
Rheological Behavior of Blends of Metallocene Catalyzed Long-Chain Branched Polyethylenes. Part I: Shear Rheological and Thermorheological Behavior



Abstract
1. Introduction
2. Experimental
2.1. Materials and Sample Preparation
2.2. Rheology
3. Results
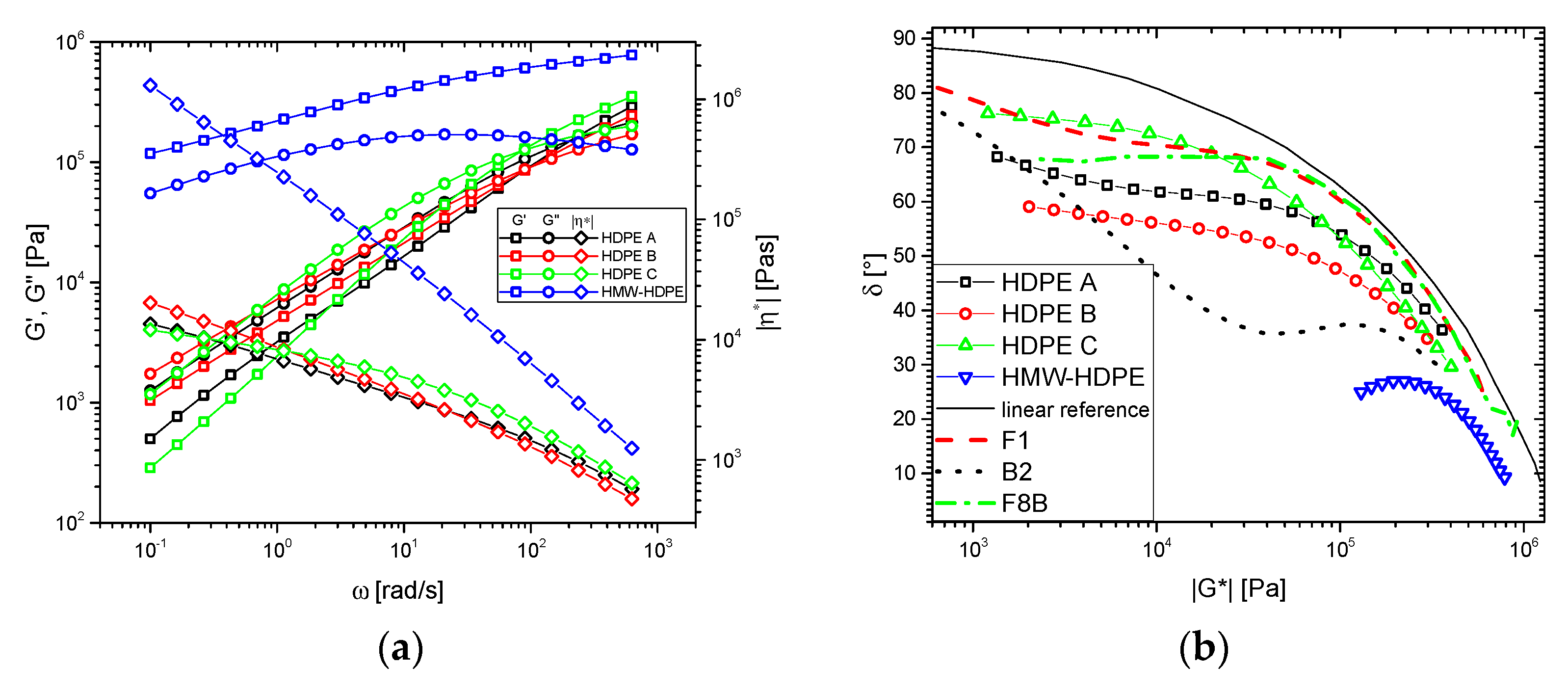
3.1. Shear Rheological Properties
3.1.1. Base Materials
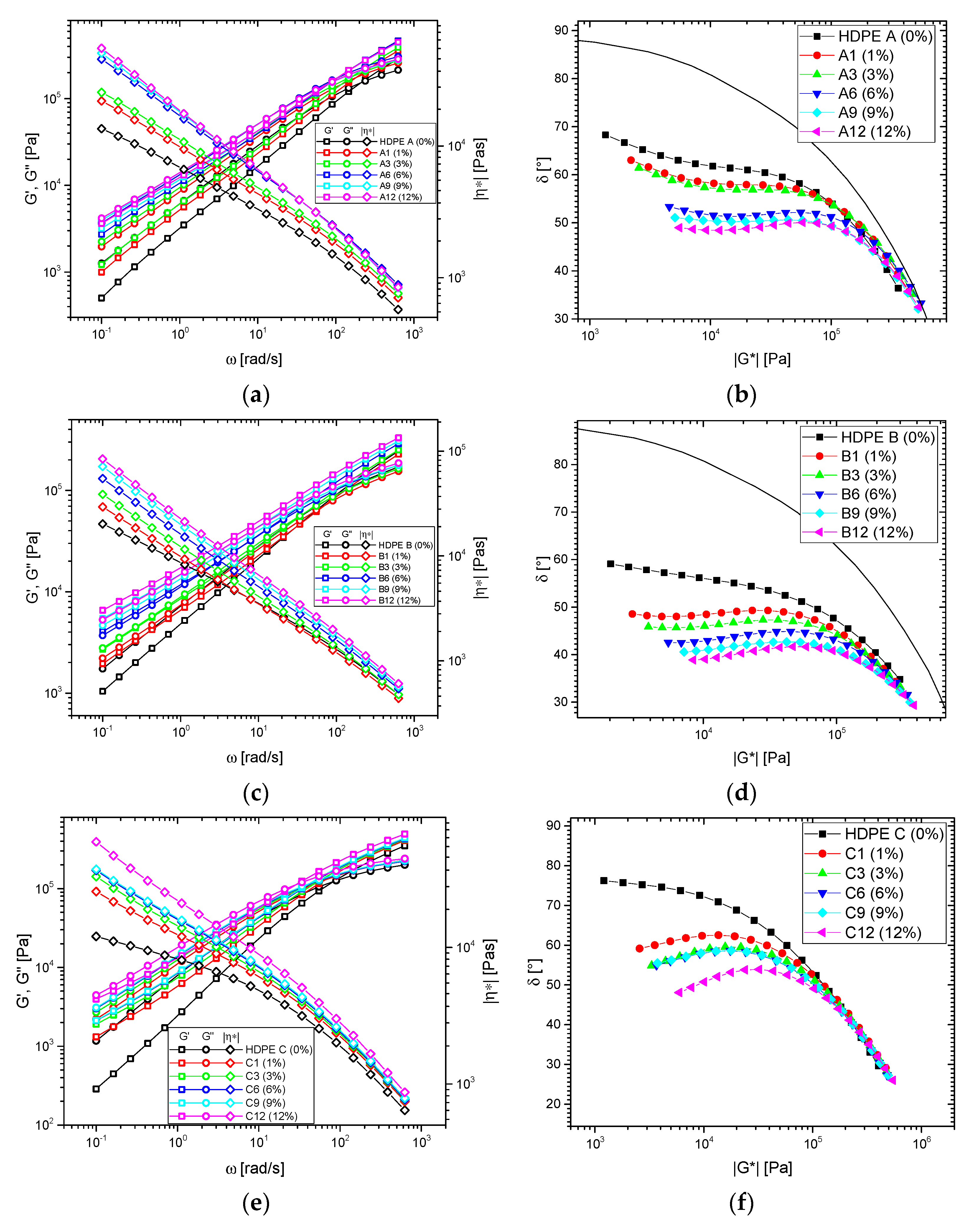
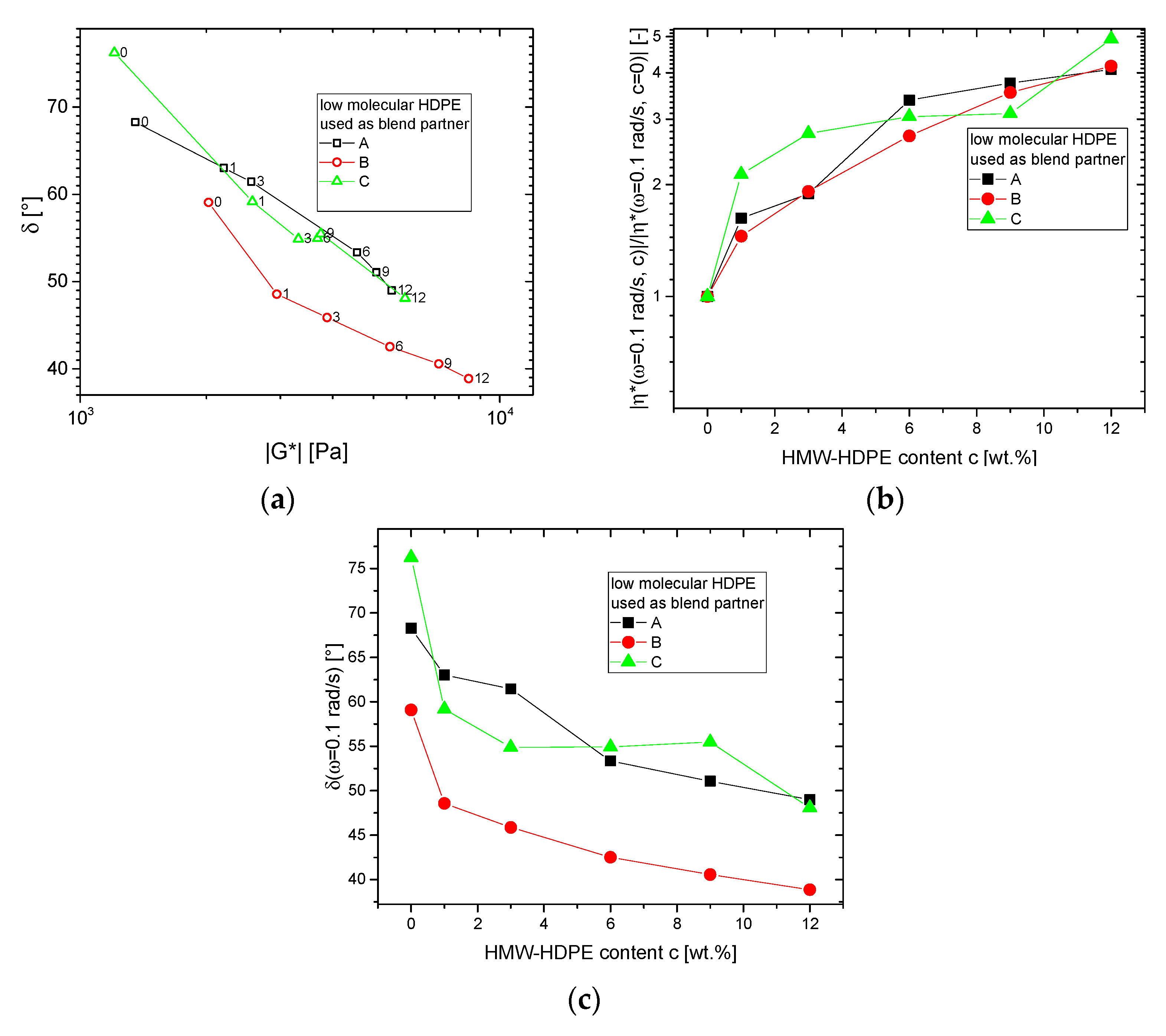
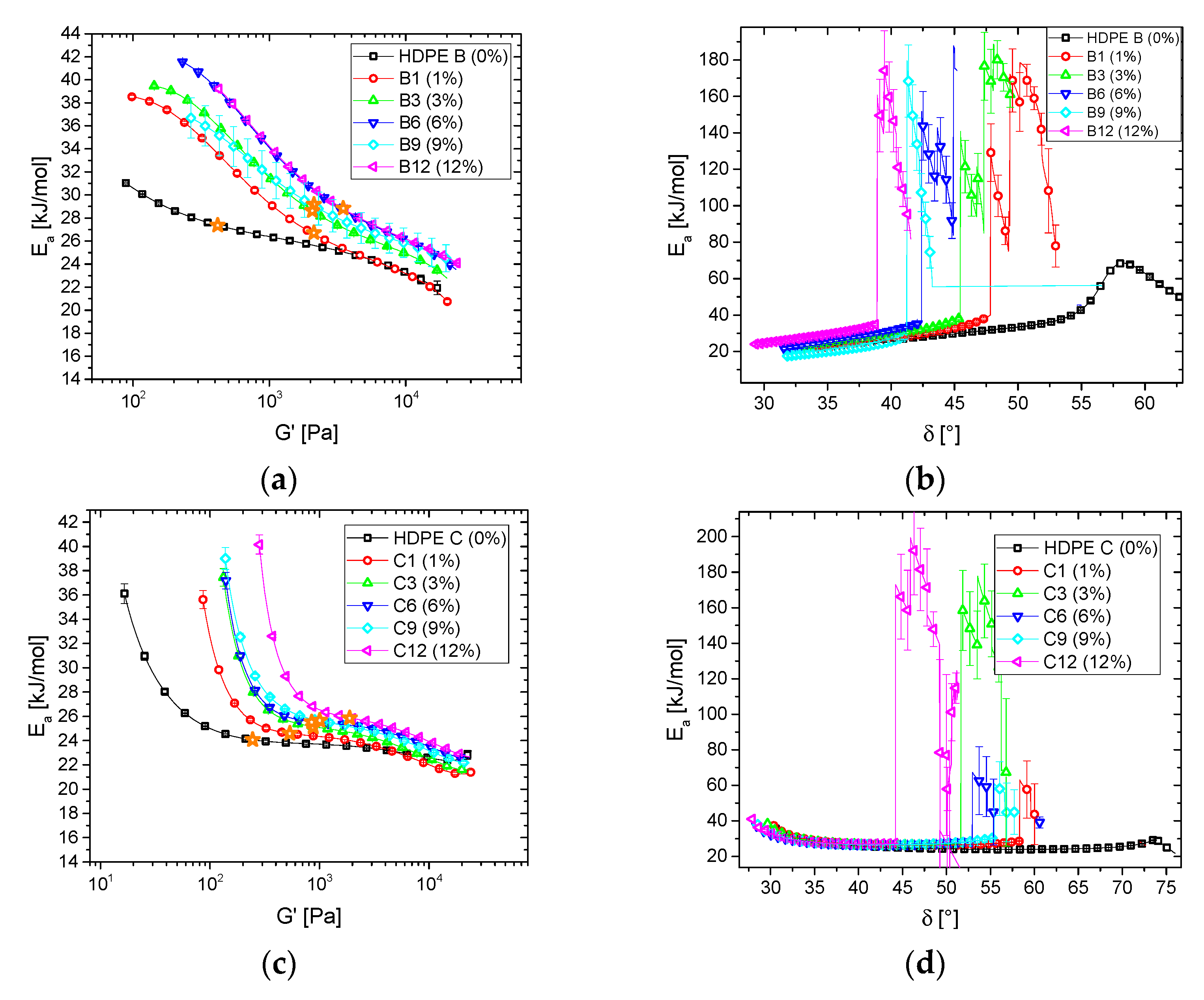
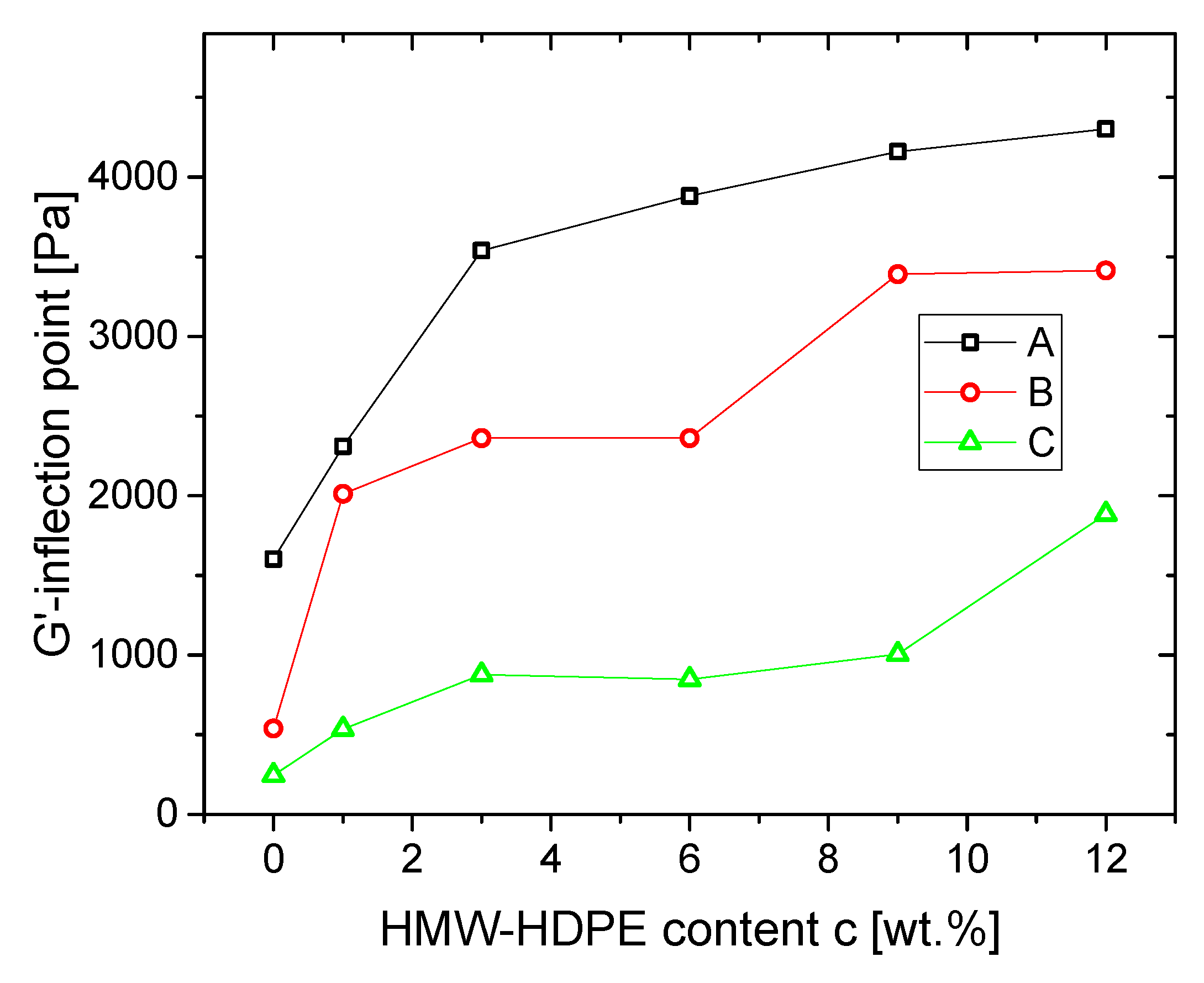
3.1.2. Blends
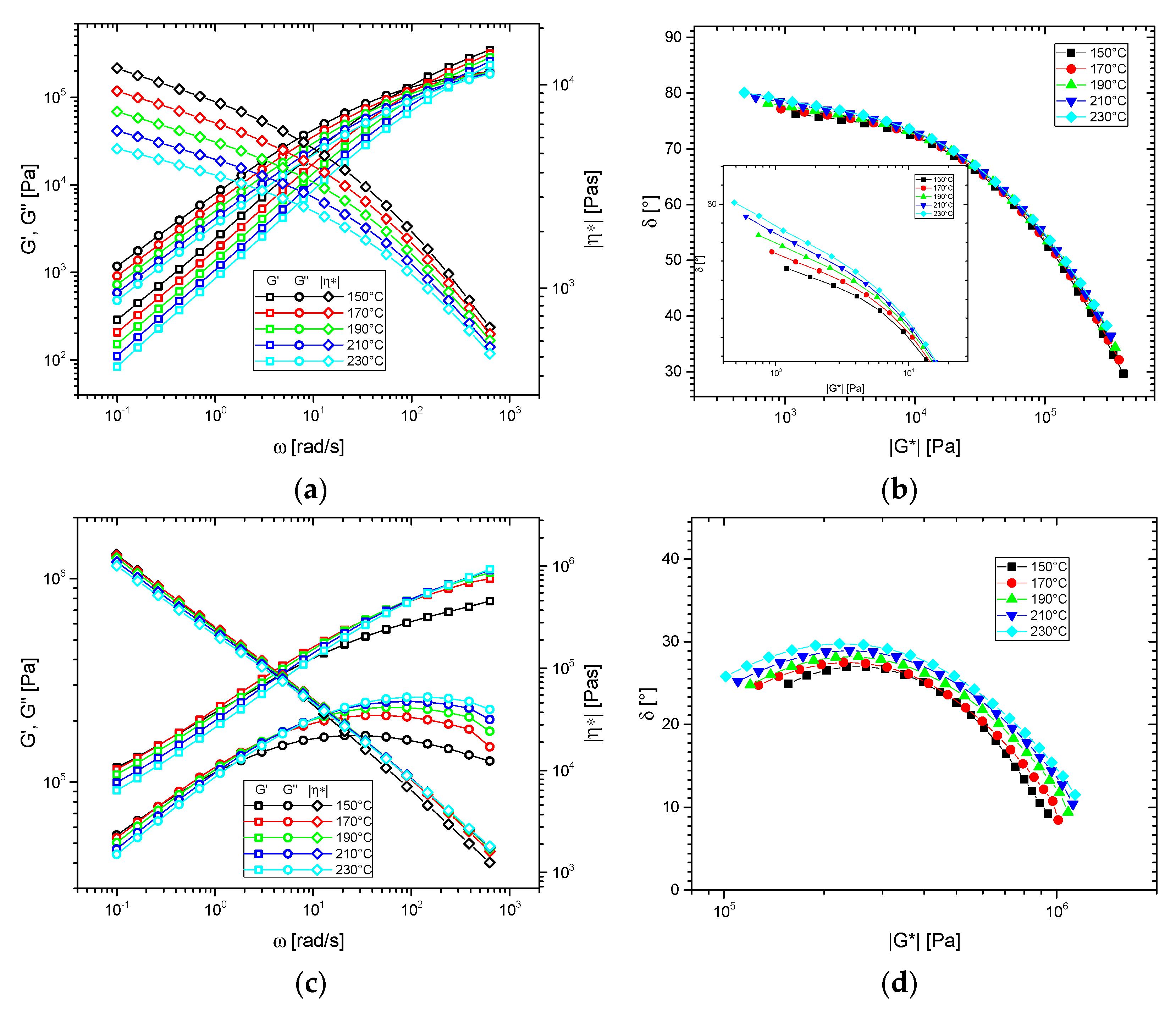
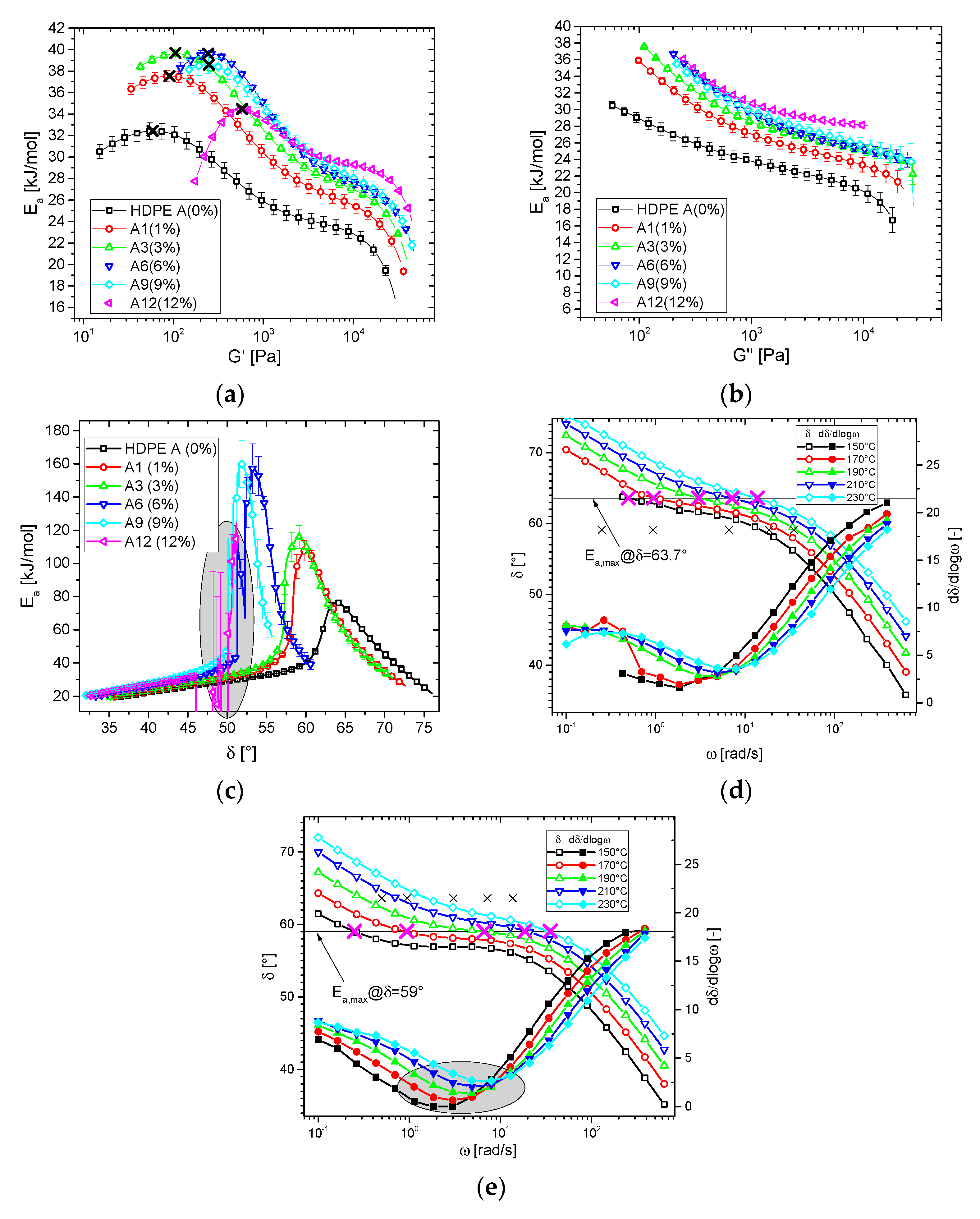
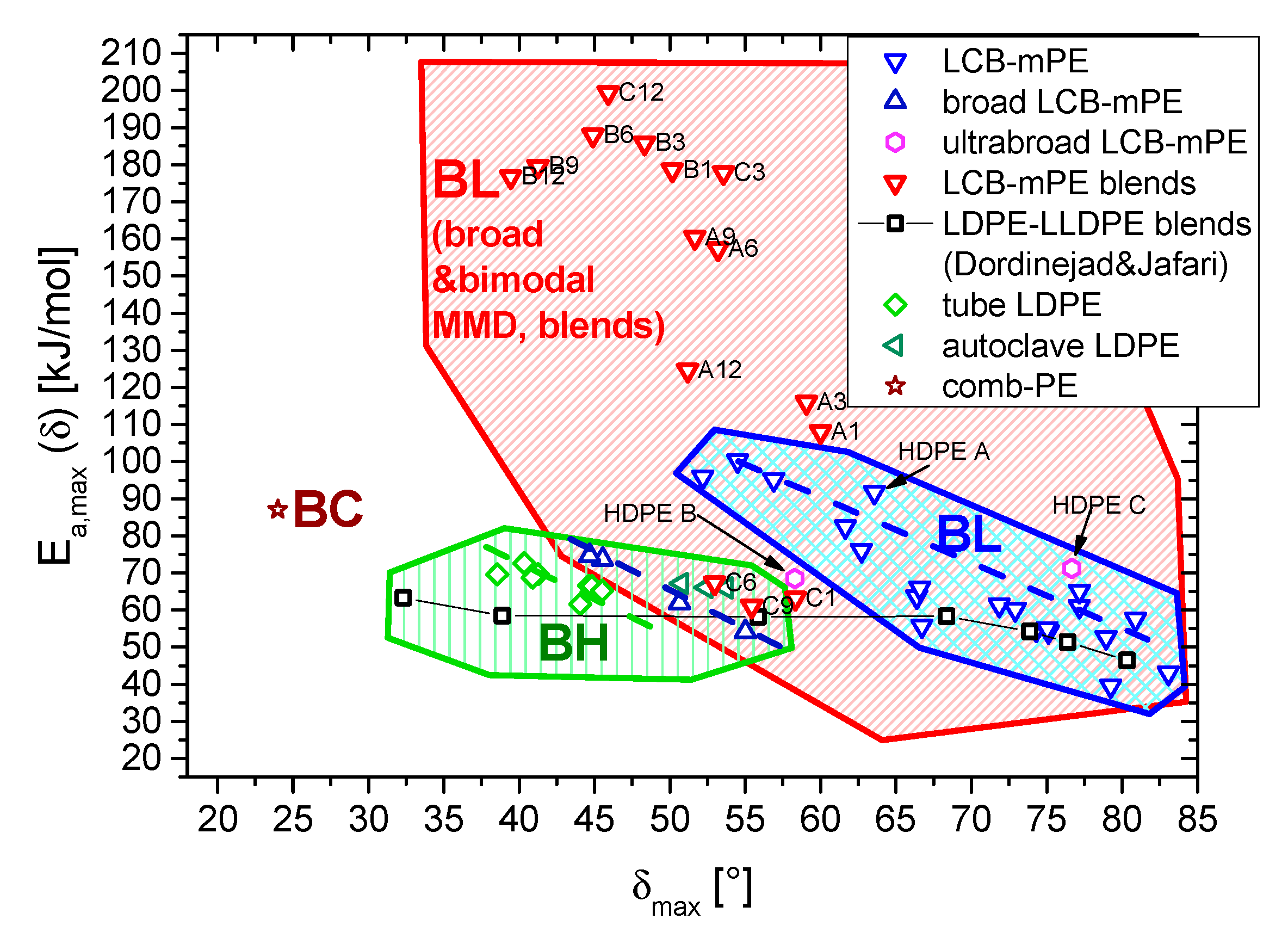
3.2. Thermorheological Behavior
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Rheological quantities | |
| Storage modulus | G′ |
| Loss modulus | G″ |
| Phase angle | δ |
| Complex viscosity function | |η*|(ω) |
| zero shear-rate viscosity | η0 |
| Magnitude of the complex modulus | |G*| |
| Angular frequency | ω |
| Relaxation strength | H |
| Relaxation time | τ |
| Hencky strain rate | |
| crossover frequency | ωc (ω of G′=G″) |
| temperature dependence according to Williams-Landel-Ferry’s law (based on free volume and not on potential barrier) | WLF-temperature dependence |
| shift factor | aT |
| Arrhenius activation energy | Ea |
| Activation energy spectrum determined from shifting δ(ω) | Ea(δ) |
| Activation energy spectrum determined from shifting G’(ω) | Ea(G′) |
| Activation energy spectrum determined from shifting G”(ω) | Ea(G″) |
| Maximum activation energy, determined from Ea(δ) | Ea,max |
| Phase angle at which Ea,max is found | δmax |
| Critical phase angle according to definition of Trinkle et al. [73] | δc |
| Slope of phase angle | dδ/dlogω |
| Material with an LCB-mPE-like thermorheologically complex behavior [62] | LCB-mPE-like behavior (BL), BL-material |
| Material with an LDPE-like thermorheologically complex behavior [62] | LDPE-like behavior (BH), BH-material |
| Material with a comb-PE-like thermorheologically complex behavior [62] | comb-PE -like behavior (BC), BC-material |
| Molecular quantities | |
| number-average molar mass | Mn |
| weight-average molar mass | Mw |
| z-average molar mass | Mz |
| Polydispersity index | Mw/Mn |
| molar mass distribution | MMD |
| long-chain branches | LCBs |
| Material types | |
| long-chain branched metallocene catalyzed polyethylene | LCB-mPE |
| long-chain branched metallocene catalyzed high densitypolyethylene | LCB-mHDPE |
| long-chain branched metallocene catalyzed linear low densitypolyethylene | LCB-mLLDPE |
| low-density polyethylene | LDPE |
| linear low density Ziegler-Natta polyethylene | ZN-LLDPE |
| high density Ziegler-Natta polyethylene | ZN-HDPE |
| Miscellaneous | |
| Logarithm (base 10) | log |
| Weight % | wt.% |
References
- Münstedt, H.; Steffl, T.; Malmberg, A. Correlation between rheological behaviour in uniaxial elongation and film blowing properties of various polyethylenes. Rheol. Acta 2005, 45, 14–22. [Google Scholar] [CrossRef]
- Münstedt, H.; Kurzbeck, S.; Stange, J. Importance of elongational properties of polymer melts for film blowing and thermoforming. Polym. Eng. Sci. 2006, 46, 1190–1195. [Google Scholar] [CrossRef]
- Hencky, H. Über die form des elastizitatsgesetzes bei ideal elastischen stoffen. Zeit. Tech. Phys. 1928, 9, 215–220. [Google Scholar]
- Stadler, F.J.; Kaschta, J.; Münstedt, H.; Becker, F.; Buback, M. Influence of molar mass distribution and long-chain branching on strain hardening of low density polyethylene. Rheol. Acta 2009, 48, 479–490. [Google Scholar] [CrossRef]
- Stadler, F.J.; Nishioka, A.; Stange, J.; Koyama, K.; Münstedt, H. Comparison of the elongational behavior of various polyolefins in uniaxial and equibiaxial flows. Rheol. Acta 2007, 46, 1003–1012. [Google Scholar] [CrossRef]
- Berry, G.C.; Fox, T.G. The viscosity of polymers and their concentrated solutions. Adv. Polym. Sci. 1968, 5, 261–357. [Google Scholar] [CrossRef]
- Stadler, F.J.; Piel, C.; Kaschta, J.; Rulhoff, S.; Kaminsky, W.; Münstedt, H. Dependence of the zero shear-rate viscosity and the viscosity function of linear high-density polyethylenes on the mass-average molar mass and polydispersity. Rheol. Acta 2006, 45, 755–764. [Google Scholar] [CrossRef]
- Stadler, F.J.; Muünstedt, H. Terminal viscous and elastic properties of linear ethene/α-olefin copolymers. J. Rheol. 2008, 52, 697. [Google Scholar] [CrossRef]
- Otegui, J.; Ramos, J.; Vega, J.F.; Martinez-Salazar, J. Effect of high molar mass species on linear viscoelastic properties of polyethylene melts. Eur. Polym. J. 2013, 49, 2748–2758. [Google Scholar] [CrossRef][Green Version]
- Ansari, M.; Hatzikiriakos, S.G.; Sukhadia, A.M.; Rohlfing, D.C. Rheology of Ziegler–Natta and metallocene high-density polyethylenes: Broad molecular weight distribution effects. Rheol. Acta 2010, 50, 17–27. [Google Scholar] [CrossRef]
- Vega, J.F.; Otegui, J.; Ramos, J.; Martínez-Salazar, J. Effect of molecular weight distribution on Newtonian viscosity of linear polyethylene. Rheol. Acta 2011, 51, 81–87. [Google Scholar] [CrossRef]
- Wasserman, S.H.; Graessley, W.W. Prediction of linear viscoelastic response for entangled polyolefin melts from molecular weight distribution. Polym. Eng. Sci. 1996, 36, 852–861. [Google Scholar] [CrossRef]
- Mavridis, H.; Shroff, R.N. Temperature-dependence of polyolefin melt rheology. Polym. Eng. Sci. 1992, 32, 1778–1791. [Google Scholar] [CrossRef]
- Carella, J.M. Comments on the paper “comparison of the rheological properties of metallocene-catalyzed and conventional high-density polyethylenes”. Macromolecules 1996, 29, 8280–8281. [Google Scholar] [CrossRef]
- Wood-Adams, P.; Costeux, S. Thermorheological behavior of polyethylene: Effects of microstructure and long chain branching. Macromolecules 2001, 34, 6281–6290. [Google Scholar] [CrossRef]
- Lohse, D.J.; Milner, S.T.; Fetters, L.J.; Xenidou, M.; Hadjichristidis, N.; Mendelson, R.A.; Garcia-Franco, C.A.; Lyon, M.K. Well-defined, model long chain branched polyethylene: 2. Melt rheological behavior. Macromolecules 2002, 35, 3066–3075. [Google Scholar] [CrossRef]
- Mohammadi, M.; Yousefi, A.A.; Ehsani, M. Thermorheological analysis of blend of high- and low-density polyethylenes. J. Polym. Res. 2012, 19. [Google Scholar] [CrossRef]
- Dordinejad, A.K.; Jafari, S.H. A qualitative assessment of long chain branching content in LLDPE, LDPE and their blends via thermorheological analysis. J. Appl. Polym. Sci. 2013, 130, 3240–3250. [Google Scholar] [CrossRef]
- Derakhshandeh, M.; Ansari, M.; Doufas, A.K.; Hatzikiriakos, S.G. Microstructure characterization of polyethylene using thermo-rheological methods. Polym. Test. 2017, 60, 68–77. [Google Scholar] [CrossRef]
- Hepperle, J.; Münstedt, H.; Haug, P.K.; Eisenbach, C.D. Rheological properties of branched polystyrenes: Linear viscoelastic behavior. Rheol. Acta 2005, 45, 151–163. [Google Scholar] [CrossRef][Green Version]
- Carella, J.M.; Gotro, J.T.; Graessley, W.W. Thermorheological effects of long-chain branching in entangled polymer melts. Macromolecules 1986, 19, 659–667. [Google Scholar] [CrossRef]
- Resch, J.A.; Keßner, U.; Stadler, F.J. Thermorheological behavior of polyethylene: A sensitive probe to molecular structure. Rheol. Acta 2011, 50, 559–575. [Google Scholar] [CrossRef]
- Auhl, D. Molekulare Struktur und Rheologische Eigenschaften von Strahlenmodifizierten Polypropylen. Ph.D. Thesis, Friedrich-Alexander Universität Erlangen-Nürnberg, Erlangen, Germany, 2006. [Google Scholar]
- Ye, Z.; AlObaidi, F.; Zhu, S. Synthesis and rheological properties of long-chain-branched isotactic polypropylenes prepared by copolymerization of propylene and nonconjugated dienes. Ind. Eng. Chem. Res. 2004, 43, 2860–2870. [Google Scholar] [CrossRef]
- Stadler, F.J.; Arikan, B.; Kaschta, J.; Kaminsky, W. Long-chain branches in syndiotactic polypropene induced by vinyl chloride. Macromol. Chem. Physic. 2010, 211, 1472–1481. [Google Scholar] [CrossRef]
- Auhl, D.; Stange, J.; Münstedt, H.; Krause, B.; Voigt, D.; Lederer, A.; Lappan, U.; Lunkwitz, K. Long-chain branched polypropylenes by electron beam irradiation and their rheological properties. Macromolecules 2004, 37, 9465–9472. [Google Scholar] [CrossRef]
- Stange, J.; Wächter, S.; Kaspar, H.; Münstedt, H. Linear rheological properties of the semi-fluorinated copolymer tetrafluoroethylene-hexafluoropropylene-vinylidenfluoride (THV) with controlled amounts of long-chain branching. Macromolecules 2007, 40, 2409–2416. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, Y.; Bai, J.; Wang, Z.; Wang, Z. Bimodal architecture and rheological and foaming properties for gamma-irradiated long-chain branched polylactides. RSC Adv. 2013, 3, 8783–8795. [Google Scholar] [CrossRef]
- Eckstein, A.; Friedrich, C.; Lobbrecht, A.; Spitz, R.; Mulhaupt, R. Comparison of the viscoelastic properties of syndio- and isotactic polypropylenes. Acta Polym. 1997, 48, 41–46. [Google Scholar] [CrossRef]
- Stadler, F.J.; Gabriel, C.; Münstedt, H. Influence of short-chain branching of polyethylenes on the temperature dependence of rheological properties in shear. Macromol Chem Physic 2007, 208, 2449–2454. [Google Scholar] [CrossRef]
- Kessner, U.; Kaschta, J.; Stadler, F.J.; le Duff, C.C.S.; Drooghaag, X.; Muünstedt, H. Thermorheological behavior of various short- and long-chain branched polyethylenes and their correlations with the molecular structure. Macromolecules 2010, 43, 7341–7350. [Google Scholar] [CrossRef]
- Gabriel, C. Einfluss der Molekularen Struktur auf das viskoelastische Verhalten von Polyethylenschmelzen. Ph.D. Thesis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany, 2001. [Google Scholar]
- Bonchev, D.; Dekmezian, A.H.; Markel, E.; Faldi, A. Topology—rheology regression models for monodisperse linear and branched polyethylenes. J. Appl. Polym. Sci. 2003, 90, 2648–2656. [Google Scholar] [CrossRef]
- Kokko, E.; Malmberg, A.; Lehmus, P.; Löfgren, B.; Seppälä, J.V. Influence of the catalyst and polymerization conditions on the long-chain branching of metallocene-catalyzed polyethenes. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 376–388. [Google Scholar] [CrossRef]
- Carella, J.M.; Graessley, W.W.; Fetters, L.J. Effects of chain microstructure on the viscoelastic properties of linear polymer melts: Polybutadienes and hydrogenated polybutadienes. Macromolecules 1984, 17, 2775–2786. [Google Scholar] [CrossRef]
- Malmberg, A.; Liimatta, J.; Lehtinen, A.; Löfgren, B. Characteristics of long chain branching in ethene polymerization with single site catalysts. Macromolecules 1999, 32, 6687–6696. [Google Scholar] [CrossRef]
- Laun, H.M. Orientation of macromolecules and elastic deformations in polymer melts. Influence of molecular structure on the reptation of molecules. Prog. Colloid Polym. Sci. 1987, 75, 111–139. [Google Scholar] [CrossRef]
- Stadler, F.J.; Bailly, C. A new method for the calculation of continuous relaxation spectra from dynamic-mechanical data. Rheol. Acta 2009, 48, 33–49. [Google Scholar] [CrossRef]
- Kaschta, J.; Stadler, F.J. Avoiding waviness of relaxation spectra. Rheol. Acta 2009, 48, 709–713. [Google Scholar] [CrossRef]
- Stadler, F.J. Effect of incomplete datasets on the calculation of continuous relaxation spectra from dynamic-mechanical data. Rheol. Acta 2010, 49, 1041–1057. [Google Scholar] [CrossRef]
- Stadler, F.J.; Kaschta, J.; Münstedt, H. Thermorheological behavior of various long-chain branched polyethylenes. Macromolecules 2008, 41, 1328–1333. [Google Scholar] [CrossRef]
- Kessner, U.; Kaschta, J.; Münstedt, H. Determination of method-invariant activation energies of long-chain branched low-density polyethylenes. J. Rheol. 2009, 53, 1001–1016. [Google Scholar] [CrossRef]
- Stadler, F.J.; Chun, Y.S.; Han, J.H.; Lee, E.; Park, S.H.; Yang, C.B.; Choi, C. Deriving comprehensive structural information on long-chain branched polyethylenes from analysis of thermo-rheological complexity. Polymer 2016, 104, 179–192. [Google Scholar] [CrossRef]
- Bai, L.; Li, Y.-M.; Yang, W.; Yang, M.-B. Rheological behavior and mechanical properties of high-density polyethylene blends with different molecular weights. J. Appl. Polym. Sci. 2010, 118, 1356–1363. [Google Scholar] [CrossRef]
- Shen, H.-W.; Xie, B.-H.; Yang, W.; Yang, M.-B. Thermal and rheological properties of polyethylene blends with bimodal molecular weight distribution. J. Appl. Polym. Sci. 2013, 129, 2145–2151. [Google Scholar] [CrossRef]
- Chen, Y.; Zou, H.; Liang, M.; Liu, P. Rheological, thermal, and morphological properties of low-density polyethylene/ultra-high-molecular-weight polyethylene and linear low-density polyethylene/ultra-high-molecular-weight polyethylene blends. J. Appl. Polym. Sci. 2013, 129, 945–953. [Google Scholar] [CrossRef]
- Chaudhuri, K.; Poddar, S.; Pol, H.; Lele, A.; Mathur, A.; Rao, G.S.S.; Jasra, R. The effect of processing conditions on the rheological properties of blends of ultra high molecular weight polyethylene with high-density polyethylene. Polym. Eng. Sci. 2019, 59, 821–829. [Google Scholar] [CrossRef]
- Dordinejad, A.K.; Jafari, S.H.; Khonakdar, H.A.; Wagenknecht, U.; Heinrich, G. Thermorheological behavior analysis of mLLDPE and mVLDPE: Correlation with branching structure. J. Appl. Polym. Sci. 2013, 129, 458–463. [Google Scholar] [CrossRef]
- Zimm, B.H.; Stockmayer, W.H. The Dimensions of chain molecules containing branches and rings. J. Chem. Phys. 1949, 17, 1301–1314. [Google Scholar] [CrossRef]
- Stadler, F.J. Detecting very low levels of long-chain branching in metallocene-catalyzed polyethylenes. Rheol. Acta 2012, 51, 821–840. [Google Scholar] [CrossRef]
- Chaudhuri, K.; Lele, A.K. Rheological quantification of the extent of dissolution of ultrahigh molecular weight polyethylene in melt-compounded blends with high density polyethylene. J. Rheol. 2020, 64, 1–12. [Google Scholar] [CrossRef]
- Stadler, F.J.; Karimkhani, V. Correlations between the Characteristic rheological quantities and molecular structure of long-chain branched metallocene catalyzed polyethylenes. Macromolecules 2011, 44, 5401–5413. [Google Scholar] [CrossRef]
- Piel, C.; Stadler, F.J.; Kaschta, J.; Rulhoff, S.; Münstedt, H.; Kaminsky, W. Structure-property relationships of linear and long-chain branched metallocene high-density polyethylenes characterized by shear rheology and SEC-MALLS. Macromol. Chem. Physic. 2006, 207, 26–38. [Google Scholar] [CrossRef]
- Stadler, F.J.; Piel, C.; Kaminsky, W.; Münstedt, H. Rheological characterization of long-chain branched polyethylenes and comparison with classical analytical methods. Macromol. Symp. 2006, 236, 209–218. [Google Scholar] [CrossRef]
- Stadler, F.J.; Piel, C.; Klimke, K.; Kaschta, J.; Parkinson, M.; Wilhelm, M.; Kaminsky, W.; Münstedt, H. Influence of type and content of various comonomers on long-chain branching of ethene/α-olefin copolymers. Macromolecules 2006, 39, 1474–1482. [Google Scholar] [CrossRef]
- Bersted, B.H. On the effects of very low-levels of long-chain branching on rheological behavior in polyethylene. J. Appl. Polym. Sci. 1985, 30, 3751–3765. [Google Scholar] [CrossRef]
- Stadler, F.J.; Mahmoudi, T. Understanding the effect of short-chain branches by analyzing viscosity functions of linear and short-chain branched polyethylenes. Korea-Aust. Rheol. J. 2012, 23, 185–193. [Google Scholar] [CrossRef]
- Yan, Z.-C.; Stadler, F.J. Classification of thermorheological complexity for linear and branched polyolefins. Rheol. Acta 2018, 57, 377–388. [Google Scholar] [CrossRef]
- Stadler, F.J.; Münstedt, H. Numerical description of shear viscosity functions of long-chain branched metallocene-catalyzed polyethylenes. J. Non-Newton. Fluid Mech. 2008, 151, 129–135. [Google Scholar] [CrossRef]
- Stadler, F.J.; Münstedt, H. Erratum to “Numerical description of shear viscosity functions of long-chain branched metallocene-catalyzed polyethylenes” [J. Non-Newton. Fluid Mech. 151 (2008) 129–135]. J. Non-Newton. Fluid Mech. 2008, 153, 2–3. [Google Scholar] [CrossRef]
- Kronig, R.D.L. On the theory of dispersion of x-rays. J. Opt. Soc. Am. Rev. Sci. Instrum. 1926, 12, 547–557. [Google Scholar] [CrossRef]
- Kramers, H.A. La diffusion de la lumiere par les atomes. Atti Cong. Intern. Fis. 1927, 2, 545–557. [Google Scholar]
- Kazatchkov, I.B.; Bohnet, N.; Goyal, S.K.; Hatzikiriakos, S.G. Influence of molecular structure on the rheological and processing behavior of polyethylene resins. Polym. Eng. Sci. 1999, 39, 804–815. [Google Scholar] [CrossRef]
- Carreau, P.J. Rheological equations from molecular network theories. Trans. Soc. Rheol. 1972, 16, 99–127. [Google Scholar] [CrossRef]
- Yasuda, K.; Armstrong, R.C.; Cohen, R.E. Shear-flow properties of concentrated-solutions of linear and star branched polystyrenes. Rheol. Acta 1981, 20, 163–178. [Google Scholar] [CrossRef]
- Stadler, F.J.; Münstedt, H. Correlations between the shape of viscosity functions and the molecular structure of long-chain branched polyethylenes. Macromol. Mater. Eng. 2009, 294, 25–34. [Google Scholar] [CrossRef]
- Münstedt, H. Dependence of the elongational behavior of polystyrene melts on molecular-weight and molecular-weight distribution. J. Rheol. 1980, 24, 847–867. [Google Scholar] [CrossRef]
- Gabriel, C.; Münstedt, H. Influence of long-chain branches in polyethylenes on linear viscoelastic flow properties in shear. Rheol. Acta 2002, 41, 232–244. [Google Scholar] [CrossRef]
- Roovers, J.; Graessley, W.W. Melt rheology of some model comb polystyrenes. Macromolecules 1981, 14, 766–773. [Google Scholar] [CrossRef]
- Stadler, F.J.; Arikan-Conley, B.A.; Kaschta, J.; Kaminsky, W.; Muünstedt, H. Synthesis and characterization of novel ethylene-graft-ethylene/propylene copolymers. Macromolecules 2011, 44, 5053–5063. [Google Scholar] [CrossRef]
- Auhl, D.; Stadler, F.J.; Münstedt, H. Rheological properties of electron beam-irradiated polypropylenes with different molar masses. Rheol. Acta 2012, 51, 979–989. [Google Scholar] [CrossRef]
- Stadler, F.J.; Mahmoudi, T. Evaluation of relaxation spectra of linear, short, and long-chain branched polyethylenes. Korea-Aust. Rheol. J. 2013, 25, 39–53. [Google Scholar] [CrossRef]
- Trinkle, S.; Friedrich, C. Van gurp-palmen-plot: A way to characterize polydispersity of linear polymers. Rheol. Acta 2001, 40, 322–328. [Google Scholar] [CrossRef]
- Trinkle, S.; Walter, P.; Friedrich, C. Van gurp-palmen plot II-Classification of long chain branched polymers by their topology. Rheol. Acta 2002, 41, 103–113. [Google Scholar] [CrossRef]
- Tobita, H. Dimensions of branched polymers formed in simultaneous long-chain branching and random scission. J. Polym. Sci. Polym. Phys. 2001, 39, 2960–2968. [Google Scholar] [CrossRef]
- Pearson, D.S.; Helfand, E. Viscoelastic properties of star-shaped polymers. Macromolecules 1984, 17, 888–895. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Mw [g/mol] | Mz [g/mol] | Mw/Mn [−] | LCB from SEC-MALLS * |
|---|---|---|---|---|
| HDPE A | 87,000 | 320,000 | 4.2 | medium |
| HDPE B | 105,000 | 1,400,000 | 15.8 | medium-strong |
| HDPE C | 180,000 | 5,400,000 | 14.1 | none |
| HMW-HDPE 1 | 460,000 | 930,000 | 3.6 | weak |
| Weight Content of HMW-HDPE 1 Added | HDPE A | HDPE B | HDPE C |
|---|---|---|---|
| 1% | A1 | B1 | C1 |
| 3% | A3 | B3 | C3 |
| 6% | A6 | B6 | C6 |
| 9% | A9 | B9 | C9 |
| 12% | A12 | B12 | C12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Shekh, M.I.; Cui, S.; Stadler, F.J. Rheological Behavior of Blends of Metallocene Catalyzed Long-Chain Branched Polyethylenes. Part I: Shear Rheological and Thermorheological Behavior. Polymers 2021, 13, 328. https://doi.org/10.3390/polym13030328
Chen C, Shekh MI, Cui S, Stadler FJ. Rheological Behavior of Blends of Metallocene Catalyzed Long-Chain Branched Polyethylenes. Part I: Shear Rheological and Thermorheological Behavior. Polymers. 2021; 13(3):328. https://doi.org/10.3390/polym13030328
Chicago/Turabian StyleChen, Chuangbi, Mehdihasan I. Shekh, Shuming Cui, and Florian J. Stadler. 2021. "Rheological Behavior of Blends of Metallocene Catalyzed Long-Chain Branched Polyethylenes. Part I: Shear Rheological and Thermorheological Behavior" Polymers 13, no. 3: 328. https://doi.org/10.3390/polym13030328
APA StyleChen, C., Shekh, M. I., Cui, S., & Stadler, F. J. (2021). Rheological Behavior of Blends of Metallocene Catalyzed Long-Chain Branched Polyethylenes. Part I: Shear Rheological and Thermorheological Behavior. Polymers, 13(3), 328. https://doi.org/10.3390/polym13030328