
Synthesis and Chemical Functionalization of Pseudo-Homogeneous Catalysts for Biodiesel Production—Oligocat

 ,
,
Abstract
:1. Introduction
1.1. Biodiesel in Brief Facts
- to be applied in second generation stock;
- being a Brønsted–Lowry acid catalyst;
- to act initially as homogeneous catalyst;
- to provide conversion yield of TAG’s to FA(X)E above 96.5 wt.% within one reaction cycle.
1.2. Biodiesel Heterogeneous Catalysts
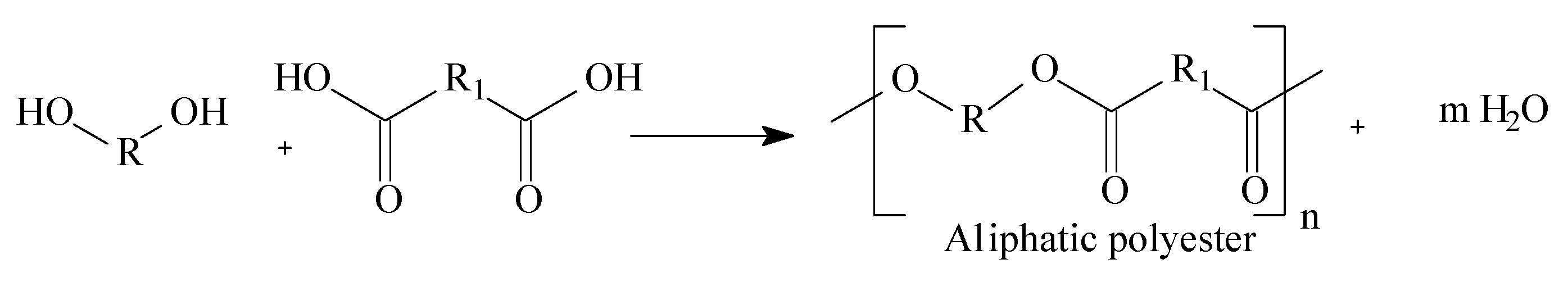
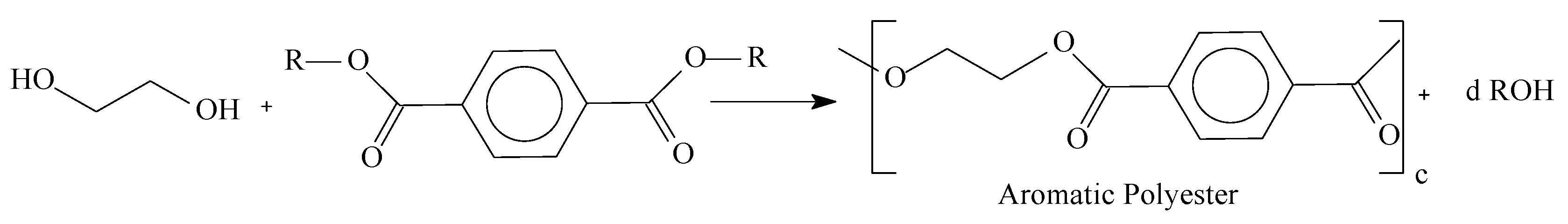
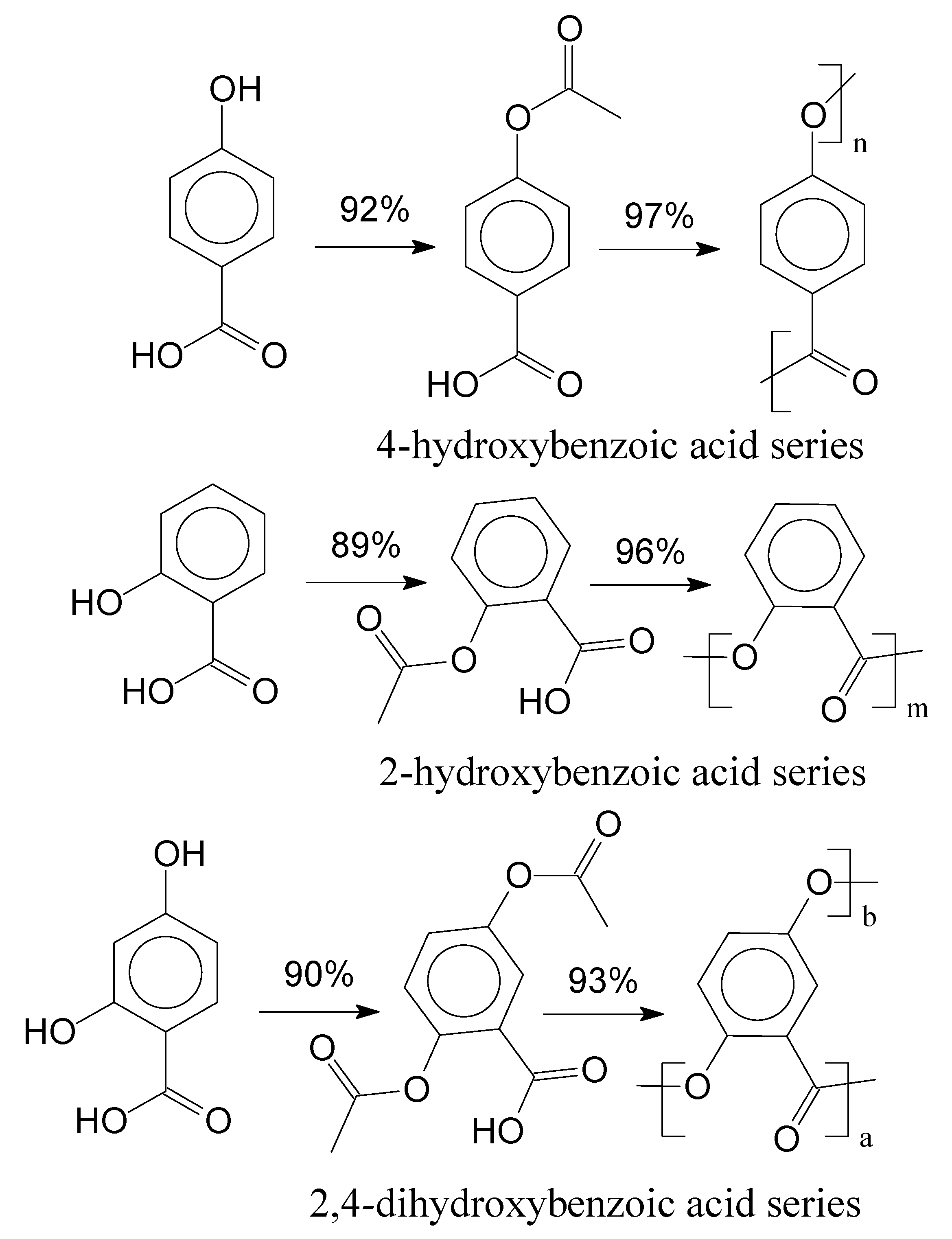
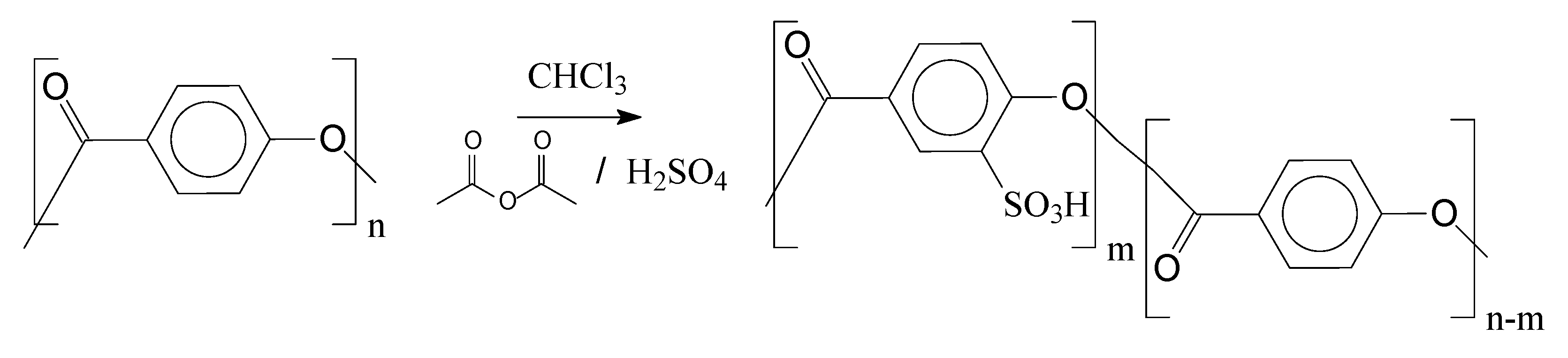
1.3. Mass Polymerization and Sulfonation Reactions to Generate Oligocat
2. Materials and Methods
2.1. Materials
2.2. Polyesters Synthesis and Sulfonation Reaction
2.2.1. Experimental Procedure 1 (EP-1)
2.2.2. Experimental Procedure 2 (EP-2)
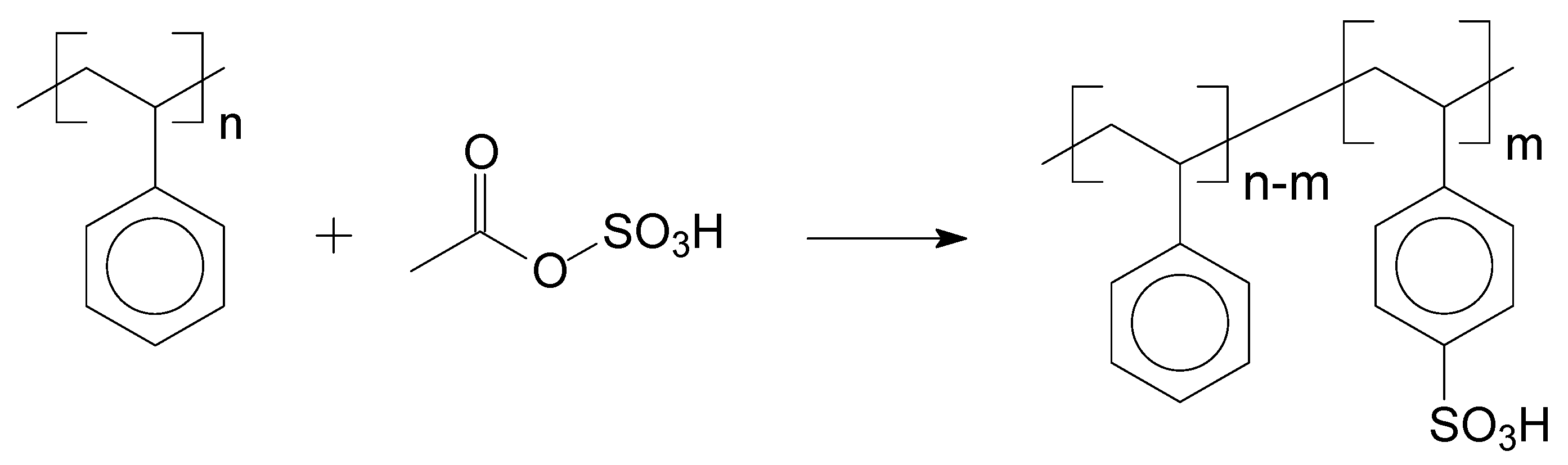
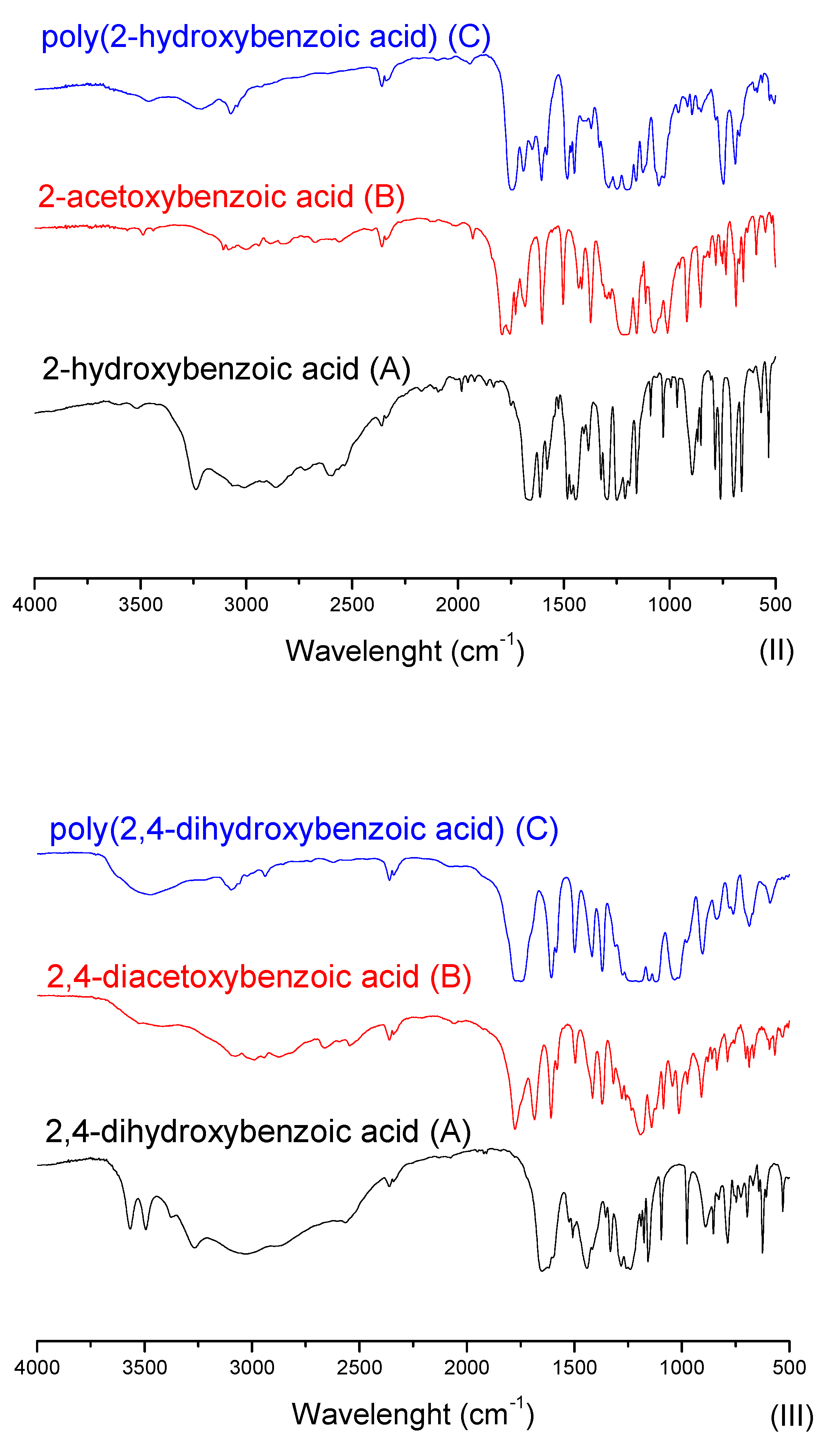
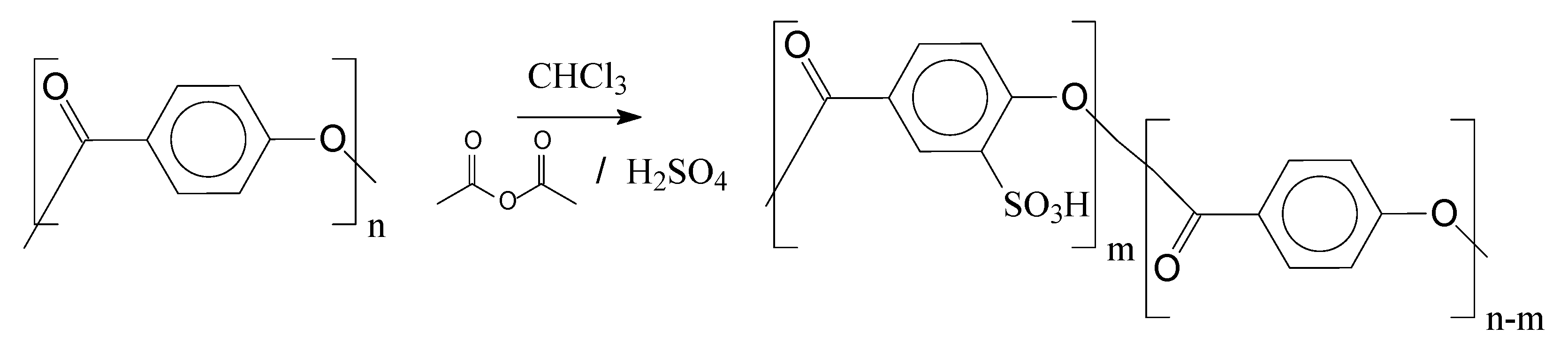
2.2.3. Sulfonation Reaction Procedure
2.3. Characterization Methods
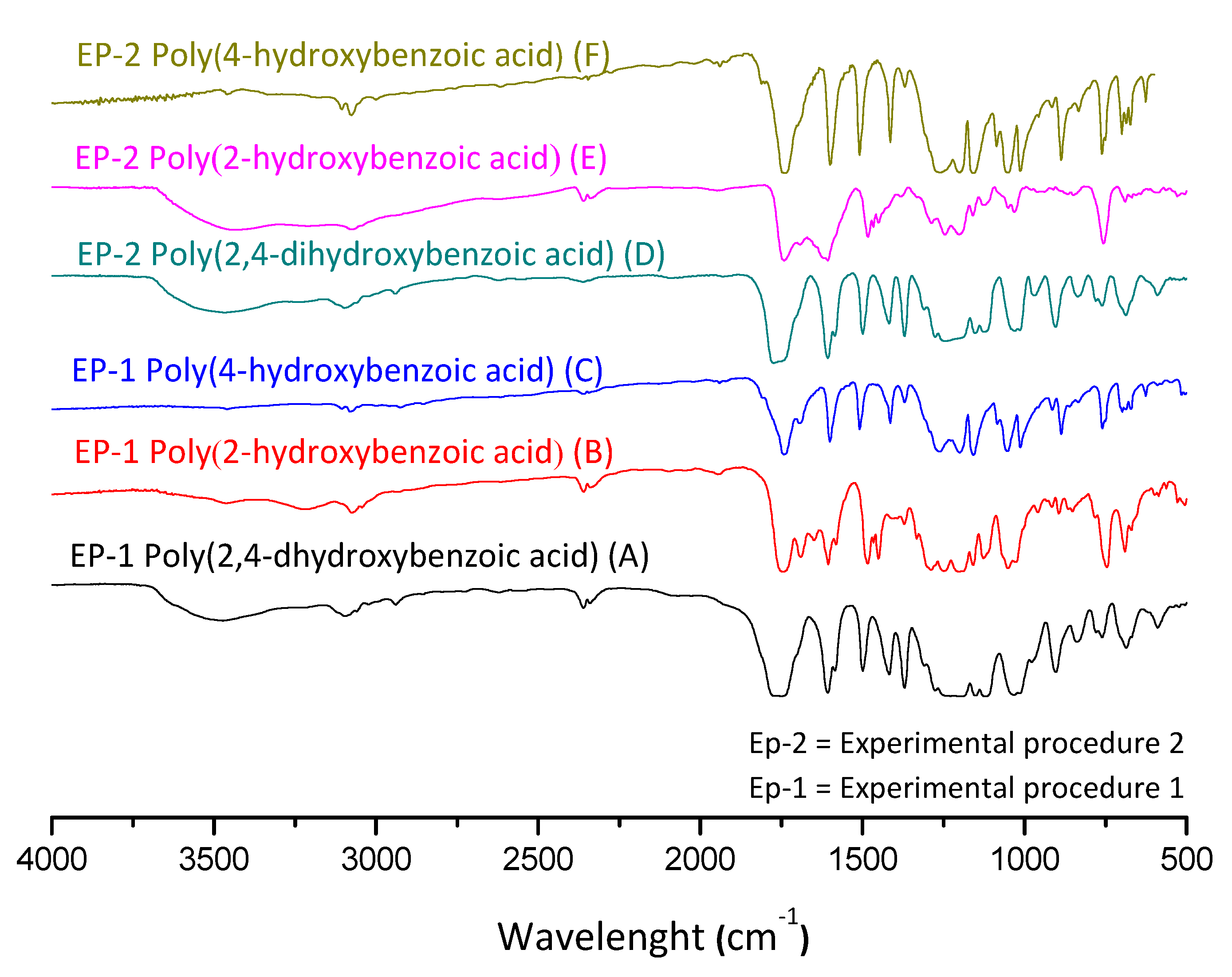
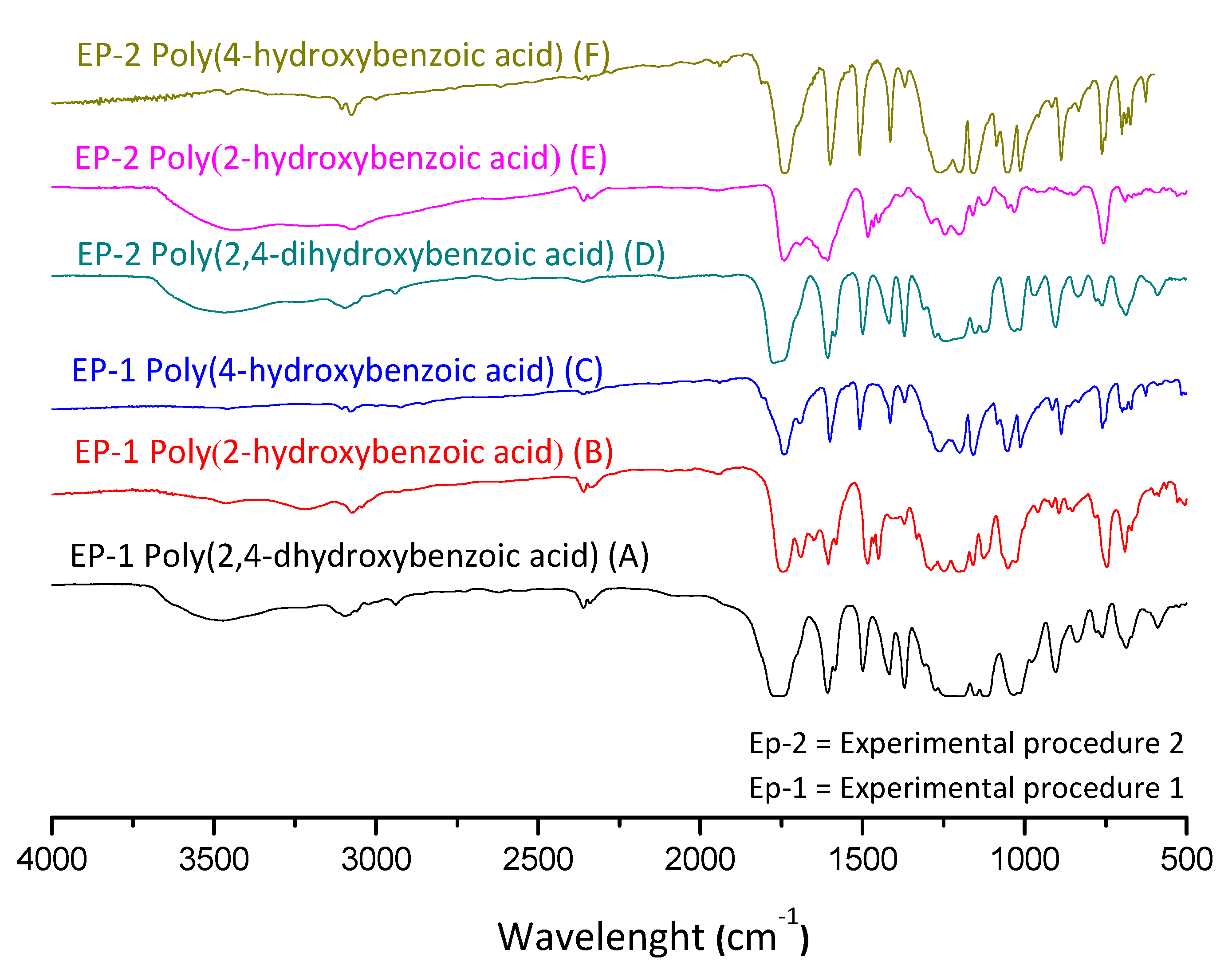
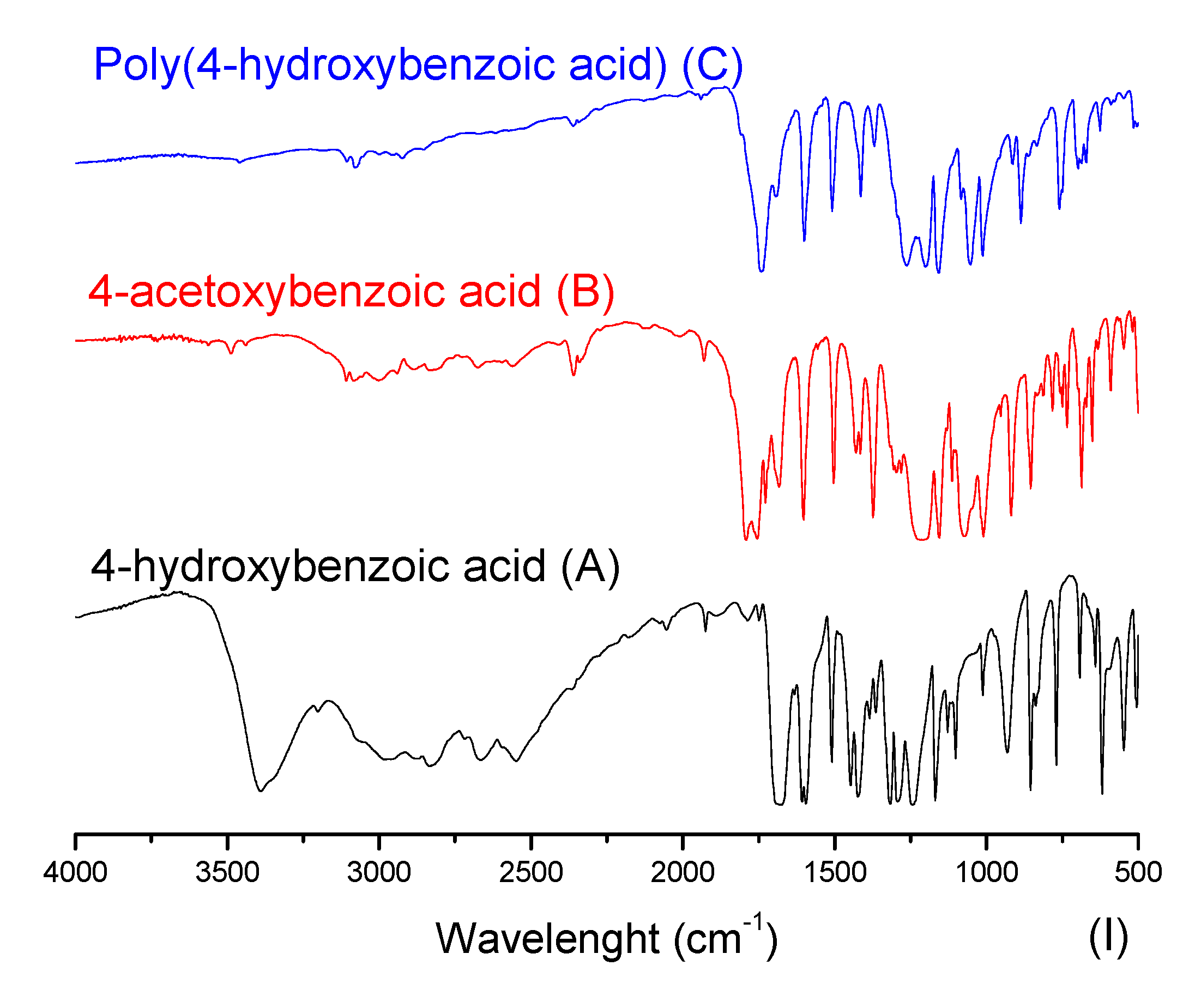
2.3.1. Fourier-Transform Infrared Spectroscopy (FTIR)
2.3.2. Gel Permeation Chromatography (GPC)
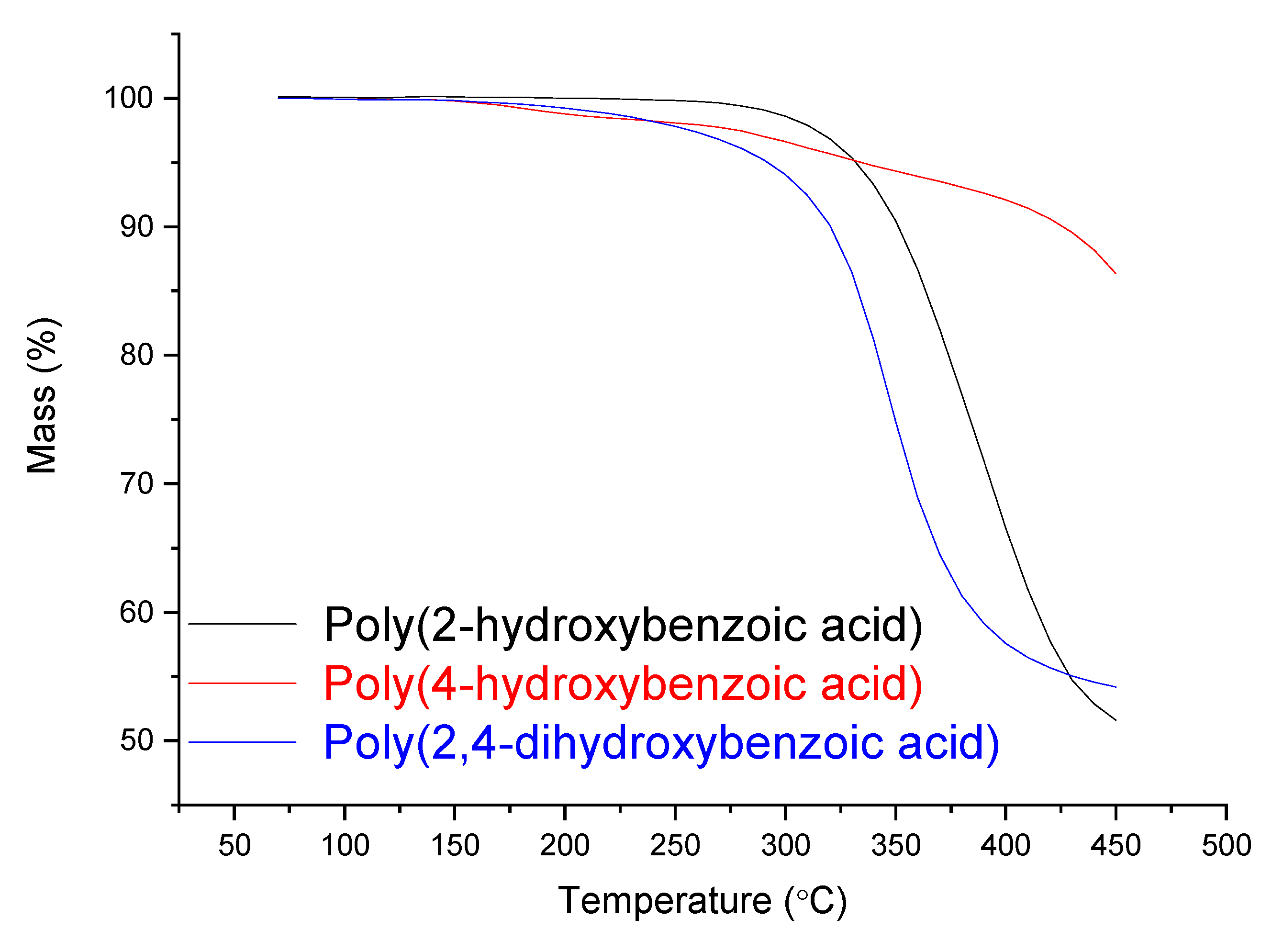
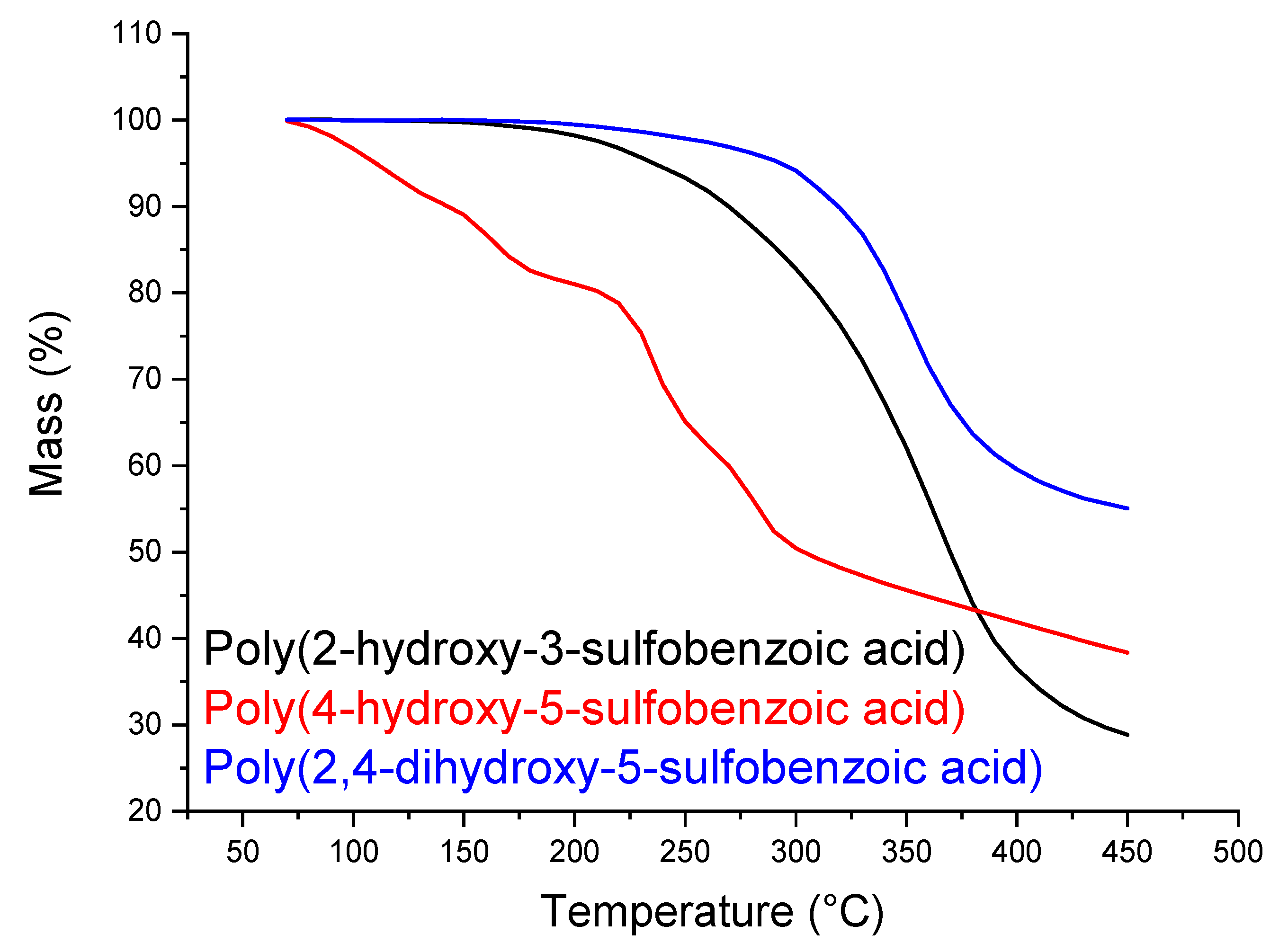
2.3.3. Thermogravimetric Analysis (TGA)
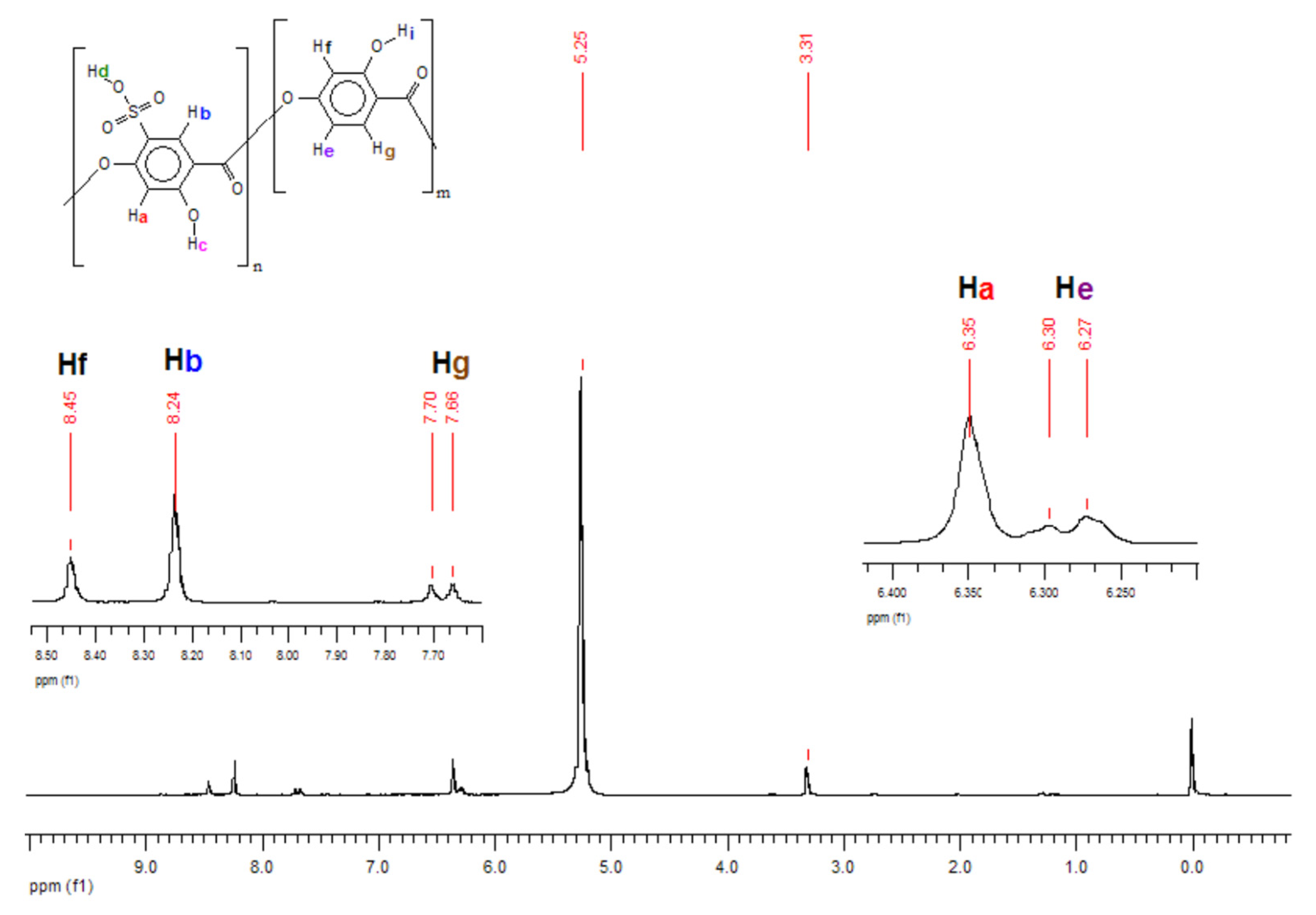
2.3.4. Nuclear Magnetic Resonance of Hydrogen
2.3.5. Qualitative Solubility Evaluation
2.3.6. Acidity Quantification through Titration
3. Results and Discussion
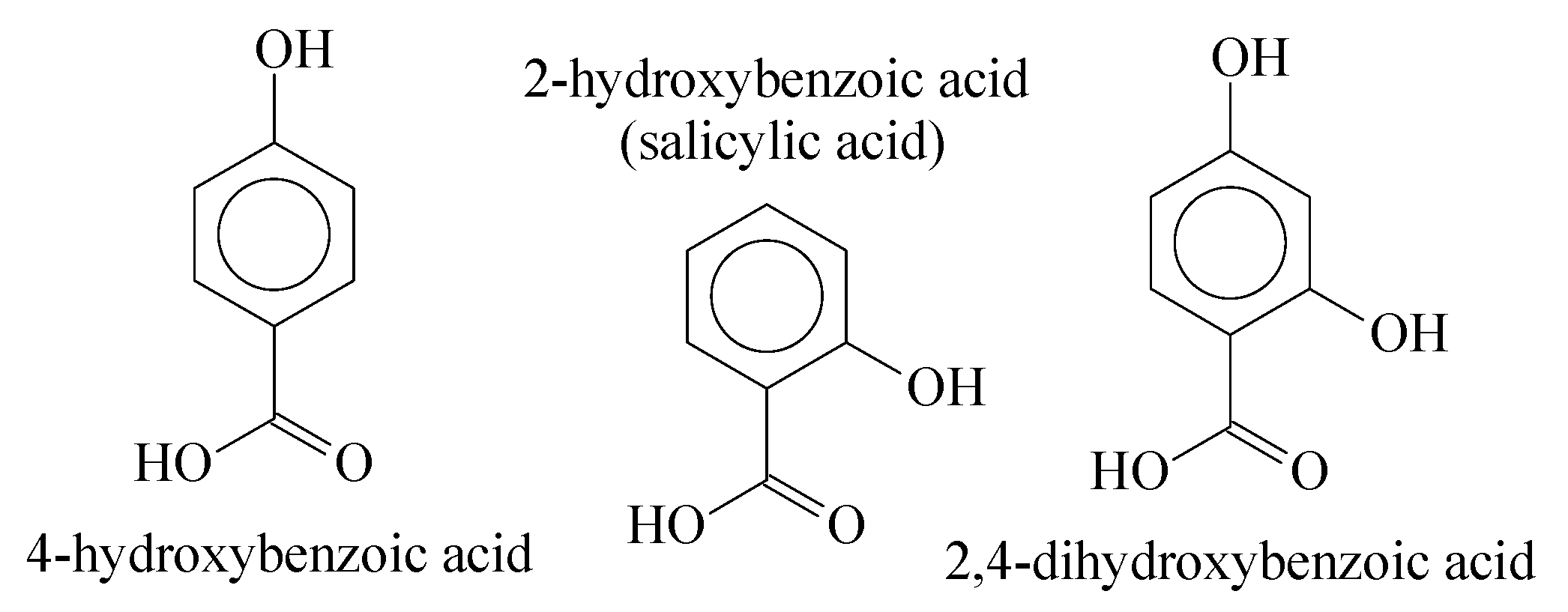
3.1. Mass Polymerization of the Aromatic Acid Compounds
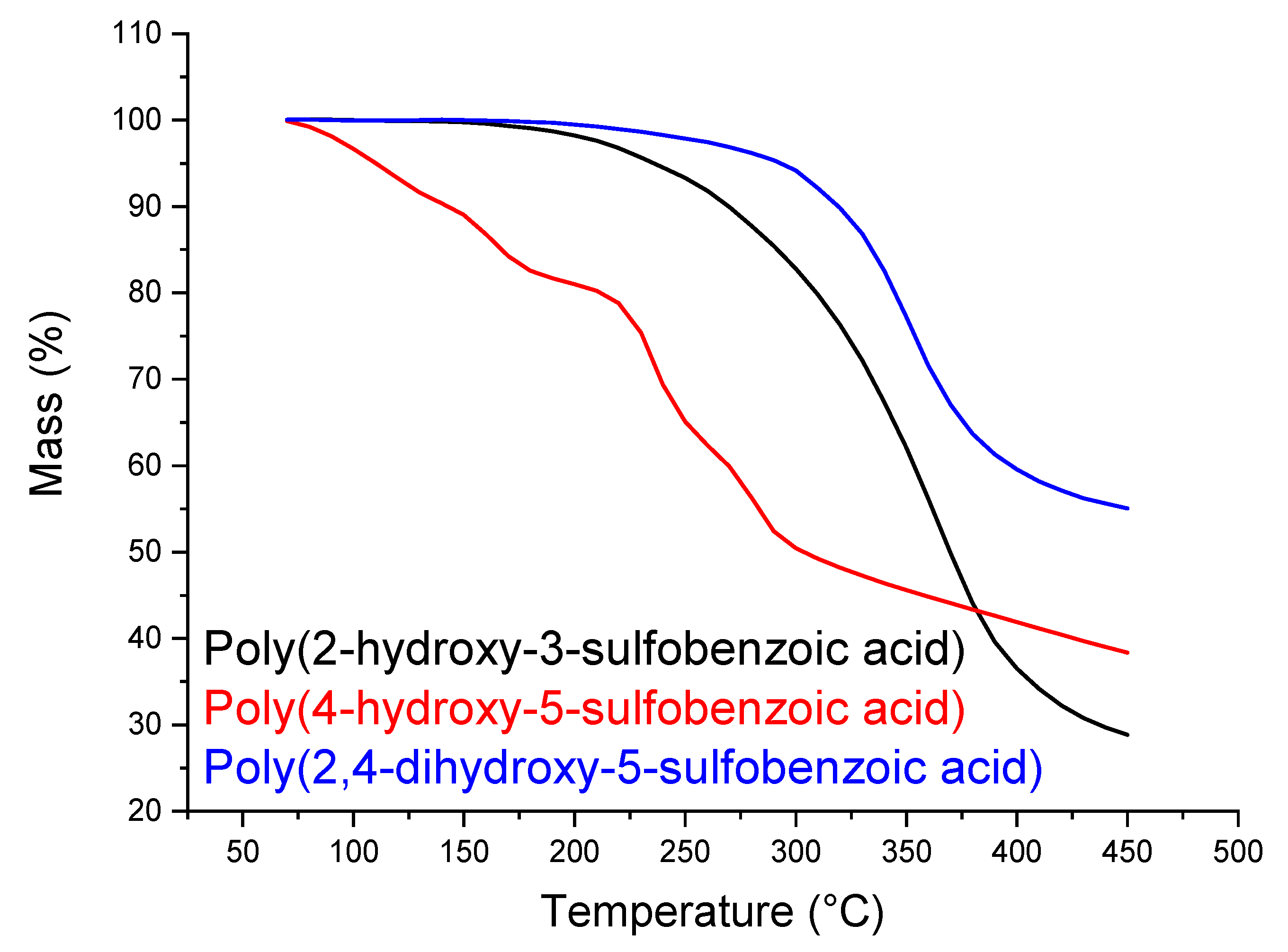
3.2. Sulfonation of the Oligomers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zailan, Z.; Tahir, M.; Jusoh, M.; Zakaria, Z.Y. A review of sulfonic group bearing porous carbon catalyst for biodiesel production. Renew. Energy 2021, 175, 430–452. [Google Scholar] [CrossRef]
- European Federation for Transport and Environment. Brief: 10 Years of EU Fuels Policy Increased EU’s Reliance on Unsustainable Biofuels. Available online: https://www.transportenvironment.org/wp-content/uploads/2021/08/Biofuels-briefing-072021.pdf (accessed on 10 November 2021).
- Vlnieska, V.; Muniz, A.S.; Oliveira, A.R.S.; César-Oliveira, M.A.F.; Kunka, D. Oligocat: Oligoesters as pseudo-homogenous catalysts for biodiesel synthesis. Polymers, 2021; submitted for publication. [Google Scholar]
- Foteinis, S.; Chatzisymeon, E.; Litinas, A.; Tsoutsos, T. Used-cooking-oil biodiesel: Life cycle assessment and comparison with first- and third-generation biofuel. Renew. Energy 2020, 153, 588–600. [Google Scholar] [CrossRef]
- Singh, D.; Sharma, D.; Soni, S.; Inda, C.S.; Sharma, S.; Sharma, P.K.; Jhalani, A. A comprehensive review of biodiesel production from waste cooking oil and its use as fuel in compression ignition engines: 3rd generation cleaner feedstock. J. Clean. Prod. 2021, 307, 127299. [Google Scholar] [CrossRef]
- Ravanipour, M.; Hamidi, A.; Mahvi, A.H. Microalgae biodiesel: A systematic review in Iran. Renew. Sustain. Energy Rev. 2021, 150, 111426. [Google Scholar] [CrossRef]
- Chhandama, M.V.L.; Satyan, K.B.; Changmai, B.; Vanlalveni, C.; Rokhum, S.L. Microalgae as a feedstock for the production of biodiesel: A review. Bioresour. Technol. Rep. 2021, 15, 100771. [Google Scholar] [CrossRef]
- Lotero, E.; Liu, Y.; Lopez, D.E.; Suwannakarn, K.; Bruce, D.A.; Goodwin, J.G. Synthesis of Biodiesel via Acid Catalysis. Ind. Eng. Chem. Res. 2005, 44, 5353–5363. [Google Scholar] [CrossRef]
- Giraldo, L.; Moreno-Piraján, J.C. Lipase supported on mesoporous materials as a catalyst in the synthesis of biodiesel from Persea americana mill oil. J. Mol. Catal. B Enzym. 2012, 77, 32–38. [Google Scholar] [CrossRef]
- Harmer, M.A.; Sun, Q. Solid acid catalysis using ion-exchange resins. Appl. Catal. A Gen. 2001, 221, 45–62. [Google Scholar] [CrossRef]
- Krishnan, S.G.; Pua, F.-L.; Zhang, F. A review of magnetic solid catalyst development for sustainable biodiesel production. Biomass Bioenergy 2021, 149, 106099. [Google Scholar] [CrossRef]
- Xie, W.; Wang, H. Immobilized polymeric sulfonated ionic liquid on core-shell structured Fe3O4/SiO2 composites: A magnetically recyclable catalyst for simultaneous transesterification and esterifications of low-cost oils to biodiesel. Renew. Energy 2020, 145, 1709–1719. [Google Scholar] [CrossRef]
- Da Conceição, L.R.V.; Carneiro, L.M.; Rivaldi, J.D.; de Castro, H.F. Solid acid as catalyst for biodiesel production via simultaneous esterification and transesterification of macaw palm oil. Ind. Crop. Prod. 2016, 89, 416–424. [Google Scholar] [CrossRef]
- Jacobson, K.; Gopinath, R.; Meher, L.C.; Dalai, A.K. Solid acid catalyzed biodiesel production from waste cooking oil. Appl. Catal. B Environ. 2008, 85, 86–91. [Google Scholar] [CrossRef]
- Ganesan, R.; Manigandan, S.; Shanmugam, S.; Chandramohan, V.; Sindhu, R.; Kim, S.-H.; Brindhadevi, K.; Pugazhendhi, A. A detailed scrutinize on panorama of catalysts in biodiesel synthesis. Sci. Total Environ. 2021, 777, 145683. [Google Scholar] [CrossRef]
- Liu, X.; Xing, S.; Yang, L.; Fu, J.; Lv, P.; Zhang, X.; Li, M.; Wang, Z. Highly active and durable Ca-based solid base catalyst for biodiesel production. Fuel 2021, 302, 121094. [Google Scholar] [CrossRef]
- De Oliveira, A.D.; De Lima, M.A.B.; Pires, L.H.D.O.; Da Silva, M.R.; Da Luz, P.T.S.; Angélica, R.S.; Filho, G.N.D.R.; Da Costa, C.E.F.; Luque, R.; Nascimento, L.A.S.D. Bentonites Modified with Phosphomolybdic Heteropolyacid (HPMo) for Biowaste to Biofuel Production. Materials 2019, 12, 1431. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Trejo-López, E.; González-Díaz, M.O.; Aguilar-Vega, M. Waste cooking oil transesterification by sulfonated polyphenylsulfone catalytic membrane: Characterization and biodiesel production yield. Renew. Energy 2022, 182, 1219–1227. [Google Scholar] [CrossRef]
- Yan, J.; Kuang, Y.; Gui, X.; Han, X.; Yan, Y. Engineering a malic enzyme to enhance lipid accumulation in Chlorella protothecoides and direct production of biodiesel from the microalgal biomass. Biomass Bioenergy 2019, 122, 298–304. [Google Scholar] [CrossRef]
- Gardy, J.; Hassanpour, A.; Lai, X.; Ahmed, M.H.; Rehan, M. Biodiesel production from used cooking oil using a novel surface functionalised TiO2 nano-catalyst. Appl. Catal. B Environ. 2017, 207, 297–310. [Google Scholar] [CrossRef]
- Alagumalai, A.; Mahian, O.; Hollmann, F.; Zhang, W. Environmentally benign solid catalysts for sustainable biodiesel production: A critical review. Sci. Total Environ. 2021, 768, 144856. [Google Scholar] [CrossRef]
- Xie, W.; Wan, F. Immobilization of polyoxometalate-based sulfonated ionic liquids on UiO-66-2COOH metal-organic frameworks for biodiesel production via one-pot transesterification-esterification of acidic vegetable oils. Chem. Eng. J. 2019, 365, 40–50. [Google Scholar] [CrossRef]
- Xie, W.; Wang, H. Grafting copolymerization of dual acidic ionic liquid on core-shell structured magnetic silica: A magnetically recyclable Brönsted acid catalyst for biodiesel production by one-pot transformation of low-quality oils. Fuel 2021, 283, 118893. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Pan, H.; Wang, A.; Xu, C.; Yang, S. Magnetically recyclable basic polymeric ionic liquids for efficient transesterification of Firmiana platanifolia L.f. oil into biodiesel. Energy Convers. Manag. 2017, 153, 462–472. [Google Scholar] [CrossRef]
- Carothers, W.H. Studies on polymerization and ring formation. I. An introduction to the general theory of condensation polymers. J. Am. Chem. Soc. 1929, 51, 2548–2559. [Google Scholar] [CrossRef]
- Edlund, U.; Albertsson, A.C. Polyesters based on diacid monomers. Adv. Drug Deliv. Rev. 2003, 55, 585–609. [Google Scholar] [CrossRef]
- Pang, K.; Kotek, R.; Tonelli, A. Review of conventional and novel polymerization processes for polyesters. Prog. Polym. Sci. 2006, 31, 1009–1037. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Wang, L.; Wang, D.; Zhu, G.; Li, J. Synthesis and properties of highly branched sulfonated poly(arylene ether)s as proton exchange membranes. Eur. Polym. J. 2011, 47, 1985–1993. [Google Scholar] [CrossRef]
- Akelah, A.; Sherrington, D.C. Recent developments in the application of functionalized polymers in organic synthesis. Polymer 1983, 24, 1369–1386. [Google Scholar] [CrossRef]
- Soldi, R.A. Síntese e caracterização de catalisadores poliméricos ácidos, a partir da reciclagem química de poliestireno, e avaliação na síntese de biodiesel em fase heterogênea. In Departamento de Química; Universidade Federal do Paraná: Curitiba, Brazil, 2006; pp. 6–12. [Google Scholar]
- Stevens, M.P. Polymer Chemistry: An Introduction; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Kučera, F.; Jančář, J. Homogeneous and heterogeneous sulfonation of polymers: A review. Polym. Eng. Sci. 1998, 38, 783–792. [Google Scholar] [CrossRef]
- Stirton, A.J.; Peterson, R.F.; Groggins, P.H. Sulfonated arylstearic acids. Ind. Eng. Chem. 1940, 32, 1136–1137. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Kaji, A.; Kitamura, K.; Takase, S.; Omote, K.; Asako, Y.; Kimura, K. Polymer electrolyte membranes derived from novel fluorine-containing poly(arylene ether ketone)s by controlled post-sulfonation. Polymer 2012, 53, 4388–4398. [Google Scholar] [CrossRef]
- Peron, J.; Ruiz, E.; Jones, D.J.; Rozière, J. Solution sulfonation of a novel polybenzimidazole: A proton electrolyte for fuel cell application. J. Membr. Sci. 2008, 314, 247–256. [Google Scholar] [CrossRef]
- Barroso-Bujans, F.; Verdejo, R.; Lozano, A.; Fierro, J.L.G.; Lopez-Manchado, M.A. Sulfonation of vulcanized ethylene–propylene–diene terpolymer membranes. Acta Mater. 2008, 56, 4780–4788. [Google Scholar] [CrossRef]
- Das, S.; Kumar, P.; Dutta, K.; Kundu, P.P. Partial sulfonation of PVdF-co-HFP: A preliminary study and characterization for application in direct methanol fuel cell. Appl. Energy 2014, 113, 169–177. [Google Scholar] [CrossRef]
- Dong, D.; Jiang, S.; Ni, Y.; Jiang, B. Syntheses and properties of thermotropic copolyesters of p-hydroxybenzoic acid. Eur. Polym. J. 2001, 37, 611–617. [Google Scholar] [CrossRef]
- Field, L.D.; Sternhell, S.; Kalman, J.R. Organic Structures from Spectra; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar]
- Lee, J.K.; Li, W.; Manthiram, A. Poly(arylene ether sulfone)s containing pendant sulfonic acid groups as membrane materials for direct methanol fuel cells. J. Membr. Sci. 2009, 330, 73–79. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
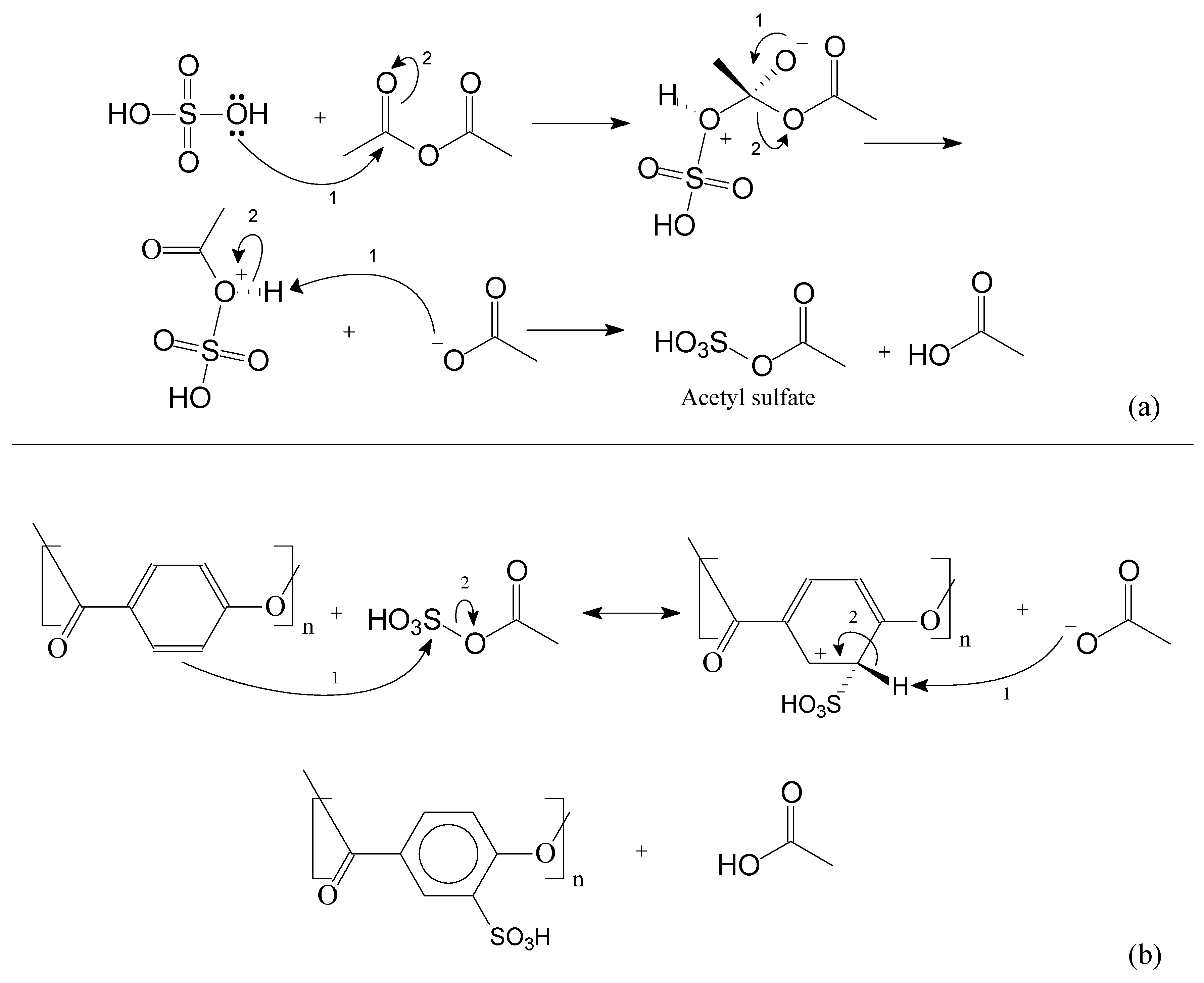{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | SO3H Content (mmol·gpolymer−1) | SO3H Substitution (mol%) |
|---|---|---|
| P4S a | 4.05 | 48.6 |
| P2S a | 3.88 | 46.6 |
| P24S b | 2.85 | 38.0 |
| Technique | SO3H Content (mmol·gpolymer−1) | SO3H Substitution (mol%) |
|---|---|---|
| 1H NMR | 4.92 | 67.3 |
| GPC | 5.06 | 68.9 |
| Titration | 2.85 | 38.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlnieska, V.; Muniz, A.S.; Oliveira, A.R.S.; César-Oliveira, M.A.F.; Kunka, D. Synthesis and Chemical Functionalization of Pseudo-Homogeneous Catalysts for Biodiesel Production—Oligocat. Polymers 2022, 14, 19. https://doi.org/10.3390/polym14010019
Vlnieska V, Muniz AS, Oliveira ARS, César-Oliveira MAF, Kunka D. Synthesis and Chemical Functionalization of Pseudo-Homogeneous Catalysts for Biodiesel Production—Oligocat. Polymers. 2022; 14(1):19. https://doi.org/10.3390/polym14010019
Chicago/Turabian StyleVlnieska, Vitor, Aline S. Muniz, Angelo R. S. Oliveira, Maria A. F. César-Oliveira, and Danays Kunka. 2022. "Synthesis and Chemical Functionalization of Pseudo-Homogeneous Catalysts for Biodiesel Production—Oligocat" Polymers 14, no. 1: 19. https://doi.org/10.3390/polym14010019
APA StyleVlnieska, V., Muniz, A. S., Oliveira, A. R. S., César-Oliveira, M. A. F., & Kunka, D. (2022). Synthesis and Chemical Functionalization of Pseudo-Homogeneous Catalysts for Biodiesel Production—Oligocat. Polymers, 14(1), 19. https://doi.org/10.3390/polym14010019