
Tandem Synthesis of Ultra-High Molecular Weight Drag Reducing Poly-α-Olefins for Low-Temperature Pipeline Transportation

 ,
, 
 and
and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Solvents and Reagents
2.2. Analysis
2.3. Oligomerization Experiments
2.4. Synthesis of New Internal Donor, 3,3-Bis(methoxymethyl)heptane (BMMH)
2.4.1. 2-Butyl-2-ethylpropane-1,3-diol
2.4.2. 2-Ethyl-2-(methoxymethyl)hexan-1-ol
2.4.3. 3,3-Bis(methoxymethyl)heptane (BMMH)
2.5. Synthesis of Titanium–Magnesium Catalyst
2.6. Polymerization Experiments
2.6.1. Preparation of Model Mixtures of Linear α-Olefins
2.6.2. Low-Temperature Polymerization
2.7. Drag Reducing Experiments
2.7.1. Sample Preparation
2.7.2. Capillary Turbulent Rheometry
3. Results and Discussion
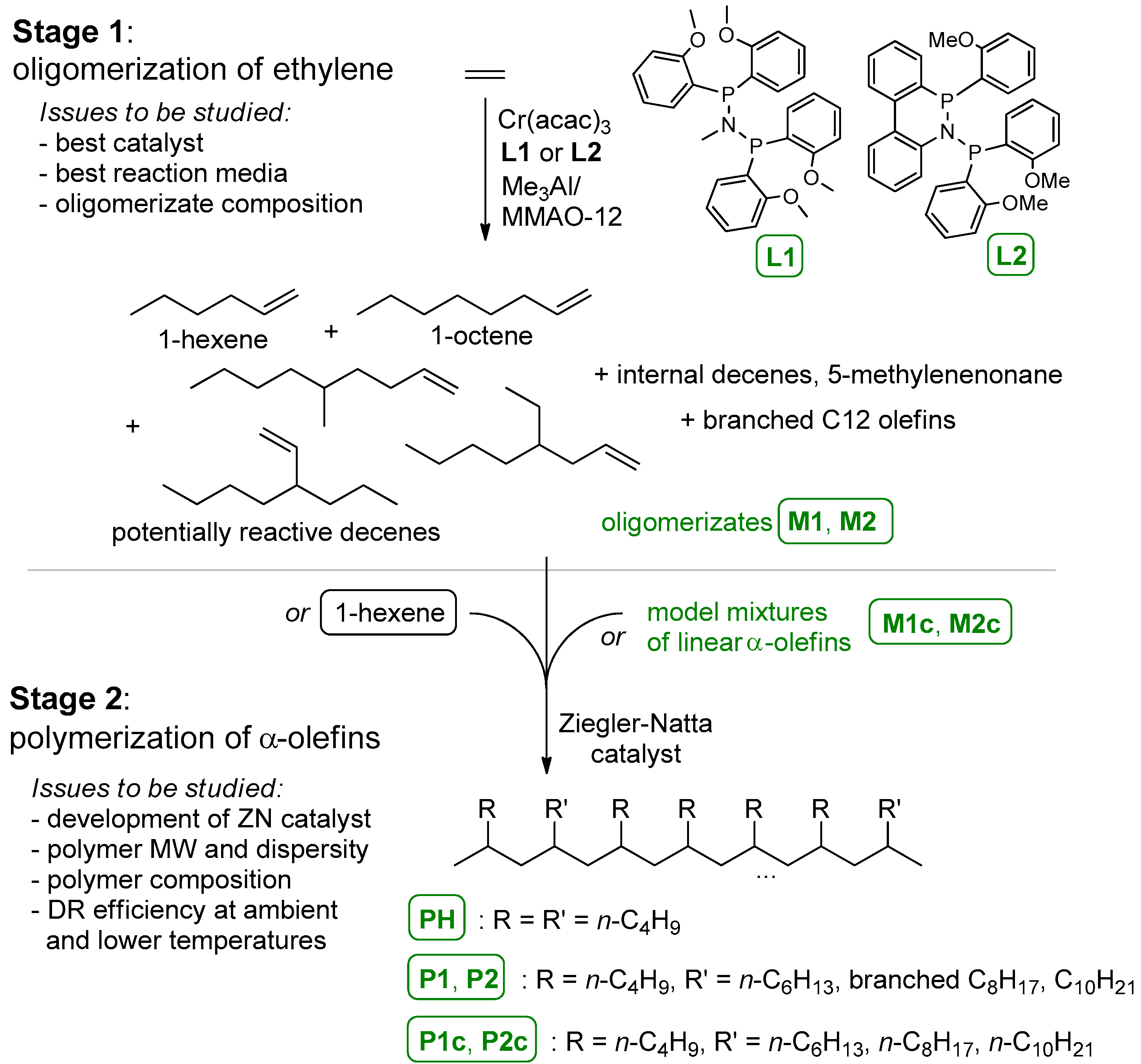
3.1. The Main Idea of the Two-Stage Synthesis of Polyolefin DRAs
3.2. Oligomerization of Ethylene
3.2.1. The Use of α-Olefins as a Reaction Media
3.2.2. Composition and Molecular Structure of the Oligomerization Products
3.3. Optimization and Preparation of Titanium–Magnesium ZN Catalyst (TMC)
3.3.1. Synthesis of Internal Donor BMMH
3.3.2. Synthesis of TMC
3.4. Polymerization and Polymer Analysis
Polymerization Experiments
3.5. Drag Reducing Efficiency
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brostow, W. Drag reduction in flow: Review of applications, mechanism and prediction. J. Ind. Eng. Chem. 2008, 14, 409–416. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Zakin, J.L.; Shi, H. Review on drag reduction and its heat transfer by additives. Adv. Mechan. Eng. 2011, 3, 478749. [Google Scholar] [CrossRef]
- Cuenca, F.G.; Marín, M.G.; Díaz, M.B.F. Energy-Savings Modeling of Oil Pipelines That Use Drag-Reducing Additives. Energy Fuels 2008, 22, 3293–3298. [Google Scholar] [CrossRef]
- Toms, B.A. Some observations on the flow of linear polymer solutions through straight tubes at large Reynolds numbers. In Proceedings of the First International Congress on Rheology, Scheveningen, The Netherlands, 21–24 September 1948; Burgers, J.M., Ed.; North Holland Publishing Co.: Amsterdam, The Netherlands, 1949. Part 2. pp. 135–142. [Google Scholar]
- Smith, R.E.; Tiederman, W.G. The mechanism of polymer thread drag reduction. Rheol. Acta 1991, 30, 103–113. [Google Scholar] [CrossRef]
- Bewersdorff, H.W.; Gyr, A.; Hoyer, K.; Tsinober, A. An investigation of possible mechanisms of heterogeneous drag reduction in pipe and channel flows. Rheol. Acta 1993, 32, 140–149. [Google Scholar] [CrossRef]
- Brostow, W.; Majumdar, S.; Singh, R.P. Drag reduction and solvation in polymer solutions. Macromol. Rapid Commun. 1999, 20, 144–147. [Google Scholar] [CrossRef]
- Holtmyer, M.D.; Chatterji, J. Study of oil soluble polymers as drag reducers. Polym. Eng. Sci. 1980, 20, 473–477. [Google Scholar] [CrossRef]
- Hong, C.H.; Jang, C.H.; Choi, H.J. Turbulent Drag Reduction with Polymers in Rotating Disk Flow. Polymers 2015, 7, 1279–1298. [Google Scholar] [CrossRef]
- Abubakar, A.; Al-Wahaibi, T.; Al-Wahaibi, Y.; Al-Hashmi, A.R.; Al-Ajmi, A. Roles of drag reducing polymers in single- and multi-phase flows. Chem. Eng. Res. Des. 2014, 92, 2153–2181. [Google Scholar] [CrossRef]
- Wang, Y. Reynolds Stress Model for Viscoelastic Drag-Reducing Flow Induced by Polymer Solution. Polymers 2019, 11, 1659. [Google Scholar] [CrossRef]
- Choi, H.J.; Jhon, M.S. Polymer-Induced Turbulent Drag Reduction. Ind. Eng. Chem. Res. 1996, 35, 2993–2998. [Google Scholar] [CrossRef]
- Ivchenko, P.V.; Nifant’ev, I.E.; Tavtorkin, A.V. Polyolefin Drag Reducing Agents (Review). Pet. Chem. 2016, 56, 775–787. [Google Scholar] [CrossRef]
- Nesyn, G.V.; Sunagatullin, R.Z.; Shibaev, V.P.; Malkin, A.Y. Drag reduction in transportation of hydrocarbon liquids: From fundamentals to engineering applications. J. Petrol. Sci. Eng. 2018, 161, 715–725. [Google Scholar] [CrossRef]
- White, C.M.; Mungal, M.G. Mechanics and prediction of turbulent drag reduction with polymer additives. Ann. Rev. Fluid Mech. 2008, 40, 235–256. [Google Scholar] [CrossRef]
- Wang, S.-N.; Graham, M.D.; Hahn, F.J.; Xi, L. Time-series and extended Karhunen–Loéve analysis of turbulent drag reduction in polymer solutions. AIChE J. 2014, 60, 1460–1475. [Google Scholar] [CrossRef]
- Jin, S.; Collins, L.R. Dynamics of dissolved polymer chains in isotropic turbulence. New J. Phys. 2007, 9, 360. [Google Scholar] [CrossRef]
- Mortazavi, S.M.M. Correlation of polymerization conditions with drag reduction efficiency of poly(1-hexene) in oil pipelines. Iran. Polym. J. 2016, 25, 731–737. [Google Scholar] [CrossRef]
- Echevskaya, L.; Matsko, M.; Nikolaeva, M.; Sergeev, S.; Zakharov, V. Kinetic features of hexene-1 polymerization over supported titanium–magnesium catalyst. Macromol. React. Eng. 2014, 8, 666–672. [Google Scholar] [CrossRef]
- Harjuhahto, H.; Virtanen, E.; Karbasi, A.K.; Rockas, L.; Liaho, T.; Karhu, E.; Eklund, M. Process for Preparing and Method of Using A Drag Reducing Agent. Patent WO0234802, 2 May 2002. [Google Scholar]
- Tavtorkin, A.N.; Gavrilenko, I.F.; Kostitsyna, N.N.; Korchagina, S.A.; Chinova, M.S. Comparison of the Turbulent Drag Reduction Effectiveness of Polymers from Higher Olefin Monomers (Hexene, Octene, Decene, Dodecene) in the Production of Hydrodynamic Drag Reducing Agents for Transportation of Hydrocarbon Liquids. Russ. J. Appl. Chem. 2020, 93, 788–793. [Google Scholar] [CrossRef]
- Mortazavi, S.M.M.; Ahmadjo, S.; Omidvar, M.; Zamani, M.R.; Fallahnezhad, R. Investigation into the effect of branch length of polyolefin and its statistical distribution on the flow improving performance. J. Polym. Res. 2021, 28, 24. [Google Scholar] [CrossRef]
- Valiev, M.I.; Hasbiullin, I.I.; Kazakov, V.V. Specifics of using drag reducing additives based on polyalphaolefins at various oil temperatures. Oil Oil Prod. Pipeline Transp. Sci. Technol. 2016, 6, 32–37. [Google Scholar]
- Kommareddi, N.S.; Ramsay, G.G.; Motler, J.F. Method of Preparing a Polymer Under Predetermined Temperature Conditions, and Apparatus Therefor. Patent US7015290, 26 August 2004. [Google Scholar]
- Balashov, A.V.; Rusinov, P.G.; Nifantiev, I.E. Drag-Reducing Agent and Method for Producing Same. Patent WO2016204654, 22 December 2016. [Google Scholar]
- Atiqullah, M.; Al-Sarkhi, A.S.; Al-Thenayan, F.M.; Al-Malki, A.R.; Xu, W.; Hossaen, A. Catalyst Composition and A Process for Making Ultra High Molecular Weight Poly (Alpha-Olefin) Drag Reducing Agents. Patent US9969826, 15 May 2018. [Google Scholar]
- Do, L.H.; Labinger, J.A.; Bercaw, J.E. Mechanistic Studies of Ethylene and α-Olefin Co-Oligomerization Catalyzed by Chromium−PNP Complexes. Organometallics 2012, 31, 5143–5149. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gong, M.; Liu, Z.; Li, Y.; Ma, Y.; Sun, Q.; Zhang, J.; Liu, B. Selective Co-Oligomerization of Ethylene and 1-Hexene by Chromium-PNP Catalysts: A DFT Study. Organometallics 2016, 35, 972–981. [Google Scholar] [CrossRef]
- Wass, D.F. Olefin Trimerization Using A Catalyst Comprising A Source of Chromium, Molybdenium or Tungsten and A Ligand Containing at Least One Phosphorus, Arsenic or Antimony Atom Bound to At Least One Hetero(Hydrocarbyl) Group. Patent US7141633, 28 November 2006. [Google Scholar]
- Carter, A.; Cohen, S.A.; Cooley, N.A.; Murphy, A.; Scutt, J.; Wass, D.F. High activity ethylene trimerisation catalysts based on diphosphine ligands. Chem. Commun. 2002, 858–859. [Google Scholar] [CrossRef] [PubMed]
- Blann, K.; Bollmann, A.; Dixon, J.; Navaling, A.; Morgan, D.; Maumela, H.; Killian, E.; Hess, F.M.; Otto, S.; Pepler, L.; et al. Tetramerization of Olefins. Patent WO2004056479, 8 July 2004. [Google Scholar]
- Agapie, T. Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Coord. Chem. Rev. 2011, 255, 861–880. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Ivchenko, P.V.; Vinogradov, A.A.; Vinogradov, A.A.; Bagrov, V.V. The Method of Oligomerization of Ethylene in Organic Solvent Media in the Presence of Chromium Catalyst and Organoaluminium Activator. Patent RU2717241, 19 March 2020. [Google Scholar]
- Sydora, O.L. Selective Ethylene Oligomerization. Organometallics 2019, 38, 997–1010. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Vinogradov, A.A.; Vinogradov, A.A.; Roznyatovsky, V.A.; Grishin, Y.K.; Ivanyuk, A.V.; Sedov, I.V.; Churakov, A.V.; Ivchenko, P.V. 5,6-Dihydrodibenzo [c,e] [1,2] azaphosphinine-based PNP ligands, Cr coordination and Cr(III) precatalysts for ethylene oligomerization. Organometallics 2018, 37, 2660–2664. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Korchagina, S.A.; Chinova, M.S.; Vinogradov, A.A.; Vinogradov, A.A.; Roznyatovsky, V.A.; Khaidapova, D.D.; Ivchenko, P.V. The synthesis of ultra-high molecular weight poly(1-hexene)s by low-temperature Ziegler-Natta precipitation polymerization in fluorous reaction media. Polymer 2018, 139, 98–106. [Google Scholar] [CrossRef]
- McGuinness, D.S.; Wasserscheid, P.; Keim, W.; Hu, C.; Englert, U.; Dixon, J.T.; Grove, C. Novel Cr-PNP complexes as catalysts for the trimerisation of ethylene. Chem. Commun. 2003, 334–335. [Google Scholar] [CrossRef]
- Wass, D.F. Chromium-catalysed ethene trimerisation and tetramerization—Breaking the rules in olefin oligomerisation. Dalton Trans. 2007, 816–819. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Zhang, X.; Wu, W.; Zhang, G.; Xu, S.; Shi, M. Switchable ethylene tri-/tetramerization with high activity: Subtle effect presented by backbone-substituent of carbon-bridged diphosphine ligands. ACS Catal. 2013, 3, 2311–2317. [Google Scholar] [CrossRef]
- Kuhlmann, S.; Blann, K.; Bollmann, A.; Dixon, J.T.; Killian, E.; Maumela, M.C.; Maumela, H.; Morgan, D.H.; Prétorius, M.; Taccardi, N.; et al. N-substituted diphosphinoamines: Toward rational ligand design for the efficient tetramerization of ethylene. J. Catal. 2007, 245, 279–284. [Google Scholar] [CrossRef]
- van Leeuwen, P.W.N.M.; Clément, N.D.; Tschan, M.J.-L. New processes for the selective production of 1-octene. Coord. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- De Wet-Roos, D.; Du Toit, A.; Joubert, D.J. Homogeneous tandem catalysis of the bis-(diphenylphosphino)-amine/chromium tetramerization catalyst with metallocene catalysts. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6847–6856. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Luo, M.; Chen, H.; Ning, Y. Preparation of ethylene/1-octene copolymers from ethylene stock with tandem catalytic system. J. Appl. Polym. Sci. 2008, 107, 3071–3075. [Google Scholar] [CrossRef]
- van Rensburg, W.J.; van den Berg, J.-A.; Steynberg, P.J. Role of MAO in Chromium-Catalyzed Ethylene Tri- and Tetramerization: A DFT Study. Organometallics 2007, 26, 1000–1013. [Google Scholar] [CrossRef]
- Agapie, T.; Labinger, J.A.; Bercaw, J.E. Mechanistic Studies of Olefin and Alkyne Trimerization with Chromium Catalysts: Deuterium Labeling and Studies of Regiochemistry Using a Model Chromacyclopentane Complex. J. Am. Chem. Soc. 2007, 129, 14281–14295. [Google Scholar] [CrossRef] [PubMed]
- Peitz, S.; Aluri, B.R.; Peulecke, N.; Müller, B.H.; Wöhl, A.; Müller, W.; Al-Hazmi, M.H.; Mosa, F.M.; Rosenthal, U. An Alternative Mechanistic Concept for Homogeneous Selective Ethylene Oligomerization of Chromium-Based Catalysts: Binuclear Metallacycles as a Reason for 1-Octene Selectivity? Chem. Eur. J. 2010, 16, 7670–7676. [Google Scholar] [CrossRef]
- Do, L.H.; Labinger, J.A.; Bercaw, J.E. Spectral studies of a Cr(PNP)-MAO system for selective ethylene trimerization catalysis: Searching for the active species. ACS Catal. 2013, 3, 2582–2585. [Google Scholar] [CrossRef]
- Britovsek, G.J.P.; McGuinness, D.S.; Wierenga, T.S.; Young, C.T. Single- and Double-Coordination Mechanism in Ethylene Tri- and Tetramerization with Cr/PNP Catalysts. ACS Catal. 2015, 5, 4152–4166. [Google Scholar] [CrossRef]
- Dixon, J.T.; Green, M.J.; Hess, F.M.; Morgan, D.H. Advances in selective ethylene trimerization—A critical overview. J. Organomet. Chem. 2004, 689, 3641–3668. [Google Scholar] [CrossRef]
- McGuinness, D.S. Olefin Oligomerization via Metallacycles: Dimerization, Trimerization, Tetramerization, and Beyond. Chem. Rev. 2011, 111, 2321–2341. [Google Scholar] [CrossRef]
- Alferov, K.A.; Belov, G.P.; Meng, Y. Chromium catalysts for selective ethylene oligomerization to 1-hexene and 1-octene: Recent results. Appl. Catal. A Gen. 2017, 542, 71–124. [Google Scholar] [CrossRef]
- Terano, M.; Soga, H.; Kimura, K. Catalyst for Polymerization of Olefins. Patent US4829037, 9 May 1989. [Google Scholar]
- Zhang, R.; Tan, Z.; Zhou, Q.; Xu, X.; Yin, S.; Li, F.; Song, W.; Yu, J. Catalyst Component for Olefin Polymerization, Preparation Method Thereof, and Catalyst Comprising Same. Patent CN107344976, 14 November 2017. [Google Scholar]
- Morini, G.; Albizzati, E.; Balbontin, G.; Mingozzi, I.; Sacchi, M.C.; Forlini, F.; Tritto, I. Microstructure Distribution of Polypropylenes Obtained in the Presence of Traditional Phthalate/Silane and Novel Diether Donors: A Tool for Understanding the Role of Electron Donors in MgCl2-Supported Ziegler-Natta Catalysts. Macromolecules 1996, 29, 5770–5776. [Google Scholar] [CrossRef]
- Denkwitz, Y.; Schuster, O.; Winter, A. High Performance Ziegler-Natta Catalyst Systems, Process for Producing Such Supported Catalysts and Use Thereof. Patent US9481741, 1 November 2016. [Google Scholar]
- Agnes, G.; Borsotti, G. Diethers Usable in the Preparation of Ziegler-Natta Catalysts and Their Preparation. Patent EP0361493, 4 April 1990. [Google Scholar]
- Borsotti, J. Process for the Preparation of Diethers. Patent EP0487035, 27 May 1992. [Google Scholar]
- Pereira, A.S.; Soares, E.S. Polymer degradation of dilute solutions in turbulent drag reducing flows in a cylindrical double gap rheometer device. J. Nonnewton. Fluid Mech. 2012, 179–180, 9–22. [Google Scholar] [CrossRef]
- Eskin, D. Applicability of a Taylor–Couette device to characterization of turbulent drag reduction in a pipeline. Chem. Eng. Sci. 2014, 116, 275–283. [Google Scholar] [CrossRef]
- Guersoni, V.C.B.; Bannwart, A.C.; Destefani, T.; Sabadini, E. Comparative Study of Drag Reducers for Light Hydrocarbon Flow. Pet. Sci. Technol. 2015, 33, 943–951. [Google Scholar] [CrossRef]
- Kim, C.A.; Lee, K.; Choi, H.J.; Kim, C.B.; Kim, K.Y.; Jhon, M.S. Universal Characteristics of Drag Reducing Polyisobutylene in Kerosene. J. Macromol. Sci. A Pure Appl. Chem. 1997, 34, 705–711. [Google Scholar] [CrossRef]
- Kouser, T.; Xiong, Y.; Yang, D. Contribution of Superhydrophobic Surfaces and Polymer Additives to Drag Reduction. ChemBioEng Rev. 2021, 8, 337–356. [Google Scholar] [CrossRef]
- Horn, A.F.; Merrill, E.W. Midpoint scission of macromolecules in dilute solution in turbulent flow. Nature 1984, 312, 140–141. [Google Scholar] [CrossRef]
- Zhang, X.; Duan, X.; Muzychka, Y. Degradation of flow drag reduction with polymer additives—A new molecular view. J. Mol. Liq. 2019, 292, 111360. [Google Scholar] [CrossRef]
- Soares, E.J. Review of mechanical degradation and de-aggregation of drag reducing polymers in turbulent flows. J. Non-Newton. Fluid Mech. 2020, 276, 104225. [Google Scholar] [CrossRef]
- Moussa, T.; Tiu, C. Factors affecting polymer degradation in turbulent pipe flow. Chem. Eng. Sci. 1994, 49, 1681–1692. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of C Atoms | Structure | Name | wt.% of the Components | ||||
|---|---|---|---|---|---|---|---|
| H | M1 | M1c | M2 | M2c | |||
| 6 |  | 1-hexene | 97.9 | 74.4 | 74.4 | 47.2 | 47.2 |
| 7 |  | toluene | 2.1 | 2.1 | 2.1 | 2.1 | 2.1 |
| 8 |  | 1-octene | – | 10.4 | 10.4 | 25.1 | 25.1 |
| 10 |  | 4-vinyloctane | – | 2.9 | – | 5.1 | – |
| 10 |  | 4-ethyl-1-octene | – | 2.1 | – | 3.9 | – |
| 10 |  | 5-methyl-1-nonene | – | 4.5 | – | 9.6 | – |
| 10 |  | 5-methylenenonane | – | 1.6 | – | 1.9 | – |
| 10 |  | 1-decene | – | 0.2 | 11.6 | 0.2 | 21.0 |
| 10 |  | 5-decene | – | 0.1 | – | 0.1 | – |
| 10 |  | 4-decene | – | 0.2 | – | 0.2 | – |
| 12 | – | C12 alkenes 1 | – | 1.5 | 1.5 2 | 4.6 | 4.6 2 |
| Polymer | Yield, % | Mp, ×106 Da 1 | Mn, ×106 Da 1 | ÐM1 | Comonomer Molar Ratio 2 | ||
|---|---|---|---|---|---|---|---|
| C6 | C8 | C10 | |||||
| PH | 97 | 4.21 | 1.94 | 2.31 | 100/100 | – | – |
| P1 | 91 | 4.74 | 3.23 | 1.61 | 85/84.6 | 8/8.7 | 3/7.8 |
| P1c | 95 | 4.32 | 2.00 | 2.31 | 83/83.4 | 8/8.9 | 6/7.8 |
| P2 | 89 | 4.92 | 2.42 | 1.55 | 61/61.2 | 20/24.4 | 9/14.5 |
| P2c | 97 | 5.45 | 1.87 | 2.91 | 60/60.0 | 24/23.9 | 16/16.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nifant’ev, I.E.; Tavtorkin, A.N.; Vinogradov, A.A.; Korchagina, S.A.; Chinova, M.S.; Borisov, R.S.; Artem’ev, G.A.; Ivchenko, P.V. Tandem Synthesis of Ultra-High Molecular Weight Drag Reducing Poly-α-Olefins for Low-Temperature Pipeline Transportation. Polymers 2021, 13, 3930. https://doi.org/10.3390/polym13223930
Nifant’ev IE, Tavtorkin AN, Vinogradov AA, Korchagina SA, Chinova MS, Borisov RS, Artem’ev GA, Ivchenko PV. Tandem Synthesis of Ultra-High Molecular Weight Drag Reducing Poly-α-Olefins for Low-Temperature Pipeline Transportation. Polymers. 2021; 13(22):3930. https://doi.org/10.3390/polym13223930
Chicago/Turabian StyleNifant’ev, Ilya E., Alexander N. Tavtorkin, Alexey A. Vinogradov, Sofia A. Korchagina, Maria S. Chinova, Roman S. Borisov, Grigory A. Artem’ev, and Pavel V. Ivchenko. 2021. "Tandem Synthesis of Ultra-High Molecular Weight Drag Reducing Poly-α-Olefins for Low-Temperature Pipeline Transportation" Polymers 13, no. 22: 3930. https://doi.org/10.3390/polym13223930
APA StyleNifant’ev, I. E., Tavtorkin, A. N., Vinogradov, A. A., Korchagina, S. A., Chinova, M. S., Borisov, R. S., Artem’ev, G. A., & Ivchenko, P. V. (2021). Tandem Synthesis of Ultra-High Molecular Weight Drag Reducing Poly-α-Olefins for Low-Temperature Pipeline Transportation. Polymers, 13(22), 3930. https://doi.org/10.3390/polym13223930