
Preparation, Characterization and In Vitro Evaluation of Eudragit S100-Coated Bile Salt-Containing Liposomes for Oral Colonic Delivery of Budesonide



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
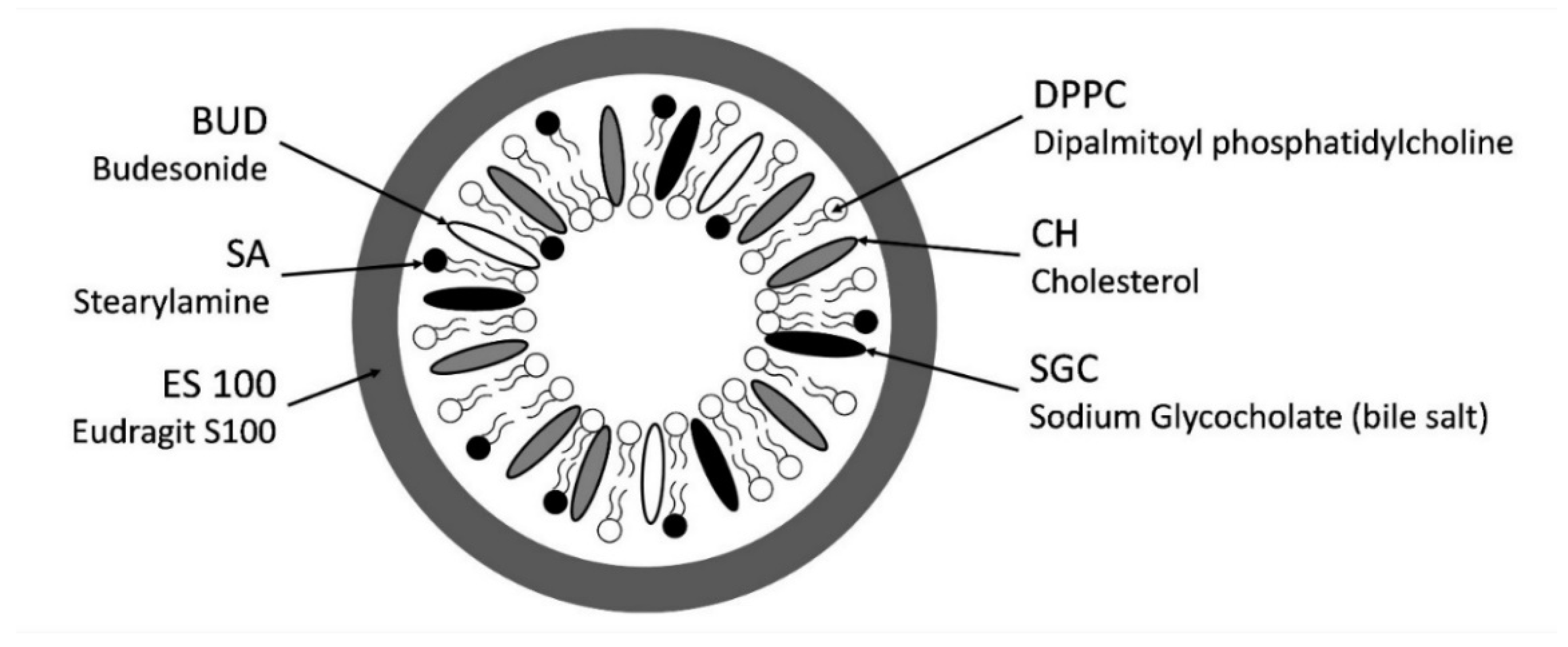
2.2.1. Formulation of Liposomes
2.2.2. Coating of Liposomes
2.2.3. Entrapment Efficiency (EE) Determination
2.2.4. Particle Size Measurements
2.2.5. Zeta Potential Measurements
2.2.6. Drug Release Study
2.2.7. Stability Study
2.2.8. Transmission Electronic Microscopy (TEM)
2.2.9. UV Assay
2.2.10. Fourier Transform Infrared Spectroscopy (FTIR)
2.2.11. Statistical Analysis
3. Results
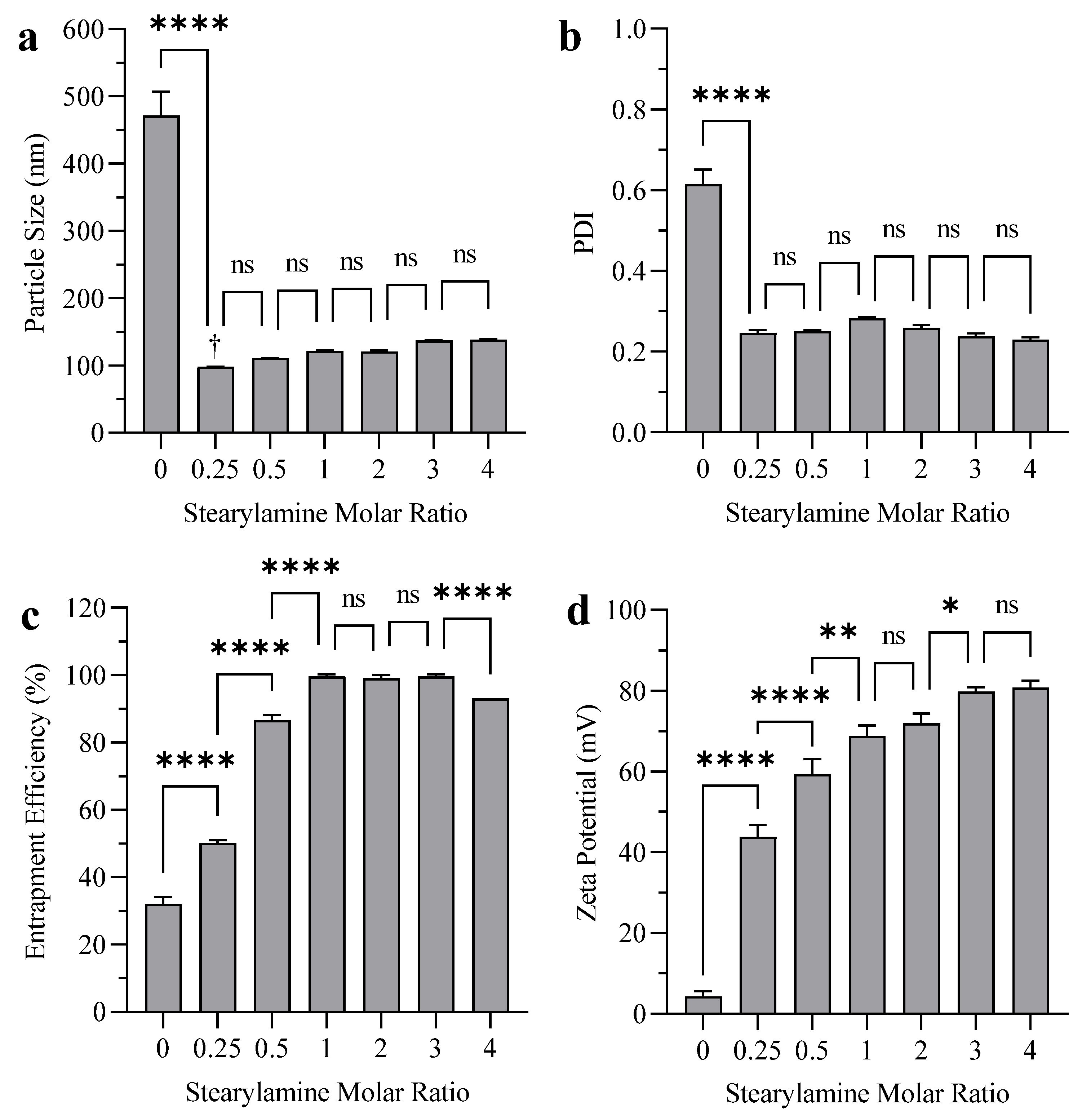
3.1. Effect of SA on Liposome Particle Size, PDI, EE, and Zeta Potential
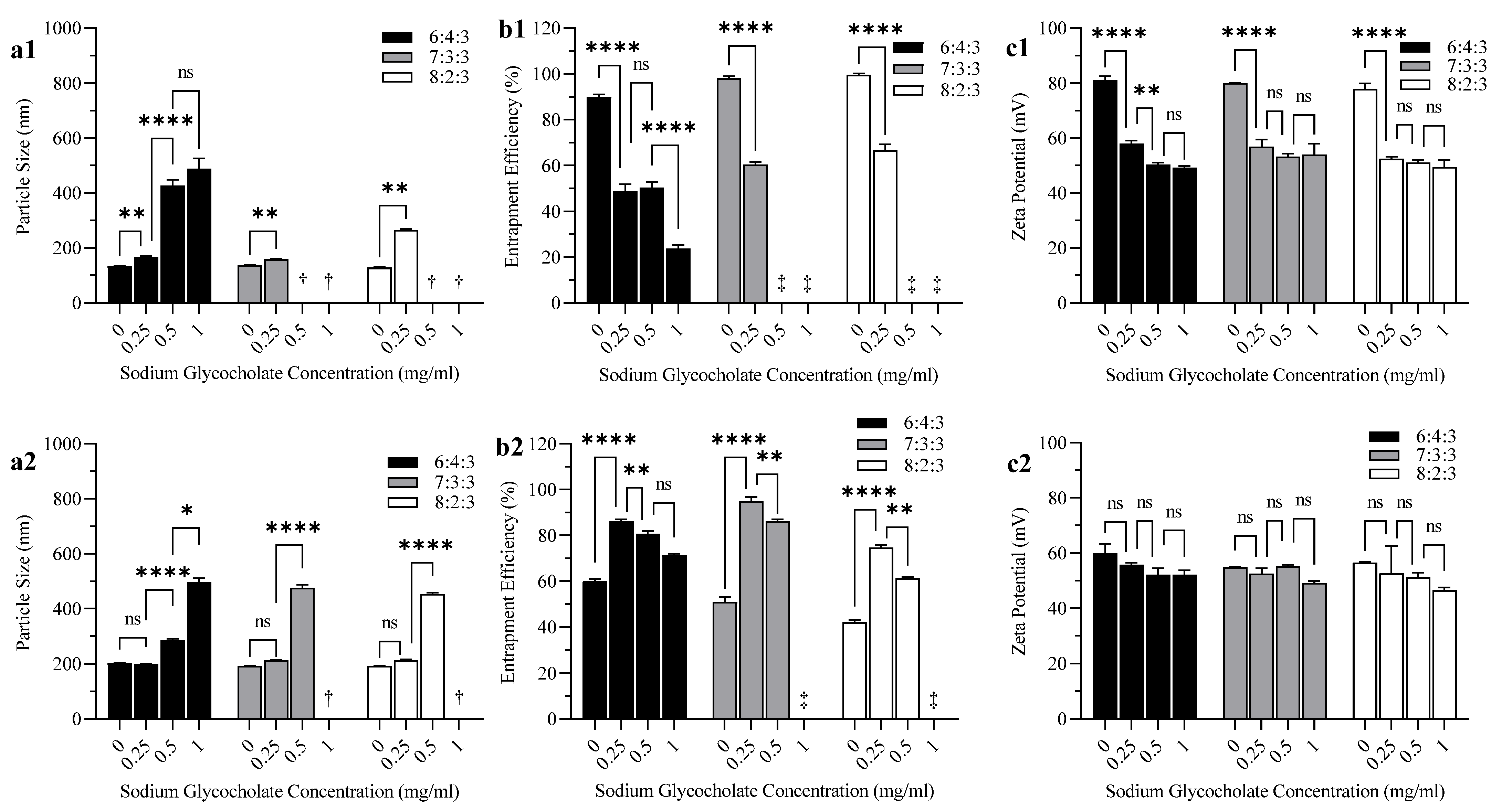
3.2. Effect of Bile Salt and CH Content on Liposome Particle Size, EE, and Zeta Potential
3.3. Effect of Drug/Lipid Weight Ratio on Liposome EE
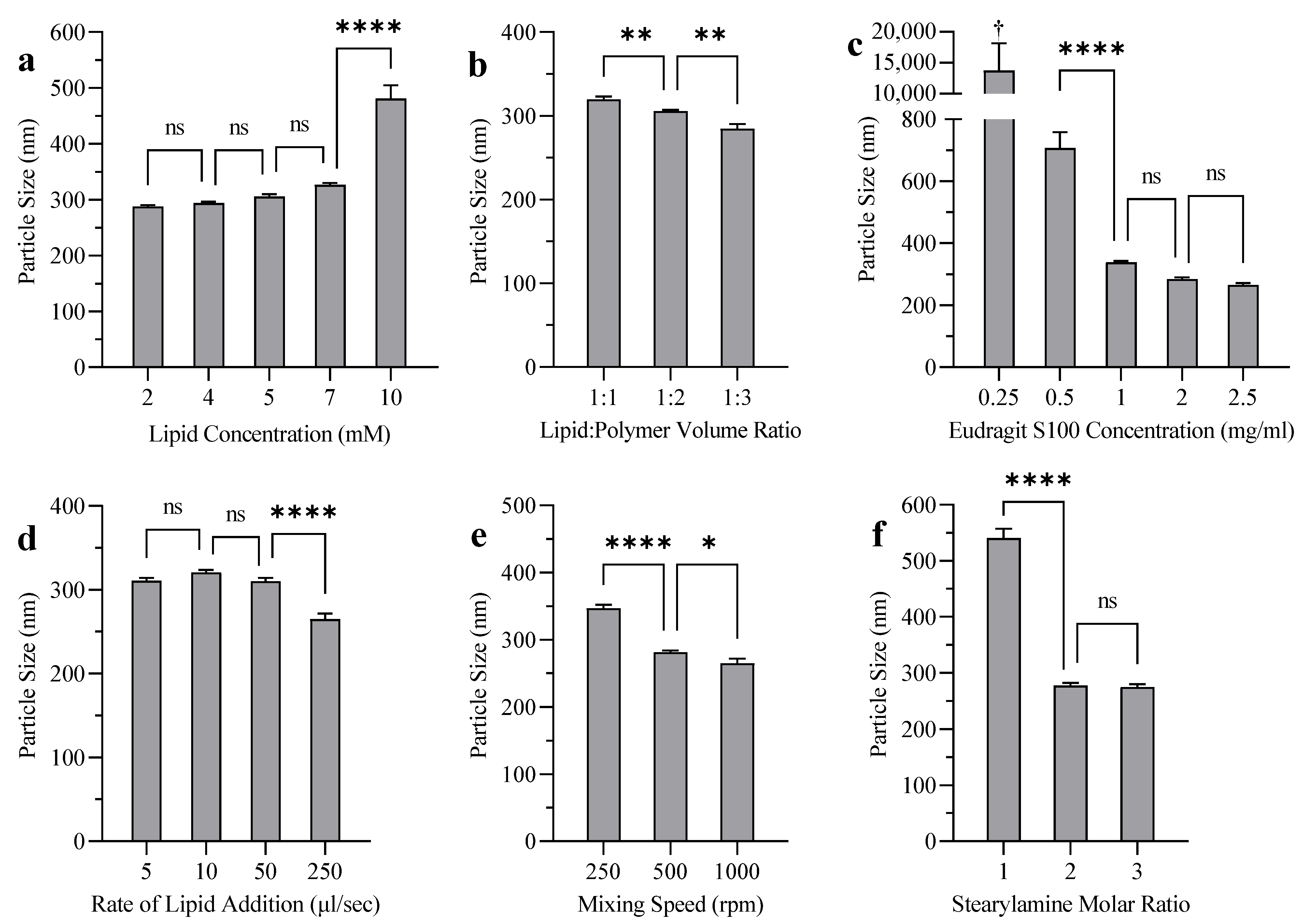
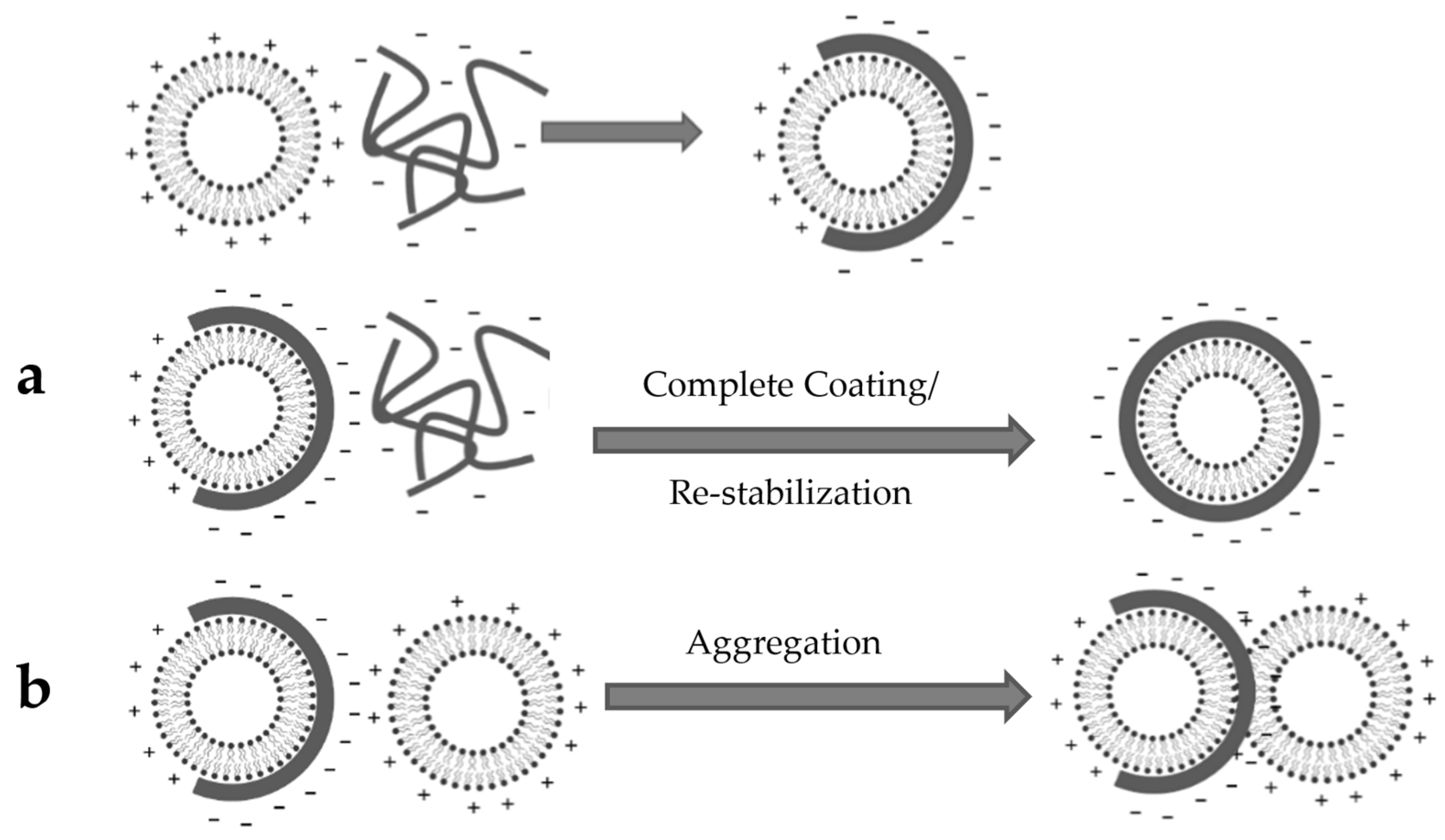
3.4. Liposome Coating
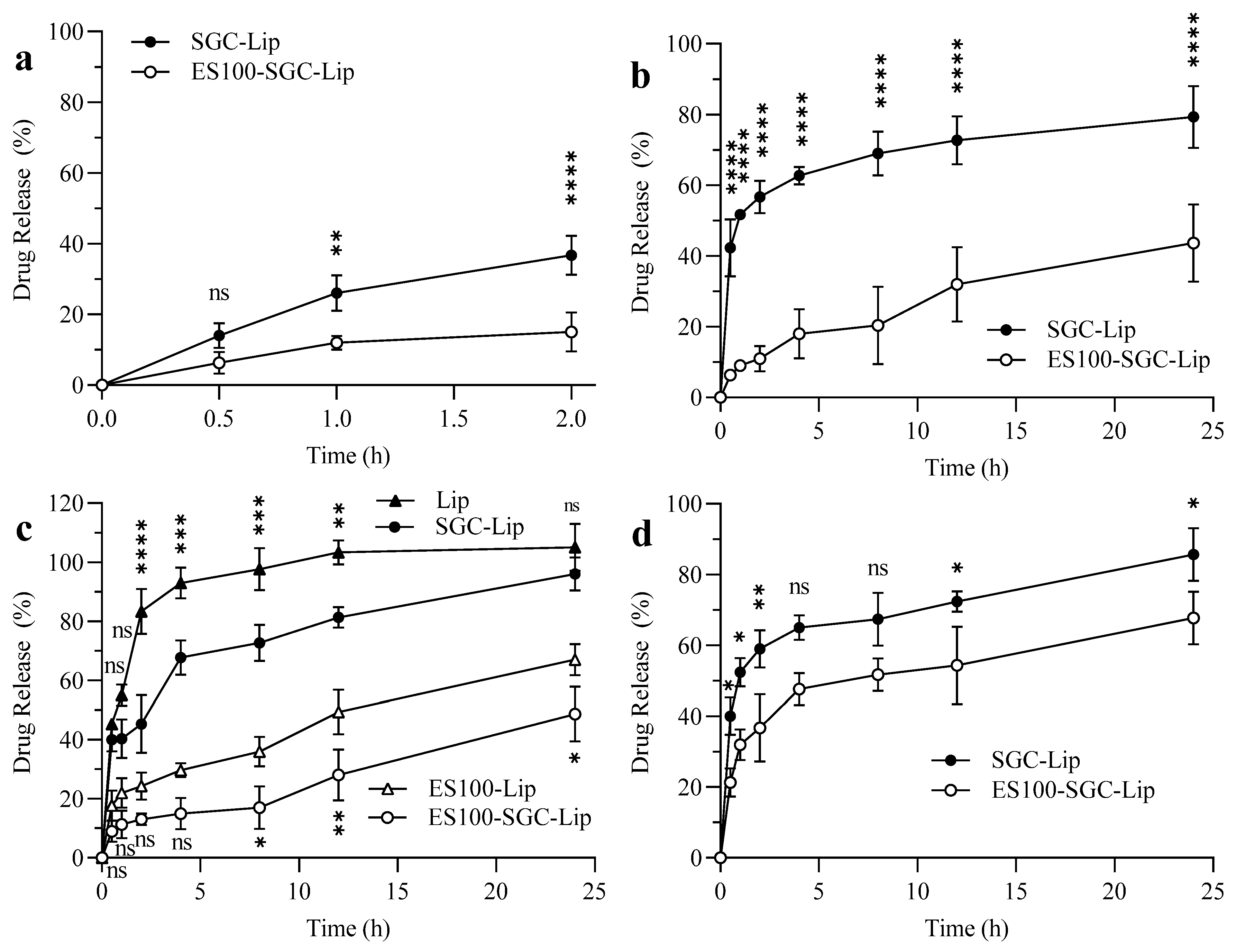
3.5. Release Data
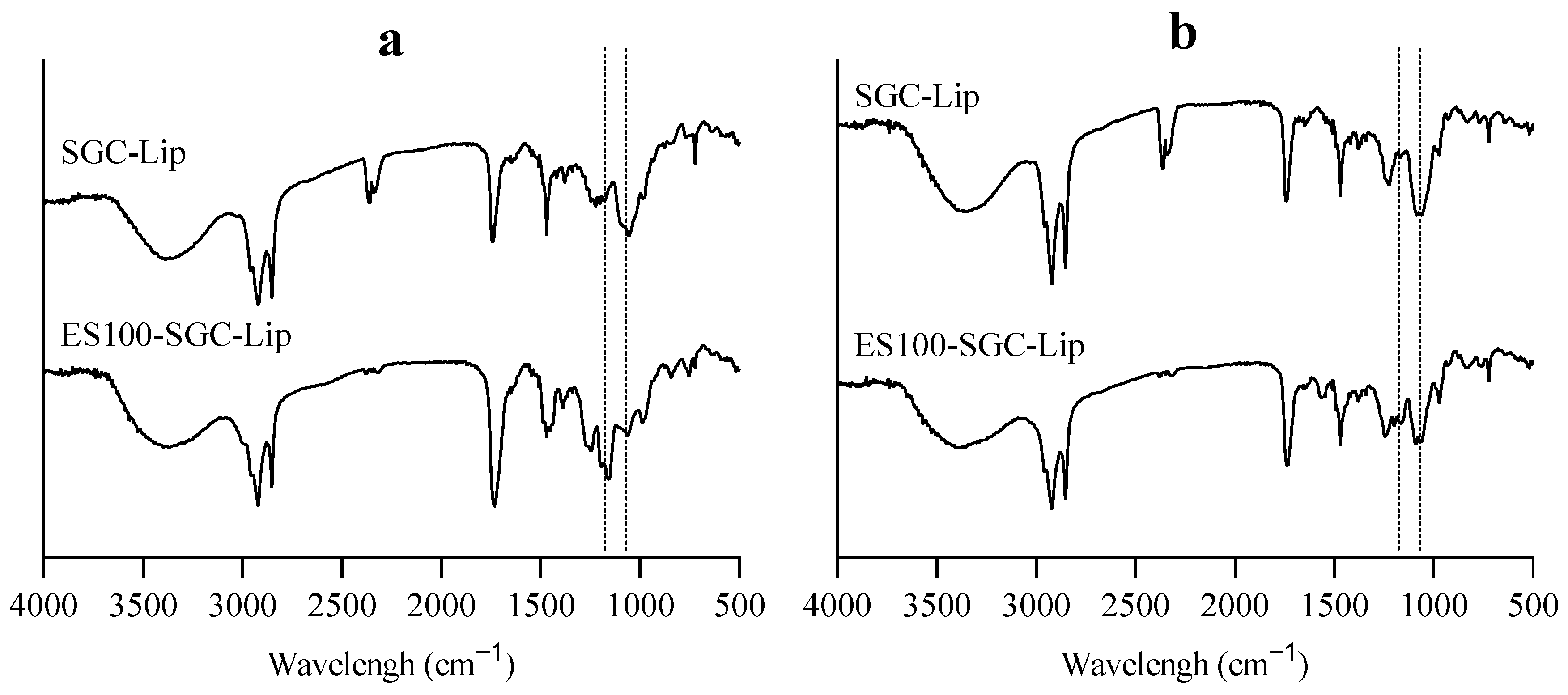
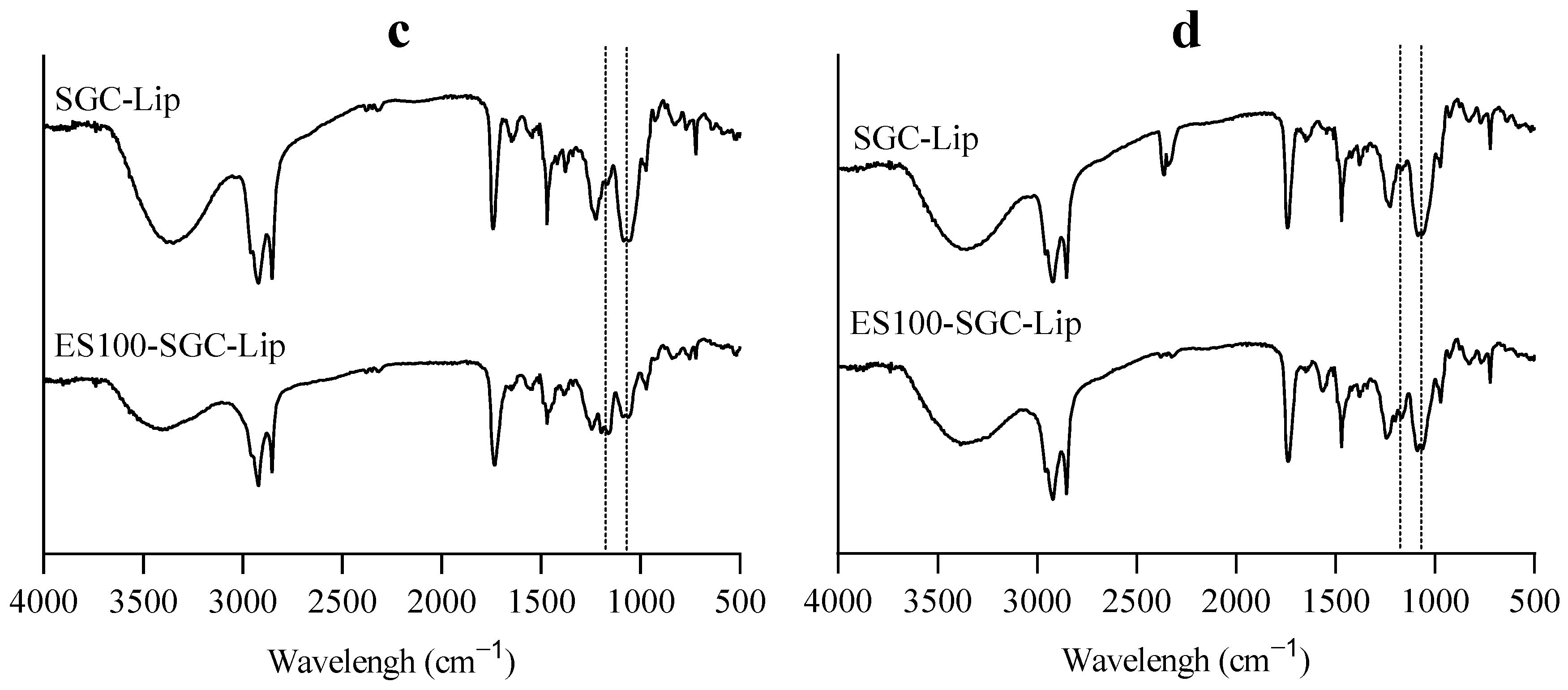
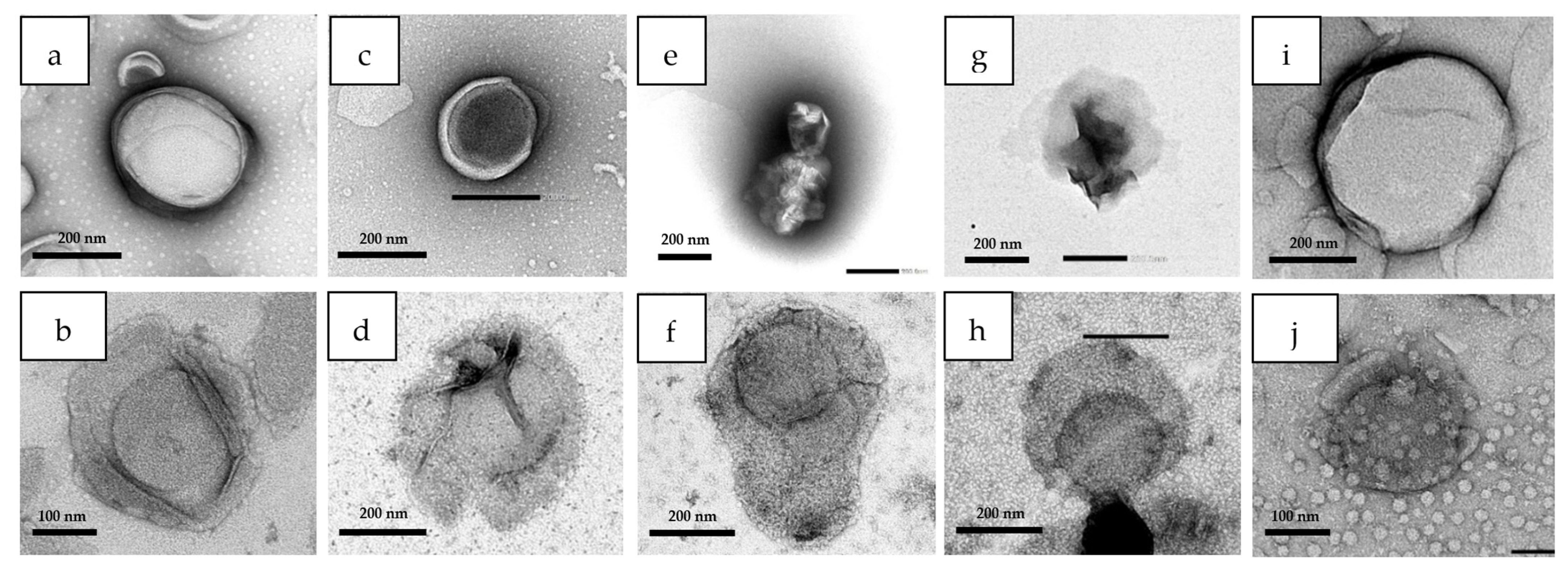
3.6. Liposomal Integrity
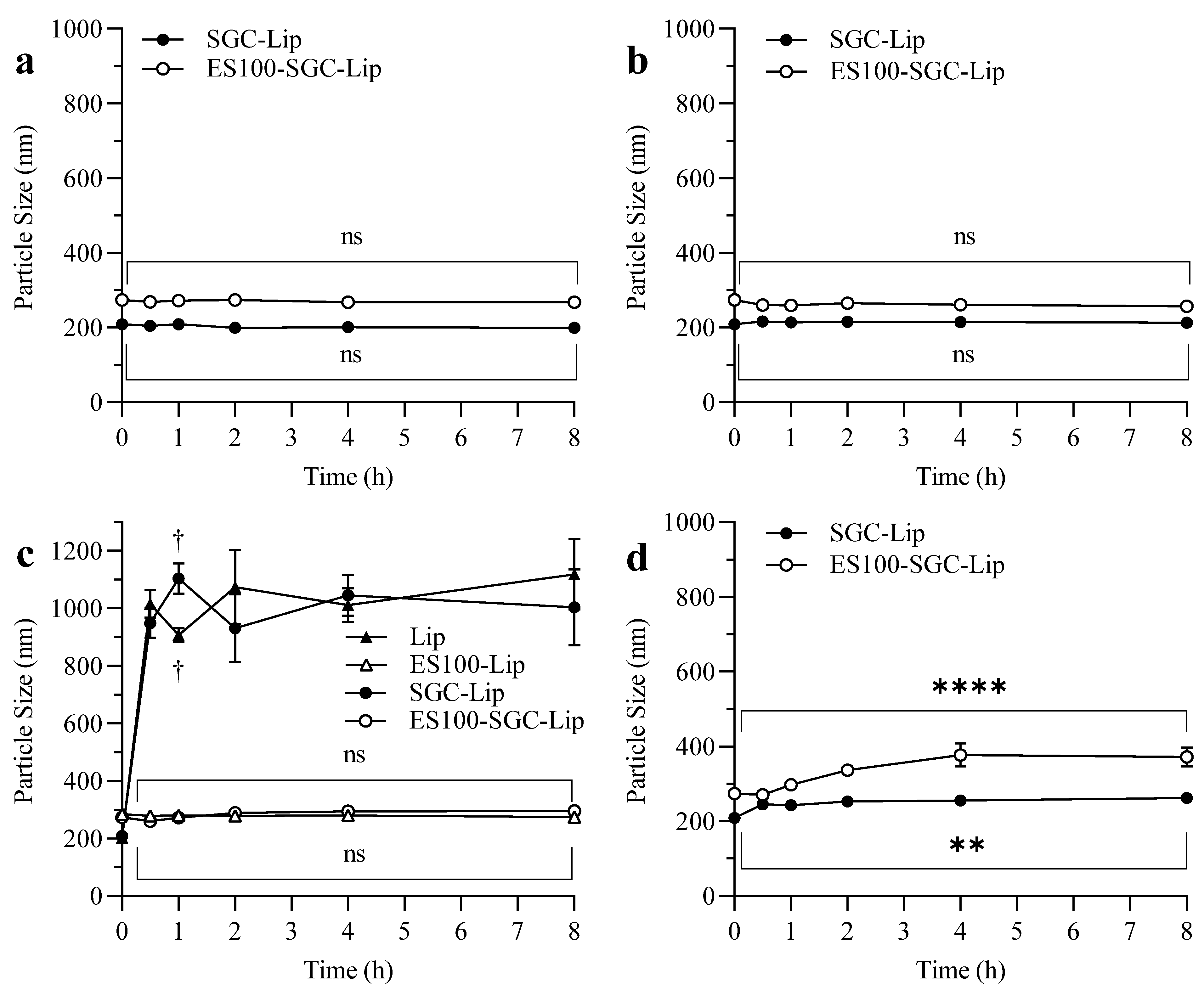
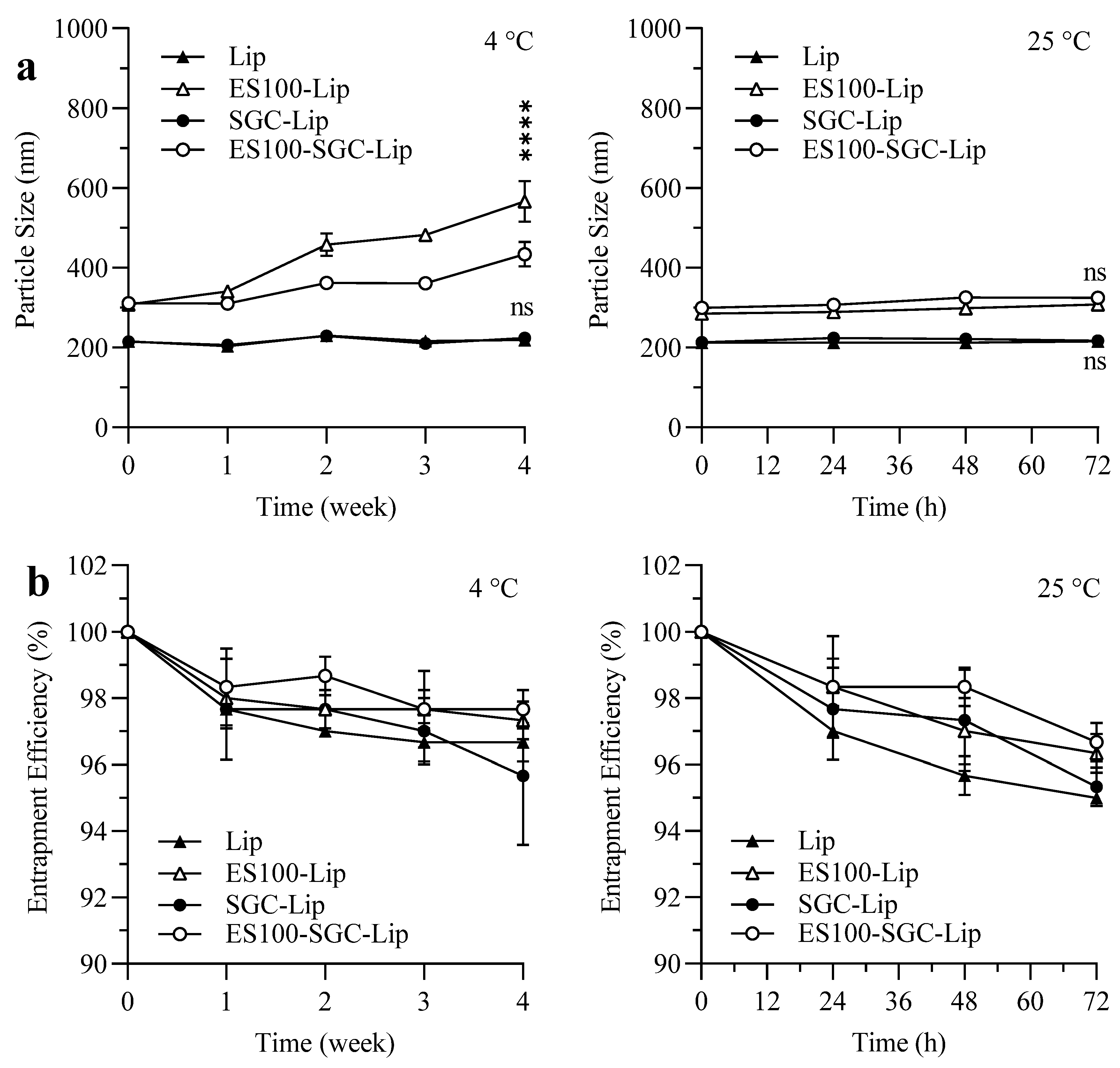
3.7. Stability Study
4. Discussion
4.1. Effect of SA on Liposome Particle Size, PDI, EE, and Zeta Potential
4.2. Effect of Bile Salt and CH Content on Liposome Particle Size, EE, and Zeta Potential
4.3. Effect of Drug/Lipid Weight Ratio on EE
4.4. Liposome Coating
4.5. Drug Release Study
4.6. Liposomal Integrity
4.7. Stability Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinha, V.R.; Singh, A.; Kumar, R.V.; Singh, S.; Kumria, R.; Bhinge, J. Oral colon-specific drug delivery of protein and peptide drugs. Crit. Rev. Ther. Drug Carr. Syst. 2007, 24, 63–92. [Google Scholar] [CrossRef] [PubMed]
- Maroni, A.; Zema, L.; Del Curto, M.D.; Foppoli, A.; Gazzaniga, A. Oral colon delivery of insulin with the aid of functional adjuvants. Adv. Drug Deliv. Rev. 2012, 64, 540–556. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, G.; Qiu, Y.; Li, M.; Liu, D.; Hu, D.; Gu, X.; Xiao, Z. Nanoparticle-based oral delivery systems for colon targeting: Principles and design strategies. Sci. Bull. 2016, 61, 670–681. [Google Scholar] [CrossRef]
- Wong, T.W.; Colombo, G.; Sonvico, F. Pectin Matrix as Oral Drug Delivery Vehicle for Colon Cancer Treatment. AAPS PharmSciTech 2011, 12, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Cattel, L.; Ceruti, M.; Dosio, F. From conventional to stealth liposomes: A new frontier in cancer chemotherapy. J. Chemother. 2004, 16, 94–97. [Google Scholar] [CrossRef]
- Tirosh, B.; Khatib, N.; Barenholz, Y.; Nissan, A.; Rubinstein, A. Transferrin as a luminal target for negatively charged liposomes in the inflamed colonic mucosa. Mol. Pharm. 2009, 6, 1083–1091. [Google Scholar] [CrossRef]
- D’Argenio, G.; Calvani, M.; Casamassimi, A.; Petillo, O.; Margarucci, S.; Rienzo, M.; Peluso, I.; Calvani, R.; Ciccodicola, A.; Caporaso, N. Experimental colitis: Decreased Octn2 and Atb0+ expression in rat colonocytes induces carnitine depletion that is reversible by carnitine-loaded liposomes. FASEB J. 2006, 20, 2544–2546. [Google Scholar] [CrossRef]
- Kesisoglou, F.; Zhou, S.Y.; Niemiec, S.; Lee, J.W.; Zimmermann, E.M.; Fleisher, D. Liposomal Formulations of Inflammatory Bowel Disease Drugs: Local versus Systemic Drug Delivery in a Rat Model. Pharm. Res. 2005, 22, 1320–1330. [Google Scholar] [CrossRef]
- Jubeh, T.T.; Barenholz, Y.; Rubinstein, A. Differential adhesion of normal and inflamed rat colonic mucosa by charged liposomes. Pharm. Res. 2004, 21, 447–453. [Google Scholar] [CrossRef]
- Park, K.; Kwon, I.C.; Park, K. Oral protein delivery: Current status and future prospect. React. Funct. Polym. 2011, 71, 280–287. [Google Scholar] [CrossRef]
- Iwanaga, K.; Ono, S.; Narioka, K.; Morimoto, K.; Kakemi, M.; Yamashita, S.; Nango, M.; Oku, N. Oral delivery of insulin by using surface coating liposomes: Improvement of stability of insulin in GI tract. Int. J. Pharm. 1997, 157, 73–80. [Google Scholar] [CrossRef]
- Takeuchi, H.; Matsui, Y.; Sugihara, H.; Yamamoto, H.; Kawashima, Y. Effectiveness of submicron-sized, chitosan-coated liposomes in oral administration of peptide drugs. Int. J. Pharm. 2005, 303, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Werle, M.; Takeuchi, H. Chitosan–aprotinin coated liposomes for oral peptide delivery: Development, characterisation and in vivo evaluation. Int. J. Pharm. 2009, 370, 26–32. [Google Scholar] [CrossRef]
- Barea, M.; Jenkins, M.; Gaber, M.; Bridson, R. Evaluation of liposomes coated with a pH responsive polymer. Int. J. Pharm. 2010, 402, 89–94. [Google Scholar] [CrossRef]
- Barea, M.; Jenkins, M.; Lee, Y.; Johnson, P.; Bridson, R. Encapsulation of liposomes within pH responsive microspheres for oral colonic drug delivery. Int. J. Biomater. 2012, 2012, 458712. [Google Scholar] [CrossRef]
- Kim, H.Y.; Cheon, J.H.; Lee, S.H.; Min, J.Y.; Back, S.-Y.; Song, J.G.; Kim, D.H.; Lim, S.-J.; Han, H.-K. Ternary nanocomposite carriers based on organic clay-lipid vesicles as an effective colon-targeted drug delivery system: Preparation and in vitro/in vivo characterization. J. Nanobiotech. 2020, 18, 17. [Google Scholar] [CrossRef]
- Birru, W.A.; Warren, D.B.; Ibrahim, A.; Williams, H.D.; Benameur, H.; Porter, C.J.; Chalmers, D.K.; Pouton, C.W. Digestion of phospholipids after secretion of bile into the duodenum changes the phase behavior of bile components. Mol. Pharm. 2014, 11, 2825–2834. [Google Scholar] [CrossRef]
- Conacher, M.; Alexander, J.; Brewer, J.M. Oral immunisation with peptide and protein antigens by formulation in lipid vesicles incorporating bile salts (bilosomes). Vaccine 2001, 19, 2965–2974. [Google Scholar] [CrossRef]
- Chia-Ming, C.; Weiner, N. Gastrointestinal uptake of liposomes. I. In vitro and in situ studies. Int. J. Pharm. 1987, 37, 75–85. [Google Scholar] [CrossRef]
- Chiang, C.-M.; Weiner, N. Gastrointestinal uptake of liposomes. II. In vivo studies. Int. J. Pharm. 1987, 40, 143–150. [Google Scholar] [CrossRef]
- Hu, S.; Niu, M.; Hu, F.; Lu, Y.; Qi, J.; Yin, Z.; Wu, W. Integrity and stability of oral liposomes containing bile salts studied in simulated and ex vivo gastrointestinal media. Int. J. Pharm. 2013, 441, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Khatri, K.; Gupta, P.N.; Goyal, A.K.; Mehta, A.; Vyas, S.P. Oral immunization against hepatitis B using bile salt stabilized vesicles (bilosomes). J. Pharm. Pharm. Sci. 2008, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Arafat, M.; Kirchhoefer, C.; Mikov, M.; Sarfraz, M.; Löbenberg, R. Nanosized liposomes containing bile salt: A vesicular nanocarrier for enhancing oral bioavailability of BCS class III drug. J. Pharm. Pharm. Sci. 2017, 20, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Lu, Y.; Hovgaard, L.; Wu, W. Liposomes containing glycocholate as potential oral insulin delivery systems: Preparation, in vitro characterization, and improved protection against enzymatic degradation. Int. J. Nanomed. 2011, 6, 1155. [Google Scholar]
- Niu, M.; Lu, Y.; Hovgaard, L.; Guan, P.; Tan, Y.; Lian, R.; Qi, J.; Wu, W. Hypoglycemic activity and oral bioavailability of insulin-loaded liposomes containing bile salts in rats: The effect of cholate type, particle size and administered dose. Eur. J. Pharm. Biopharm. 2012, 81, 265–272. [Google Scholar] [CrossRef]
- Aburahma, M.H. Bile salts-containing vesicles: Promising pharmaceutical carriers for oral delivery of poorly water-soluble drugs and peptide/protein-based therapeutics or vaccines. Drug Deliv. 2016, 23, 1847–1867. [Google Scholar] [CrossRef]
- Guan, P.; Lu, Y.; Qi, J.; Niu, M.; Lian, R.; Hu, F.; Wu, W. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int. J. Nanomed. 2011, 6, 965. [Google Scholar]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- De Leo, V.; Milano, F.; Mancini, E.; Comparelli, R.; Giotta, L.; Nacci, A.; Longobardi, F.; Garbetta, A.; Agostiano, A.; Catucci, L. Encapsulation of Curcumin-Loaded Liposomes for Colonic Drug Delivery in a pH-Responsive Polymer Cluster Using a pH-Driven and Organic Solvent-Free Process. Molecules 2018, 23, 739. [Google Scholar] [CrossRef]
- Jain, D.; Panda, A.K.; Majumdar, D.K. Eudragit S100 entrapped insulin microspheres for oral delivery. Aaps Pharmscitech 2005, 6, E100–E107. [Google Scholar] [CrossRef] [PubMed]
- Mattheolabakis, G.; Nie, T.; Constantinides, P.P.; Rigas, B. Sterically stabilized liposomes incorporating the novel anticancer agent phospho-ibuprofen (MDC-917): Preparation, characterization, and in vitro/in vivo evaluation. Pharm. Res. 2012, 29, 1435–1443. [Google Scholar] [CrossRef] [PubMed]
- Pchelintsev, N.A.; Adams, P.D.; Nelson, D.M. Critical Parameters for Efficient Sonication and Improved Chromatin Immunoprecipitation of High Molecular Weight Proteins. PLoS ONE 2016, 11, e0148023. [Google Scholar] [CrossRef]
- Joshi, M.R.; Misra, A. Liposomal budesonide for dry powder inhaler: Preparation and stabilization. Aaps Pharmscitech 2001, 2, 44–53. [Google Scholar] [CrossRef]
- Henriksen, I.; Smistad, G.; Karlsen, J. Interactions between liposomes and chitosan. Int. J. Pharm. 1994, 101, 227–236. [Google Scholar] [CrossRef]
- Henriksen, I.; Våagen, S.R.; Sande, S.A.; Smistad, G.; Karlsen, J. Interactions between liposomes and chitosan II: Effect of selected parameters on aggregation and leakage. Int. J. Pharm. 1997, 146, 193–203. [Google Scholar] [CrossRef]
- Sunamoto, J.; Iwamoto, K.; Kondo, H.; Shinkai, S. Liposomal membranes VI. Polysaccharide-induced aggregation of multilamellar liposomes of egg lecithin. J. Biochem. 1980, 88, 1219–1226. [Google Scholar] [CrossRef]
- Pamunuwa, G.; Karunaratne, V.; Karunaratne, D.N. Effect of Lipid Composition onIn VitroRelease and Skin Deposition of Curcumin Encapsulated Liposomes. J. Nanomater. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- Yokoyama, S.; Inagaki, A.; Tsuchiya, K.; Sakai, H.; Imura, T.; Ohkubo, T.; Tsubaki, N.; Abe, M. Stearylamine changes the liposomal shape from MLVs to LUVs. J. Oleo Sci. 2005, 54, 251–254. [Google Scholar] [CrossRef]
- Mehanna, M.M.; El-Kader, N.A.; Samaha, M.W. Liposomes as potential carriers for ketorolac ophthalmic delivery: Formulation and stability issues. Braz. J. Pharm. Sci. 2017, 53, 216127. [Google Scholar] [CrossRef]
- Gupta, P.N.; Mishra, V.; Singh, P.; Rawat, A.; Dubey, P.; Mahor, S.; Vyas, S.P. Tetanus toxoid-loaded transfersomes for topical immunization. J. Pharm. Pharm. 2005, 57, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Abdelbary, A.A.; Abd-Elsalam, W.H.; Al-Mahallawi, A.M. Fabrication of novel ultradeformable bilosomes for enhanced ocular delivery of terconazole: In vitro characterization, ex vivo permeation and in vivo safety assessment. Int. J. Pharm. 2016, 513, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Vargha-Butler, E.; Hurst, E. Study of liposomal drug delivery systems 1. Surface characterization of steroid loaded MLV liposomes. Colloids Surf. B Biointerfaces 1995, 3, 287–295. [Google Scholar] [CrossRef]
- Duangjit, S.; Opanasopit, P.; Rojanarata, T.; Ngawhirunpat, T. Characterization and In Vitro Skin Permeation of Meloxicam-Loaded Liposomes versus Transfersomes. J. Drug Deliv. 2011, 2011, 418316. [Google Scholar] [CrossRef] [PubMed]
- Salama, H.A.; Mahmoud, A.A.; Kamel, A.O.; Abdel Hady, M.; Awad, G.A. Brain delivery of olanzapine by intranasal administration of transfersomal vesicles. J. Liposome Res. 2012, 22, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Demetzos, C. Differential Scanning Calorimetry (DSC): A tool to study the thermal behavior of lipid bilayers and liposomal stability. J. Liposome Res. 2008, 18, 159–173. [Google Scholar] [CrossRef]
- Bhardwaj, U.; Burgess, D.J. Physicochemical properties of extruded and non-extruded liposomes containing the hydrophobic drug dexamethasone. Int. J. Pharm. 2010, 388, 181–189. [Google Scholar] [CrossRef]
- Sun, J.; Deng, Y.; Wang, S.; Cao, J.; Gao, X.; Dong, X. Liposomes incorporating sodium deoxycholate for hexamethylmelamine (HMM) oral delivery: Development, characterization, and in vivo evaluation. Drug Deliv. 2010, 17, 164–170. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Nir, S.; Ohki, S. Permeability properties of phospholipid membranes: Effect of cholesterol and temperature. Biochim. Biophys. Acta (BBA) Biomembr. 1972, 266, 561–583. [Google Scholar] [CrossRef]
- Gregoriadis, G.; Davis, C. Stability of liposomes invivo and invitro is promoted by their cholesterol content and the presence of blood cells. Biochem. Biophys. Res. Commun. 1979, 89, 1287–1293. [Google Scholar] [CrossRef]
- Momeni, A.; Rasoolian, M.; Momeni, A.; Navaei, A.; Emami, S.; Shaker, Z.; Mohebali, M.; Khoshdel, A. Development of liposomes loaded with anti-leishmanial drugs for the treatment of cutaneous leishmaniasis. J. Liposome Res. 2013, 23, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Stanculescu, I.; Manea, M.; Chiosa, V.; Mandravel, C. A theoretical approach to the molecular interaction between cholesterol and 2-pyridine aldoxime methyl chloride (2-Pam). An. Univ. Bucur. Chim. 2006, 1, 101–105. [Google Scholar]
- Lin, H.; Yoo, J.W.; Roh, H.J.; Lee, M.K.; Chung, S.J.; Shim, C.K.; Kim, D.D. Transport of anti-allergic drugs across the passage cultured human nasal epithelial cell monolayer. Eur. J. Pharm. Sci. 2005, 26, 203–210. [Google Scholar] [CrossRef]
- He, Y.; Luo, L.; Liang, S.; Long, M.; Xu, H. Influence of probe-sonication process on drug entrapment efficiency of liposomes loaded with a hydrophobic drug. Int. J. Polym. Mater. Polym. Biomater. 2018, 68, 193–197. [Google Scholar] [CrossRef]
- Narayan, R.; Singh, M.; Ranjan, O.; Nayak, Y.; Garg, S.; Shavi, G.V.; Nayak, U.Y. Development of risperidone liposomes for brain targeting through intranasal route. Life Sci. 2016, 163, 38–45. [Google Scholar] [CrossRef]
- Begum, M.Y.; Abbulu, K.; Sudhakar, M. Preparation, Characterization and In-Vitro Release Study of Flurbiprofen Loaded Stealth Liposomes. Chem. Sci. Trans. 2012, 1, 201–209. [Google Scholar] [CrossRef][Green Version]
- Gregory, J. Rates of flocculation of latex particles by cationic polymers. J. Colloid Interface Sci. 1973, 42, 448–456. [Google Scholar] [CrossRef]
- Mabire, F.; Audebert, R.; Quivoron, C. Flocculation properties of some water-soluble cationic copolymers toward silica suspensions: A semiquantitative interpretation of the role of molecular weight and cationicity through a “patchwork” model. J. Colloid Interface Sci. 1984, 97, 120–136. [Google Scholar] [CrossRef]
- Claesson, P.M.; Ninham, B.W. pH-dependent interactions between adsorbed chitosan layers. Langmuir 1992, 8, 1406–1412. [Google Scholar] [CrossRef]
- Demarger-André, S.; Domard, A. Chitosan behaviours in a dispersion of undecylenic acid. Structural parameters. Carbohydr. Polym. 1994, 24, 177–184. [Google Scholar] [CrossRef]
- yon Smoluchowski, M. Versuch einer mathematischen Theorie der Koagulationskinetik kolloider L6sungen. Z. Phys. Chem. 1917, 92, 129–168. [Google Scholar]
- Aisha, A.F.; Majid, A.M.S.A.; Ismail, Z. Preparation and characterization of nano liposomes of Orthosiphon stamineusethanolic extract in soybean phospholipids. BMC Biotechnol. 2014, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Chawla, A.; Sharma, P.; Pawar, P. Formulation and in vitro evaluation of Eudragit S-100 coated naproxen matrix tablets for colon-targeted drug delivery system. J. Adv. Pharm. Technol. Res. 2013, 4, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Han, J.; Ye, A.; Liu, W.; Xu, X.; Yao, Y.; Li, K.; Kong, Y.; Wei, F.; Zhou, W. Structural characterization and biological fate of lactoferrin-loaded liposomes during simulated infant digestion. J. Sci. Food Agric. 2019, 99, 2677–2684. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Guha, P.; Bhattarai, R.; Nahak, P.; Karmakar, G.; Chettri, P.; Panda, A.K. Influence of Lipid Composition, pH, and Temperature on Physicochemical Properties of Liposomes with Curcumin as Model Drug. J. Oleo Sci. 2016, 65, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Varum, F.; Al-Gousous, J.; Hofmann, M.; Page, S.; Langguth, P. In Vitro Methodologies for Evaluating Colon-Targeted Pharmaceutical Products and Industry Perspectives for Their Applications. Pharmaceutics 2022, 14, 291. [Google Scholar] [CrossRef]
- Sabot, A.; Krause, S. Simultaneous quartz crystal microbalance impedance and electrochemical impedance measurements. Investigation into the degradation of thin polymer films. Anal. Chem. 2002, 74, 3304–3311. [Google Scholar] [CrossRef]
- Klein, S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. AAPS J 2010, 12, 397–406. [Google Scholar] [CrossRef]
- Ramalho, M.J.; Loureiro, J.A.; Pereira, M.C. Poly(lactic-co-glycolic acid) Nanoparticles for the Encapsulation and Gastrointestinal Release of Vitamin B9 and Vitamin B12. ACS Appl. Nano Mater. 2021, 4, 6881–6892. [Google Scholar] [CrossRef]
- Jones, R.N.; Ramsay, D.; Keir, D.; Dobriner, K. The intensities of carbonyl bands in the infrared spectra of Steroids1. J. Am. Chem. Soc. 1952, 74, 80–88. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DPPC:CH:SA Molar Ratio | DPPC (mg) | CH (mg) | SA (mg) | BUD (mg) |
|---|---|---|---|---|
| 7:3:0 | 11.0 | 2.3 | 0.0 | 0.25 |
| 7:3:0.25 | 10.7 | 2.3 | 0.1 | 0.25 |
| 7:3:0.5 | 10.5 | 2.2 | 0.3 | 0.25 |
| 7:3:1 | 10.0 | 2.1 | 0.5 | 0.25 |
| 7:3:2 | 9.2 | 1.9 | 0.9 | 0.25 |
| 7:3:3 | 8.5 | 1.8 | 1.2 | 0.25 |
| 7:3:4 | 7.9 | 1.7 | 1.5 | 0.25 |
| 8:2:3 | 9.7 | 1.2 | 1.2 | 0.25 |
| 6:4:3 | 7.2 | 2.4 | 1.2 | 0.25 |
| Ingredient | SGF | FaSSIF | FeSSIF | PBS |
|---|---|---|---|---|
| NaCl | 2 g | 6.19 g | 11.87 g | 8 g |
| KCl | 0.2 g | |||
| NaOH pellet | 0.35 g | 4.04 g | ||
| Glacial acetic acid | 144 mM | |||
| STC | 10 mM | |||
| NaH2PO4 | 3.44 g | |||
| NaH2PO4 | 1.15 g | |||
| KH2PO4 | 0.2 g | |||
| HCl conc. | 7 mL | |||
| H2O qs. | 1000 mL | 1000 mL | 1000 mL | 1000 mL |
| pH | 1.2 | 6.5 | 5 | 7.4 |
| No. | Coating Variables | Lipid Concentration (mM) | Liposome/Polymer Volume Ratio | Polymer Concentration (mg/mL) | Rate of Lipid Addition (µL/s) | Mixing Speed (rpm) | SA Molar Ratio |
|---|---|---|---|---|---|---|---|
| 1. | Lipid Concentration (mM) | 2, 4, 5, 6, 7, 10 | 1:4 | 2 | 250 | 1000 | 3 |
| 2. | Liposome/Polymer Volume Ratio | 5 | 1:1, 1:2, 1:3, 1:4 | 2 | 250 | 1000 | 3 |
| 3. | Polymer Concentration (mg/mL) | 5 | 1:1 | 0.25, 0.5, 1, 2, 2.5 | 250 | 1000 | 3 |
| 4. | Rate of Lipid Addition (µL/s) | 5 | 1:1 | 2.5 | 5, 10, 50, 250 | 1000 | 3 |
| 5. | Mixing Speed (rpm) | 5 | 1:1 | 2.5 | 250 | 250, 500, 1000 | 3 |
| 6. | SA Molar Ratio | 5 | 1:1 | 2.5 | 250 | 1000 | 1, 2, 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alghurabi, H.; Tagami, T.; Ogawa, K.; Ozeki, T. Preparation, Characterization and In Vitro Evaluation of Eudragit S100-Coated Bile Salt-Containing Liposomes for Oral Colonic Delivery of Budesonide. Polymers 2022, 14, 2693. https://doi.org/10.3390/polym14132693
Alghurabi H, Tagami T, Ogawa K, Ozeki T. Preparation, Characterization and In Vitro Evaluation of Eudragit S100-Coated Bile Salt-Containing Liposomes for Oral Colonic Delivery of Budesonide. Polymers. 2022; 14(13):2693. https://doi.org/10.3390/polym14132693
Chicago/Turabian StyleAlghurabi, Hamid, Tatsuaki Tagami, Koki Ogawa, and Tetsuya Ozeki. 2022. "Preparation, Characterization and In Vitro Evaluation of Eudragit S100-Coated Bile Salt-Containing Liposomes for Oral Colonic Delivery of Budesonide" Polymers 14, no. 13: 2693. https://doi.org/10.3390/polym14132693
APA StyleAlghurabi, H., Tagami, T., Ogawa, K., & Ozeki, T. (2022). Preparation, Characterization and In Vitro Evaluation of Eudragit S100-Coated Bile Salt-Containing Liposomes for Oral Colonic Delivery of Budesonide. Polymers, 14(13), 2693. https://doi.org/10.3390/polym14132693