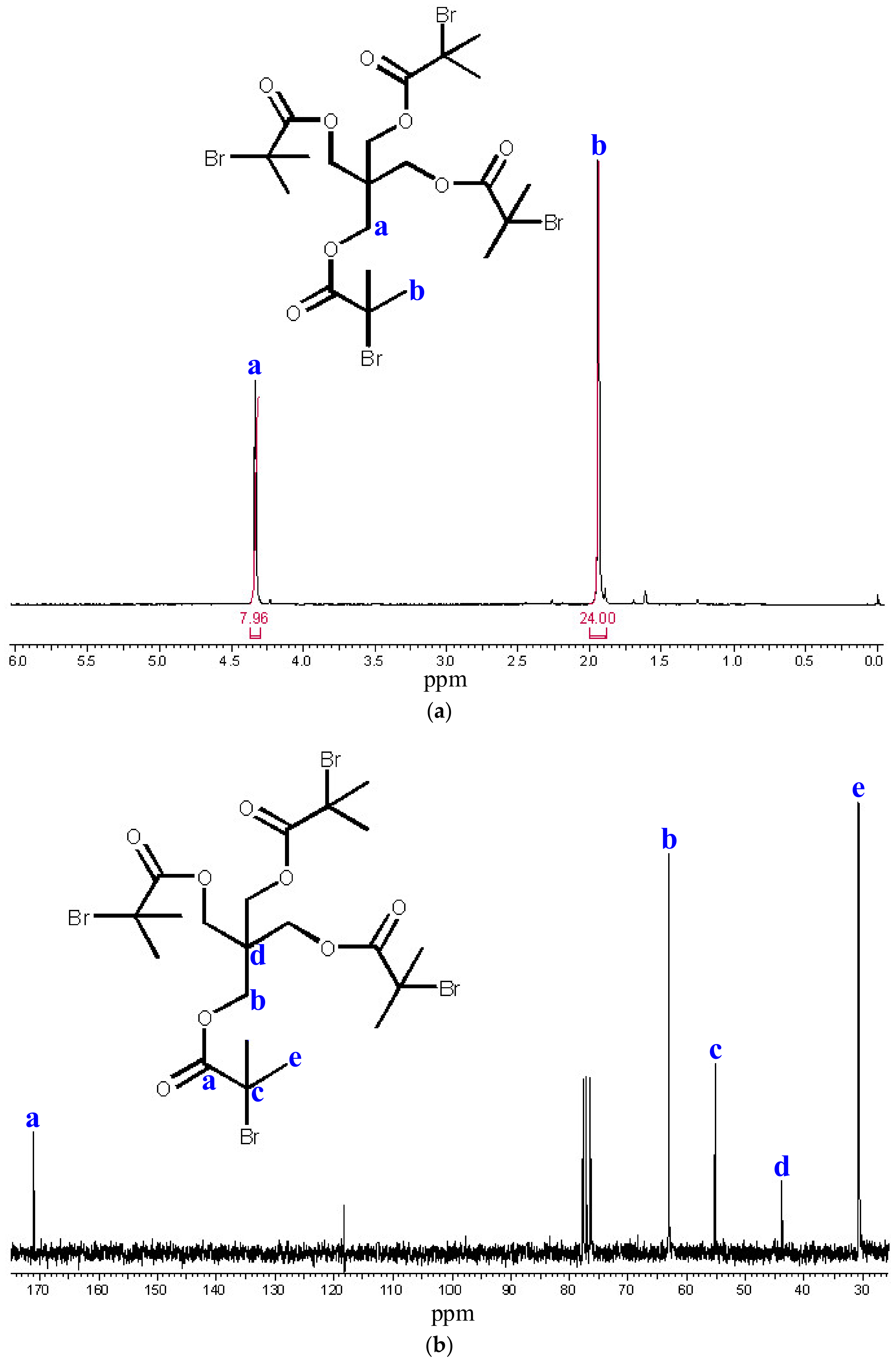
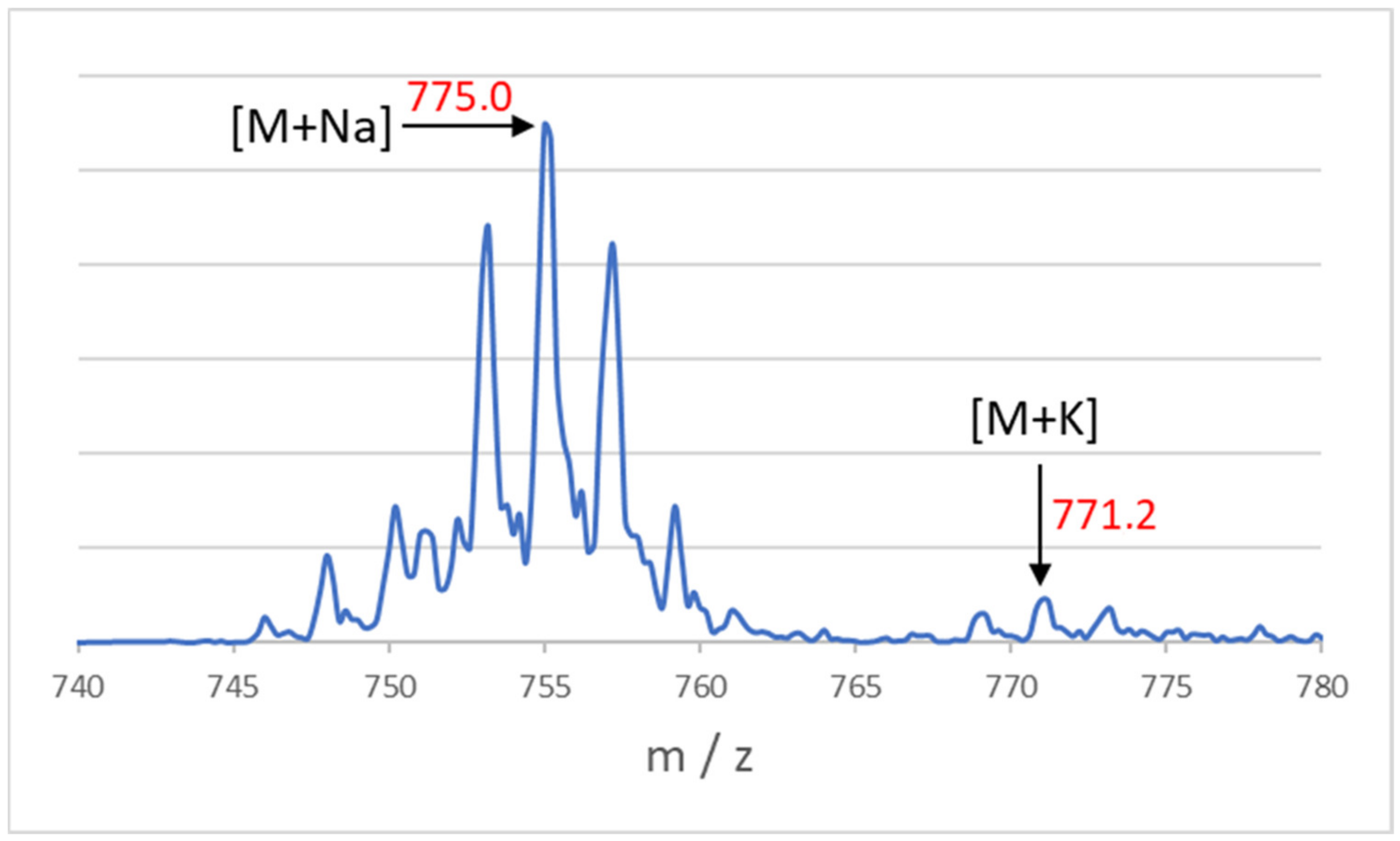

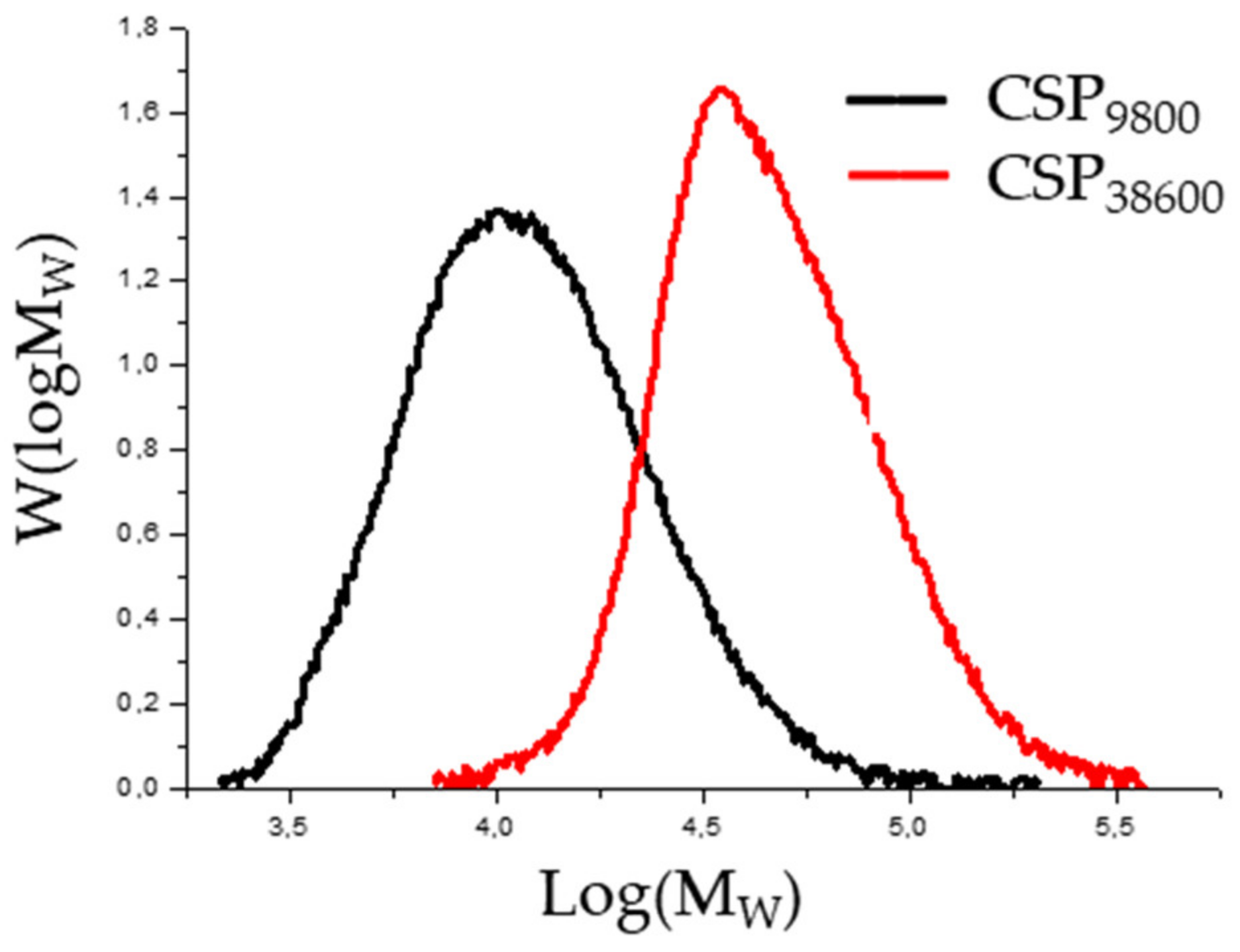


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Molar Ratio | Weight (g) | Measured Quantity (g) |
|---|---|---|---|
| Pentaerythritol | 5 | 6.8 | 6.77 |
| 2-Bromo-2-methylpropionic acid | 25 | 42 | 42.40 |
| p-toluenesulfonic acid | 1 | 2 | 2.03 |
| Toluene (solvent) | - | 173 | 172.98 |
| Name | Molar Ratio | Weight (g) | Measured Quantity (g) |
|---|---|---|---|
| l-ascorbic acid | 6 | 2.1134 | 2.1510 |
| CuBr | 0.4 | 0.1148 | 0.1142 |
| HMTETA | 0.4 | 0.5530 | 0.5530 |
| Initiator | 1 | 1.4642 | 1.4658 |
| n-butyll acrylate | 33 | 8.5358 | 8.5523 |
| Anisole | 80 | 17.3303 | 17.3687 |
| Name | Molar Ratio | Weight (g) | Measured Quantity (g) |
|---|---|---|---|
| l-ascorbic acid | 6 | 0.4227 | 0.4224 |
| CuBr | 0.4 | 0.0229 | 0.0264 |
| HMTETA | 0.4 | 0.1106 | 0.1106 |
| Initiator | 1 | 0.2929 | 0.2926 |
| n-butyll acrylate | 190 | 9.7071 | 9.7218 |
| Anisole | 80 | 19.7084 | 19.7475 |
| Name | Molar Ratio | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|
| Me3SiN3 | 20 | 0.9137 | 1.043 | 1.10 mL |
| TBAF | 20 | - | 7.945 | 8.00 mL |
| THF | 479 | 13.82 | 15.54 | 15.50 mL |
| CSBA9800 | 1 | 3.885 | - | 3.885 g |
| Name | Molar Ratio | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|
| Me3SiN3 | 20 | 0.2392 | 0.2731 | 0.28 mL |
| TBAF | 20 | - | 2.08 | 2.10 mL |
| THF | 1978 | 14.26 | 16.04 | 16.00 mL |
| CSBA38600 | 1 | 4.010 | - | 4.010 g |
| Name | Molar Ratio | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|
| Propargyl bromide | 20 | 7.9299 | 7.2 | 7.4 mL |
| NaOH | 20 | 2.6667 | - | 2.7 g |
| PEG1500 | 1 | 5 | - | 5.0034 g |
| Toluene solvent | - | 21.625 | 25 | 25 mL |
| Name | Molar Ratio | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|
| Propargyl bromide | 20 | 1.9939 | 1.86 | 1.9 mL |
| NaOH | 20 | 0.6667 | - | 0.7 g |
| PEG6000 | 1 | 5 | - | 5.0 g |
| Toluene solvent | - | 21.625 | 25 | 25 mL |
| Name | Molar Ratio | Mol (mmol) | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|---|
| CSBA9800-azide | 1 | 0.05 | 500 | - | 507 mg |
| PEG1500-alkyne | 2 | 0.1 | 150 | - | 151 mg |
| l-ascorbic acid | 2 | 0.1 | 20 | - | 20 mg |
| PMDETA | 4 | 0.2 | 35 | 0.042 | 0.15 mL |
| CuCl | 4 | 0.2 | 20 | - | 20 mg |
| Toluene | - | - | - | 1 | - |
| Reaction conditions | 35 °C, 17 h | ||||
| Name | Molar Ratio | Mol (mmol) | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|---|
| CSBA38600-azide | 1 | 0.013 | 500 | - | 507 mg |
| PEG1500-alkyne | 2 | 0.026 | 39 | - | 39 mg |
| l-ascorbic acid | 0.2 | 0.0026 | 4.6 | - | 8.8 mg |
| PMDETA | 0.4 | 0.0052 | 0.011 | 0.022 mL | |
| CuCl | 0.4 | 0.0052 | 5 | - | 5.3 mg |
| Toluene | - | - | - | 1 | - |
| Reaction conditions | room temperature, 51 h | ||||
| Name | Molar Ratio | Mol (mmol) | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|---|
| CSBA9800-azide | 1 | 0.05 | 500 | - | 500 mg |
| PEG6000-alkyne | 2 | 0.102 | 612 | - | 612 mg |
| l-ascorbic acid | 0.2 | 0.0102 | 1.8 | - | 7.4 mg |
| PMDETA | 0.4 | 0.0204 | 3.5 | 0.042 | 0.06 mL |
| CuCl | 0.4 | 0.0204 | 2 | - | 20 mg |
| Toluene | - | - | - | 2 | - |
| Reaction conditions | room temperature, 20 h | ||||
| Name | Molar Ratio | Mol (mmol) | Weight (mg) | Volume (mL) | Measured Quantity |
|---|---|---|---|---|---|
| CSBA38600-azide | 1 | 0.013 | 500 | - | 507 mg |
| PEG6000-alkyne | 2 | 0.026 | 39 | - | 39 mg |
| l-ascorbic acid | 0.2 | 0.0026 | 4.6 | - | 8.8 mg |
| PMDETA | 0.4 | 0.0052 | - | 0.011 | 0.022 ml |
| CuCl | 0.4 | 0.0052 | 5 | - | 5.3 mg |
| Toluene | - | - | - | 1 | - |
| Reaction conditions | room temperature, 20 h | ||||
| Sample | Mn 1 (g/mol) | Mw 2 (g/mol) | Mw/Mn 3 | Yield (%) | Polymerization Degree |
|---|---|---|---|---|---|
| CSBA9800 | 9800 | 15,400 | 1.57 | 78 | 18 |
| CSBA38600 | 38,600 | 55,000 | 1.42 | 80 | 74 |
| Name | Mn 1 (g/mol) | Mw 2 (g/mol) | Mw/Mn 3 | Yield (%) | Polymerization Degree |
|---|---|---|---|---|---|
| PEG1500 | 1408 | 1760 | 1.25 | - | 34 |
| PEG1500-alkin | 1606 | 2120 | 1.32 | 51 | 34 |
| PEG6000 | 5590 | 8450 | 1.51 | - | 136 |
| PEG6000-alkin | 6030 | 7760 | 1.29 | 67 | 136 |
| Code of the Amphiphilic Conetwork | Components | Temperature (°C) | Reaction Time (°C) | Color | Remark |
|---|---|---|---|---|---|
| T1 | CSBA9800-azide PEG1500-alkyne | 35 | 17 | Yellow | Rubbery, sticky |
| T2 | CSBA38600-azide PEG1500-alkyne | 25 | 51 | Blue | Rubbery, very sticky |
| T3 | CSBA9800-azide PEG6000-alkyne | 50 | 45 | Pale blue | Solid, easy to cut |
| T4 | CSBA38600-azide PEG6000-alkyne | 50 | 192 | Pale green | Solid, easy to cut |
| Code of the Amphiphilic Conetwork | C (m/m%) | H (m/m%) | BA mol% | PEG mol% |
|---|---|---|---|---|
| T1 | 61.1 | 9.5 | 51.5 | 48.5 |
| T2 | 64.3 | 9.3 | 80.4 | 19.6 |
| T3 | 58.4 | 9.3 | 20.4 | 79.6 |
| T4 | 62.7 | 9.8 | 54.3 | 45.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illés, G.; Németh, C.; Hidas, K.I.; Surányi, J.; Tóth, A.; Pajor, F.; Póti, P. Synthesis of New Type Polymers by Quasi-Living Atom Transfer Radical Polymerization. Polymers 2022, 14, 2795. https://doi.org/10.3390/polym14142795
Illés G, Németh C, Hidas KI, Surányi J, Tóth A, Pajor F, Póti P. Synthesis of New Type Polymers by Quasi-Living Atom Transfer Radical Polymerization. Polymers. 2022; 14(14):2795. https://doi.org/10.3390/polym14142795
Chicago/Turabian StyleIllés, Gergely, Csaba Németh, Karina Ilona Hidas, József Surányi, Adrienn Tóth, Ferenc Pajor, and Péter Póti. 2022. "Synthesis of New Type Polymers by Quasi-Living Atom Transfer Radical Polymerization" Polymers 14, no. 14: 2795. https://doi.org/10.3390/polym14142795
APA StyleIllés, G., Németh, C., Hidas, K. I., Surányi, J., Tóth, A., Pajor, F., & Póti, P. (2022). Synthesis of New Type Polymers by Quasi-Living Atom Transfer Radical Polymerization. Polymers, 14(14), 2795. https://doi.org/10.3390/polym14142795