
Controlling Morphology and Physio-Chemical Properties of Stimulus-Responsive Polyurethane Foams by Altering Chemical Blowing Agent Content





Abstract
:
1. Introduction
2. Materials and Methods
2.1. SMP Foam Fabrication
2.2. Chemical and Physical Characterization
2.2.1. Scanning Electron Microscopy (SEM)
2.2.2. Fourier-Transform Infrared (FTIR) Spectroscopy
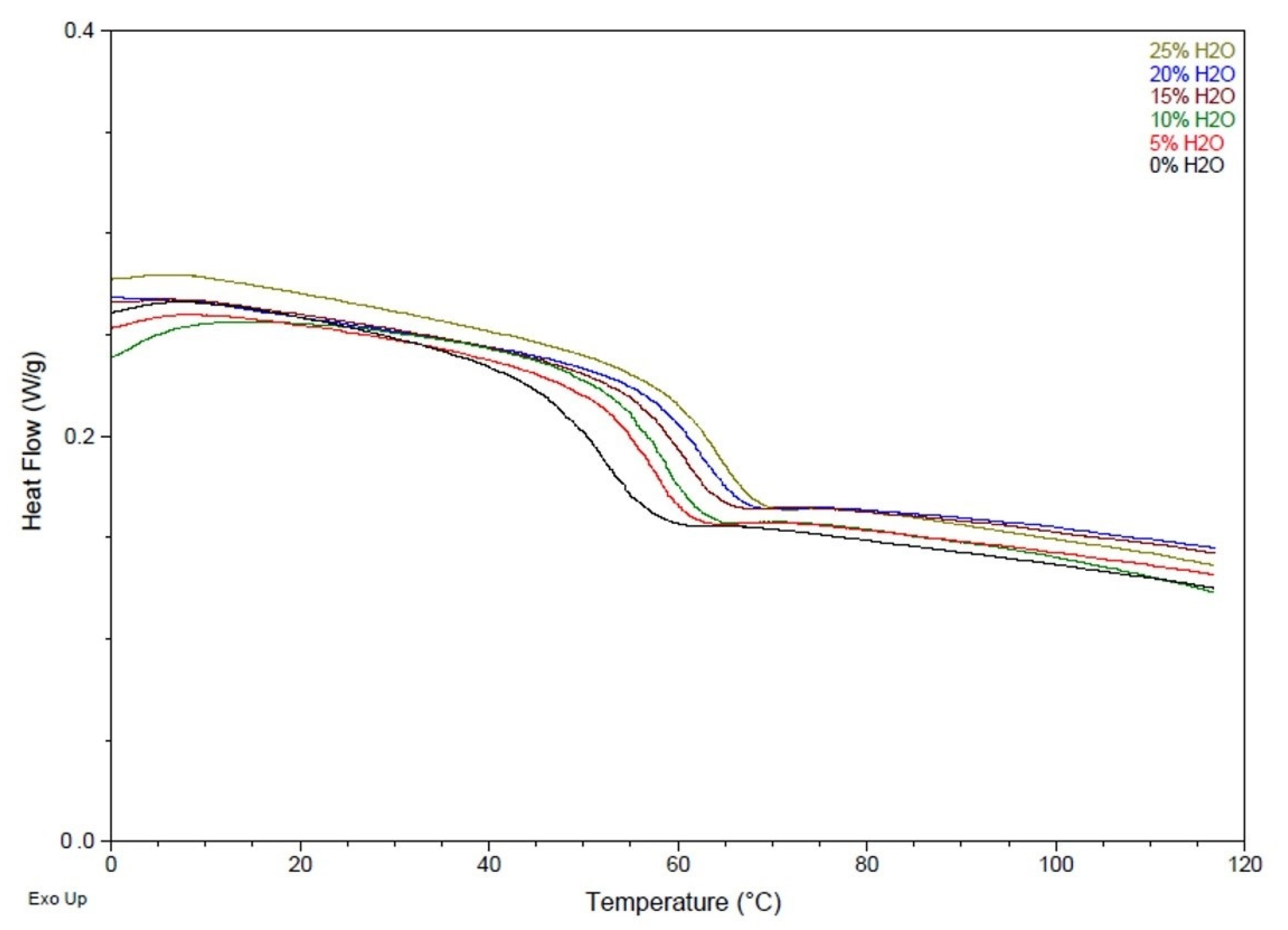
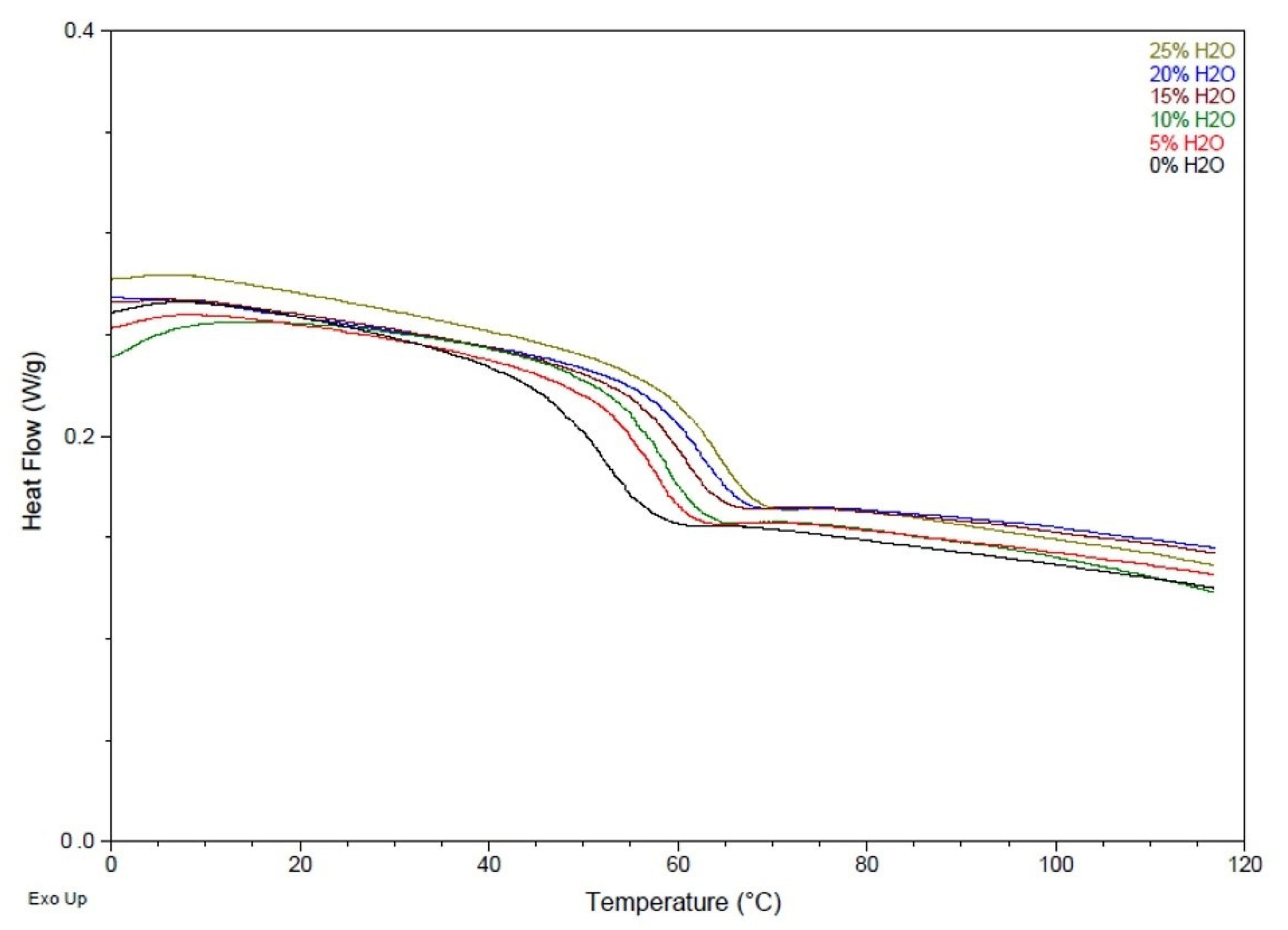
2.2.3. Differential Scanning Calorimetry (DSC)
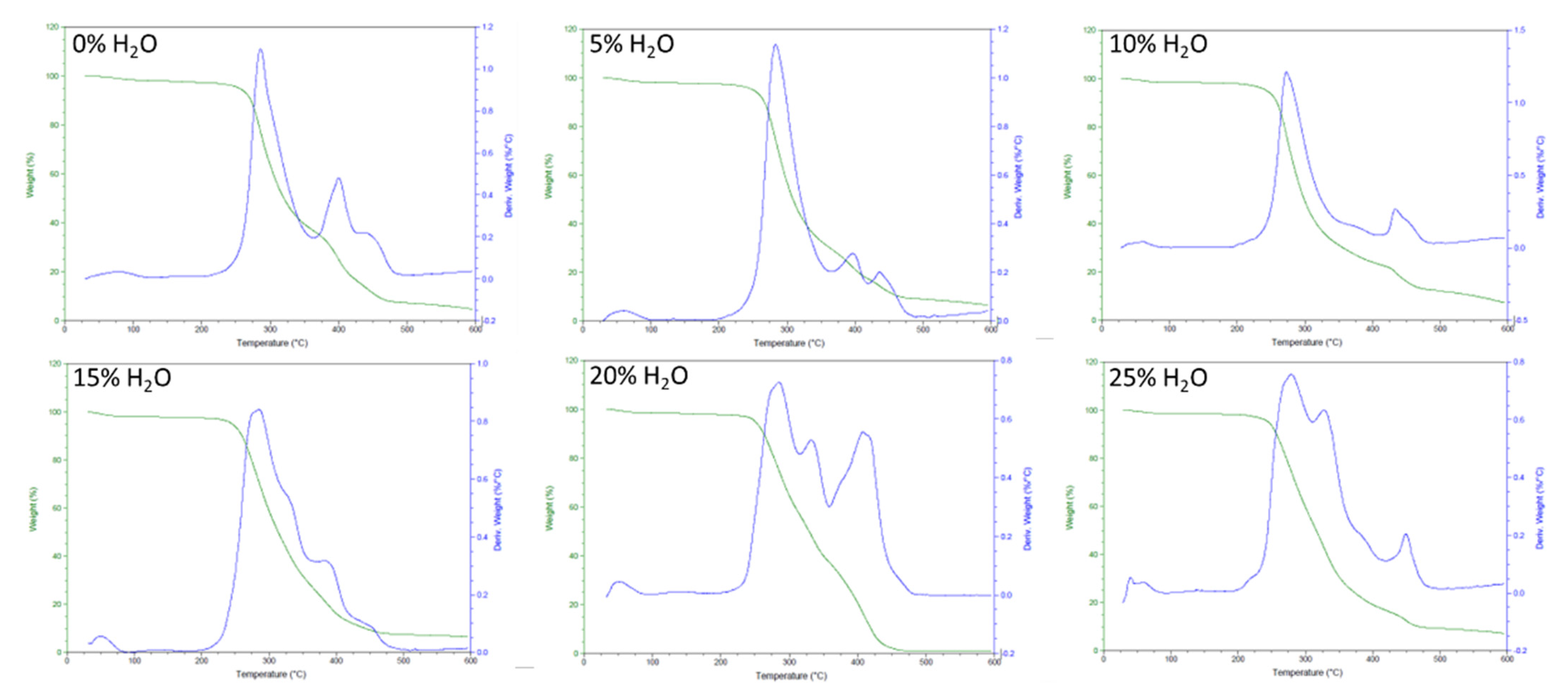
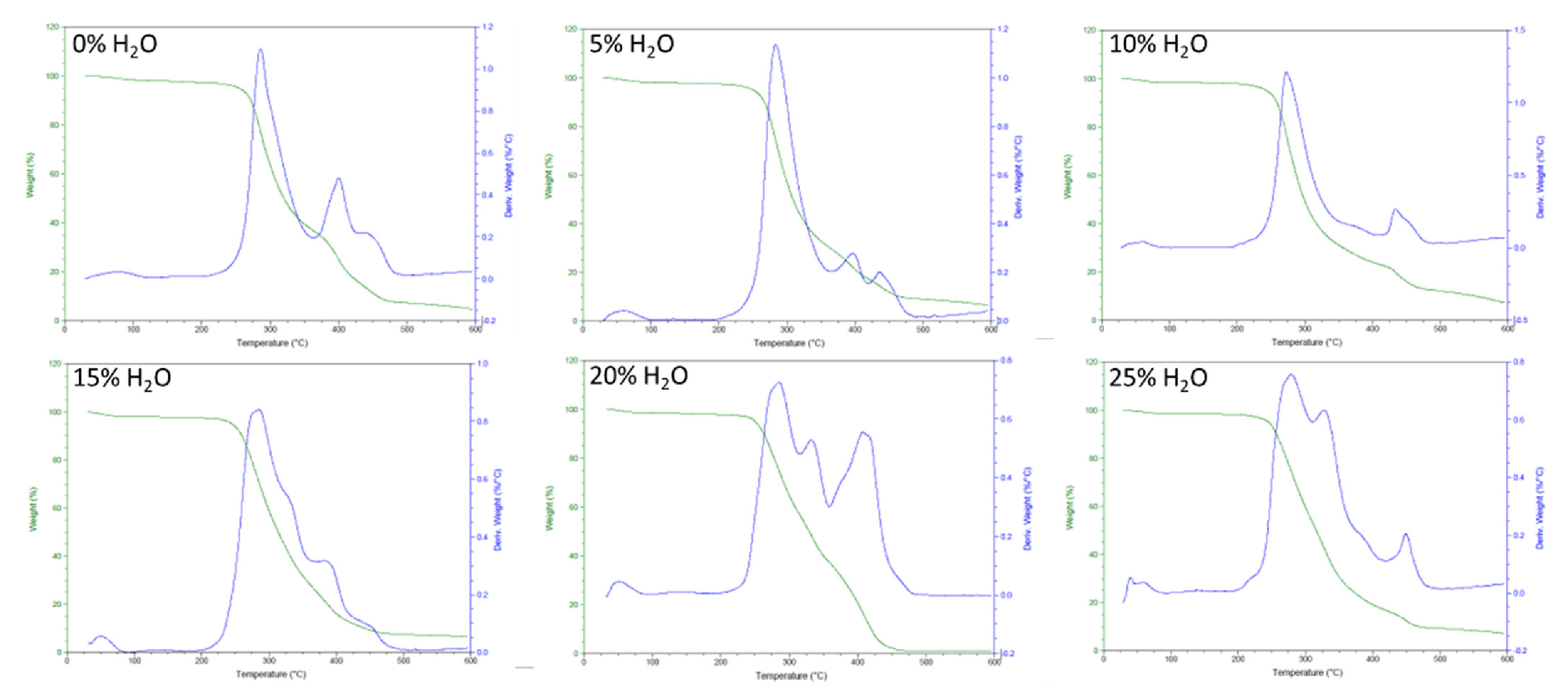
2.2.4. Thermogravimetric Analysis (TGA)
2.2.5. Tensile Testing
2.2.6. Density
2.3. In Vitro Shape Memory Behavior
2.3.1. Volume Recovery and Expansion
2.3.2. Actuation Profiles
3. Results
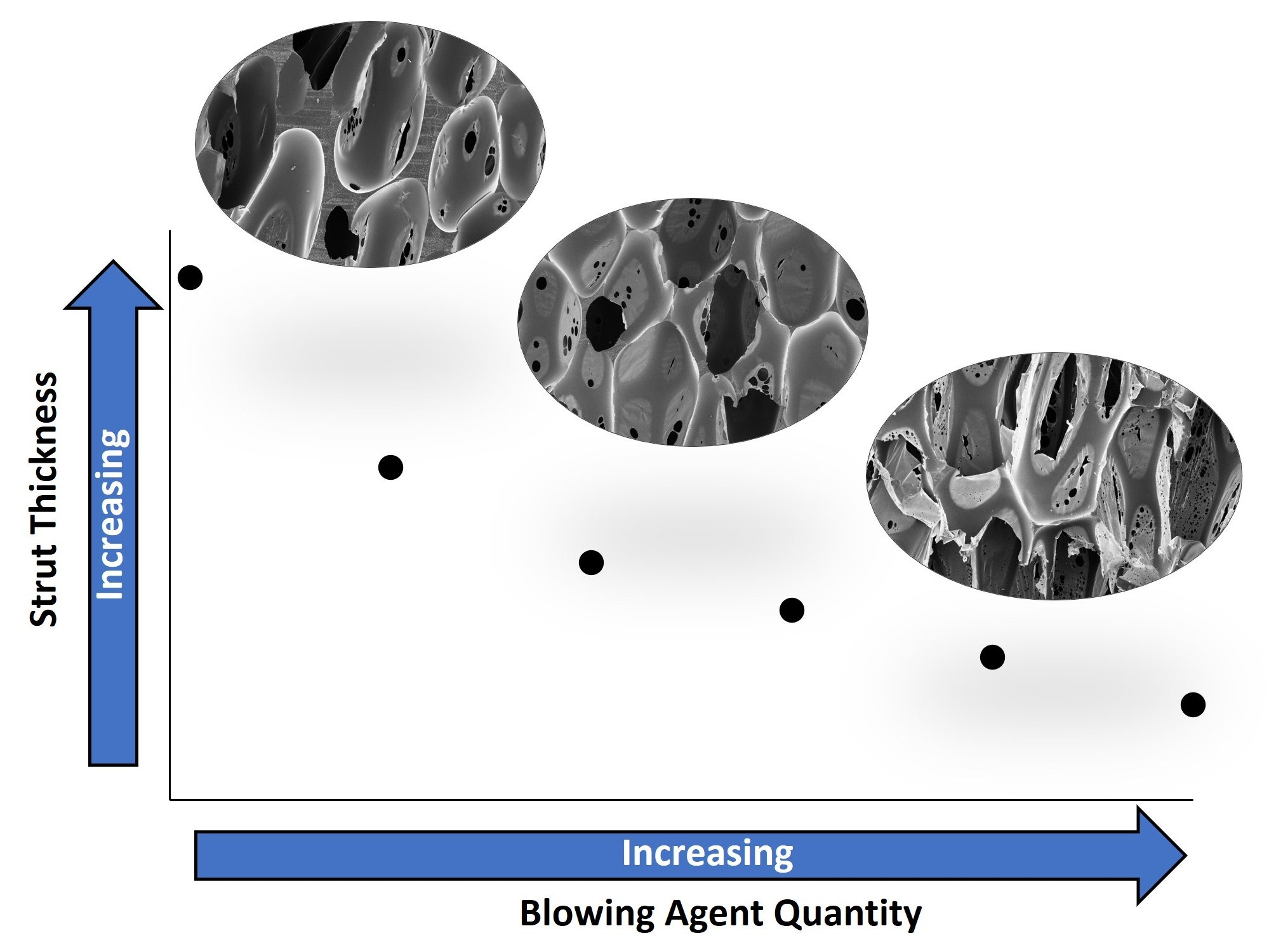
3.1. Pore Structure and Foam Density
3.2. Chemical and Thermal Characterization
3.3. Mechanical Characterization
3.4. In vitro Shape Memory Behavior
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jessen, S.L.; Friedemann, M.C.; Ginn-Hedman, A.M.; Graul, L.M.; Jokerst, S.; Robinson, C.B.; Landsman, T.L.; Clubb, F.J., Jr.; Maitland, D.J. Microscopic Assessment of Healing and Effectiveness of a Foam-Based Peripheral Occlusion Device. ACS Biomater. Sci. Eng. 2020, 6, 2588–2599. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.N.; Clubb, F.J.; Wilson, T.S.; Miller, M.W.; Fossum, T.W.; Hartman, J.; Tuzun, E.; Singhal, P.; Maitland, D.J. In vivo response to an implanted shape memory polyurethane foam in a porcine aneurysm model. J. Biomed. Mater. Res. A 2014, 102, 1231–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, P.S.W.; Miller, M.W.; Maitland, D.J. In Vitro Study of Transcatheter Delivery of a Shape Memory Polymer Foam Embolic Device for Treating Cerebral Aneurysms. J. Med. Devices 2013, 7, 020932. [Google Scholar] [CrossRef]
- Hasan, S.M.; Nash, L.D.; Maitland, D.J. Porous shape memory polymers: Design and applications. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 1300–1318. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.; Friess, F.; Krus, M.; Zolanvari, S.M.H.; Grün, G.; Kröber, H.; Pretsch, T. Shape Memory Polymer Foam with Programmable Apertures. Polymers 2020, 12, 1914. [Google Scholar] [CrossRef] [PubMed]
- Hearon, K.; Singhal, P.; Horn, J.; Small, W.t.; Olsovsky, C.; Maitland, K.C.; Wilson, T.S.; Maitland, D.J. Porous Shape Memory Polymers. Polym. Rev. (Phila. Pa.) 2013, 53, 41–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, J.; Hwang, W.; Jessen, S.L.; Keller, B.K.; Miller, M.W.; Tuzun, E.; Hartman, J.; Clubb, F.J., Jr.; Maitland, D.J. Comparison of shape memory polymer foam versus bare metal coil treatments in an in vivo porcine sidewall aneurysm model. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 1892–1905. [Google Scholar] [CrossRef]
- Herting, S.M.; Ding, Y.; Boyle, A.J.; Dai, D.; Nash, L.D.; Asnafi, S.; Jakaitis, D.R.; Johnson, C.R.; Graul, L.M.; Yeh, C.; et al. In vivo comparison of shape memory polymer foam-coated and bare metal coils for aneurysm occlusion in the rabbit elastase model. J. Biomed. Mater. Res. B Appl. Biomater. 2019, 107, 2466–2475. [Google Scholar] [CrossRef]
- Jessen, S.L.; Friedemann, M.C.; Mullen, A.E.; Ginn-Hedman, A.M.; Herting, S.M.; Maitland, D.J.; Clubb, F.J., Jr. Micro-CT and histopathology methods to assess host response of aneurysms treated with shape memory polymer foam-coated coils versus bare metal coil occlusion devices. J. Biomed. Mater. Res. B Appl. Biomater. 2020, 108, 2238–2249. [Google Scholar] [CrossRef]
- Dalton, E.; Chai, Q.; Shaw, M.W.; McKenzie, T.J.; Mullins, E.S.; Ayres, N. Hydrogel-coated polyurethane/urea shape memory polymer foams. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1389–1395. [Google Scholar] [CrossRef]
- Pfau, M.R.; McKinzey, K.G.; Roth, A.A.; Graul, L.M.; Maitland, D.J.; Grunlan, M.A. Shape memory polymer (SMP) scaffolds with improved self-fitting properties. J. Mater. Chem. B 2021, 9, 3826–3837. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Loftus, I.; Ratnam, L.; Das, R.; Mailli, L.; Hamady, M.S.; Lobotesis, K. Clinical experience with a shape memory polymer peripheral vascular embolisation plug: A case series. CVIR Endovasc. 2021, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.S.; van Schaik, P.M.; Martens, J.M.; Reijnen, M. Embolization of the false lumen using IMPEDE-FX embolization plugs as part of treatment of an infrarenal post-dissection aneurysm: A case report. CVIR Endovasc. 2020, 3, 91. [Google Scholar] [CrossRef] [PubMed]
- Hasan, S.M.; Raymond, J.E.; Wilson, T.S.; Keller, B.K.; Maitland, D.J. Effects of Isophorone Diisocyanate on the Thermal and Mechanical Properties of Shape-Memory Polyurethane Foams. Macromol. Chem. Phys. 2014, 215, 2420–2429. [Google Scholar] [CrossRef]
- Singhal, P.; Boyle, A.; Brooks, M.L.; Infanger, S.; Letts, S.D.; Small, W.D.; Maitland, D.J.D.; Wilson, T.S.D. Controlling the Actuation Rate of Low-Density Shape-Memory Polymer Foams in Water. Macromol. Chem. Phys. 2013, 214, 1204–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herting, S.M.; Monroe, M.B.B.; Weems, A.C.; Briggs, S.T.; Fletcher, G.K.; Blair, S.E.; Hatch, C.J.; Maitland, D.J. In vitro cytocompatibility testing of oxidative degradation products. J. Bioact. Compat. Polym. 2021, 36, 197–211. [Google Scholar] [CrossRef]
- Al-Moameri, H.H.; Nabhan, B.J.; Wasmi, M.T.; Abdulrehman, M.A. Impact of blowing agent-blends on polyurethane foams thermal and mechanical properties. AIP Conf. Proc. 2020, 2213, 020177. [Google Scholar]
- Modesti, M.; Adriani, V.; Simioni, F. Chemical and physical blowing agents in structural polyurethane foams: Simulation and characterization. Polym. Eng. Sci. 2000, 40, 2046–2057. [Google Scholar] [CrossRef]
- Bernardini, J.; Licursi, D.; Anguillesi, I.; Cinelli, P.; Coltelli, M.; Antonetti, C.; Venezia, A.M.; Galletti, A.; Lazzeri, A. Exploitation of Arundo donax L. Hydrolysis Residue for the Green Synthesis of Flexible Polyurethane Foams. BioResources 2017, 12, 3630–3655. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.M.; Lee, S.J.; Kim, B.K. Shape memory polyurethane foams. Express Polym. Lett. 2012, 6, 63–69. [Google Scholar] [CrossRef]
- Singhal, P.; Rodriguez, J.N.; Small, W.; Eagleston, S.; Van de Water, J.; Maitland, D.J.; Wilson, T.S. Ultra Low Density and Highly Crosslinked Biocompatible Shape Memory Polyurethane Foams. J. Polym. Sci. B Polym. Phys. 2012, 50, 724–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, S.M.; Thompson, R.S.; Emery, H.; Nathan, A.L.; Weems, A.C.; Zhou, F.; Monroe, M.B.; Maitland, D.J. Modification of Shape Memory Polymer Foams Using Tungsten, Aluminum Oxide, and Silicon Dioxide Nanoparticles. RSC Adv. 2016, 6, 918–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzęda, D.; Węgrzyk, G.; Leszczyńska, M.; Szczepkowski, L.; Gloc, M.; Ryszkowska, J. Viscoelastic Polyurethane Foams for Use as Auxiliary Materials in Orthopedics. Materials 2022, 15, 133. [Google Scholar] [CrossRef] [PubMed]
- Okrasa, M.; Leszczyńska, M.; Sałasińska, K.; Szczepkowski, L.; Kozikowski, P.; Majchrzycka, K.; Ryszkowska, J. Viscoelastic Polyurethane Foams for Use in Seals of Respiratory Protective Devices. Materials 2021, 14, 1600. [Google Scholar] [CrossRef]
- Hasan, S.M.; Fletcher, G.K.; Monroe, M.B.B.; Wierzbicki, M.A.; Nash, L.D.; Maitland, D.J. Shape Memory Polymer Foams Synthesized Using Glycerol and Hexanetriol for Enhanced Degradation Resistance. Polymers 2020, 12, 2290. [Google Scholar] [CrossRef]
- ASTM D3574-17; A. International, Standard Test Methods for Flexible Cellular Materials—Slab, Bonded, and Molded Urethane Foams, Test A-Density Test. 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Water Content (mol%) | Weight Percent (%) | |||
|---|---|---|---|---|
| HDI | HPED | TEA | Water | |
| 25 | 64.97 | 13.83 | 9.41 | 3.47 |
| 20 | 62.42 | 16.05 | 10.90 | 2.67 |
| 15 | 60.11 | 18.01 | 12.25 | 1.94 |
| 10 | 57.94 | 19.81 | 13.58 | 1.26 |
| 5 | 55.88 | 21.64 | 14.74 | 0.59 |
| 0 | 53.83 | 23.38 | 15.91 | 0.00 |
| Water Content (mol%) | Axial Pore Size (µm) | Transverse Pore Size (µm) | Strut Thickness (µm) | Density (g/cc) |
|---|---|---|---|---|
| 0 | 913 ± 117 | 479 ± 110 | 121 ± 31 | 0.169 ± 0.002 |
| 5 | 1107 ± 181 | 685 ± 139 | 101 ± 9 | 0.056 ± 0.001 |
| 10 | 1021 ± 165 | 659 ± 100 | 80 ± 26 | 0.035 ± 0.001 |
| 15 | 1188 ± 170 | 700 ± 105 | 89 ± 8 | 0.027 ± 0.001 |
| 20 | 1457 ± 214 | 848 ± 91 | 71 ± 11 | 0.021 ± 0.001 |
| 25 | 1670 ± 287 | 844 ± 105 | 62 ± 7 | 0.017 ± 0.001 |
| Water Content (mol %) | Strain at Break (%) | Tensile Modulus (MPa) | Tg ( °C) | Volume Recovery (%) | Volume Expansion (x) |
|---|---|---|---|---|---|
| 0 | 14 ± 1 | 12.12 ± 1.62 | 52 ± 1 | 106 ± 12 | 5 ± 1 |
| 5 | 36 ± 8 | 1.74 ± 0.06 | 57 ± 1 | 81 ± 7 | 17 ± 3 |
| 10 | 49 ± 6 | 0.92 ± 0.04 | 59 ± 1 | 100 ± 6 | 27 ± 3 |
| 15 | 76 ± 13 | 0.66 ± 0.04 | 62 ± 1 | 102 ± 4 | 27 ± 3 |
| 20 | 73 ± 7 | 0.31 ± 0.06 | 63 ± 1 | 104 ± 6 | 28 ± 3 |
| 25 | 83 ± 6 | 0.14 ± 0.02 | 64 ± 1 | 108 ± 6 | 23 ± 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, S.M.; Touchet, T.; Jayadeep, A.; Maitland, D.J. Controlling Morphology and Physio-Chemical Properties of Stimulus-Responsive Polyurethane Foams by Altering Chemical Blowing Agent Content. Polymers 2022, 14, 2288. https://doi.org/10.3390/polym14112288
Hasan SM, Touchet T, Jayadeep A, Maitland DJ. Controlling Morphology and Physio-Chemical Properties of Stimulus-Responsive Polyurethane Foams by Altering Chemical Blowing Agent Content. Polymers. 2022; 14(11):2288. https://doi.org/10.3390/polym14112288
Chicago/Turabian StyleHasan, Sayyeda Marziya, Tyler Touchet, Aishwarya Jayadeep, and Duncan J. Maitland. 2022. "Controlling Morphology and Physio-Chemical Properties of Stimulus-Responsive Polyurethane Foams by Altering Chemical Blowing Agent Content" Polymers 14, no. 11: 2288. https://doi.org/10.3390/polym14112288
APA StyleHasan, S. M., Touchet, T., Jayadeep, A., & Maitland, D. J. (2022). Controlling Morphology and Physio-Chemical Properties of Stimulus-Responsive Polyurethane Foams by Altering Chemical Blowing Agent Content. Polymers, 14(11), 2288. https://doi.org/10.3390/polym14112288