
A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex


Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Synthesis
2.2. Cyclic Voltammetry and EQCM Studies
2.3. In Situ Conductance Measurements
2.4. Scanning Electron Microscopy Measurements
2.5. UV-vis-NIR Spectroscopy and In Situ Spectroelectrochemical Studies
3. Results
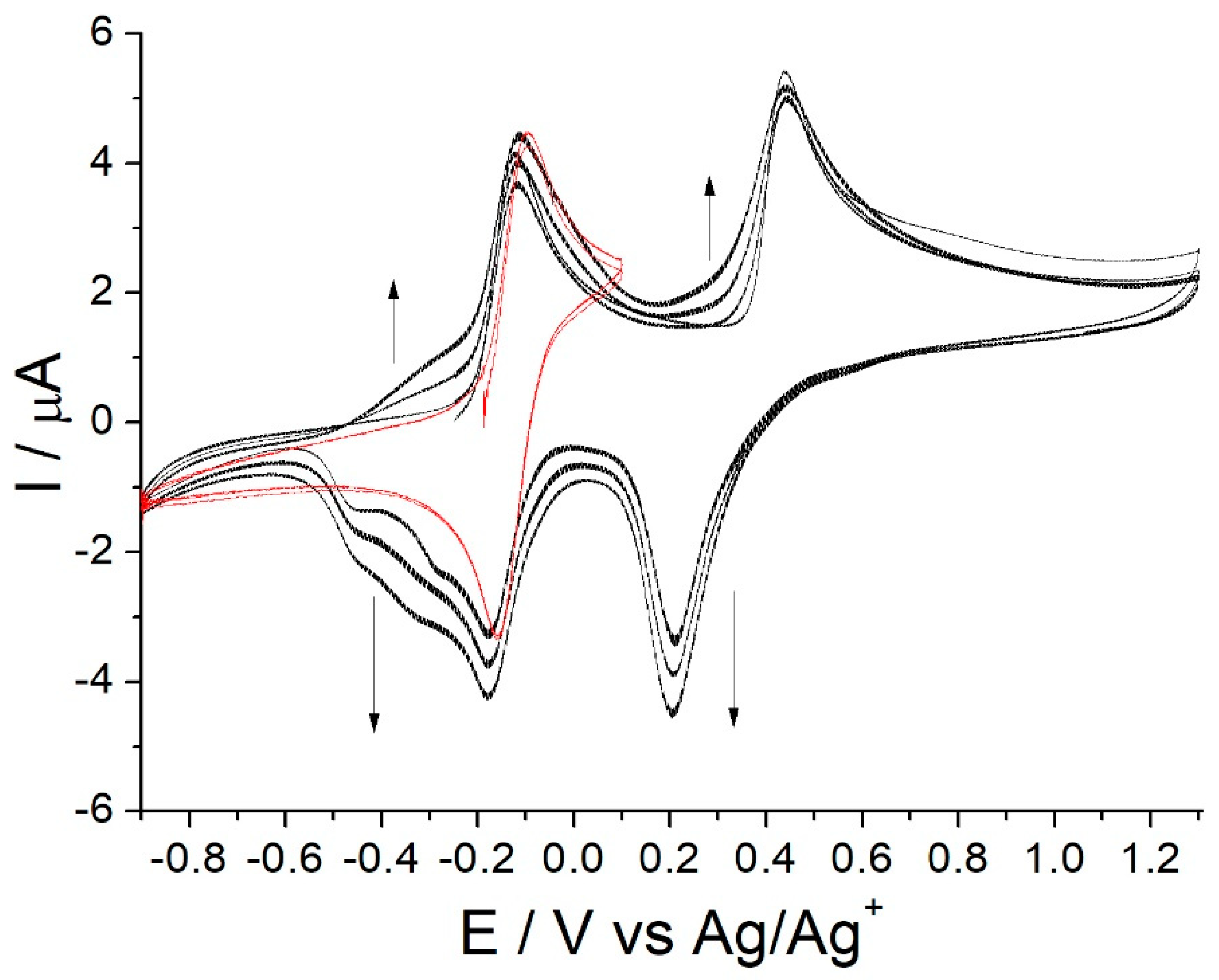
3.1. Oxidative Electrochemistry of [Co(Amben)]
3.2. EQCM Study of the Electro-Oxidative Polymerization of [Co(Amben)]
3.3. CV and EQCM Studies of Thin Poly-[Co(Amben)] Films
3.4. In Situ UV-vis-NIR Spectroelectrochemical Study of Poly-[Co(Amben)] Oxidation
3.5. In Situ Conductance and SEM Studies of Thick Poly-[Co(Amben)] Films
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heinze, J.; Frontana-Uribe, B.A.; Ludwigs, S. Electrochemistry of Conducting Polymers—Persistent Models and New Concepts. Chem. Rev. 2010, 110, 4724–4771. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Recent advances in the functional applications of conducting metallopolymers. Coord. Chem. Rev. 2018, 377, 237–258. [Google Scholar] [CrossRef]
- Clarke, R.M.; Herasymchuk, K.; Storr, T. Electronic structure elucidation in oxidized metal–salen complexes. Coord. Chem. Rev. 2017, 352, 67–82. [Google Scholar] [CrossRef]
- Chepurnaya, I.A.; Karushev, M.P.; Alekseeva, E.V.; Lukyanov, D.A.; Levin, O.V. Redox-conducting polymers based on metal-salen complexes for energy storage applications. Pure Appl. Chem. 2020, 92, 1239–1258. [Google Scholar] [CrossRef]
- Nunes, M.; Araújo, M.; Fonseca, J.; Moura, C.; Hillman, R.; Freire, C. High-Performance Electrochromic Devices Based on Poly[Ni(salen)]-Type Polymer Films. ACS Appl. Mater. Interfaces 2016, 8, 14231–14243. [Google Scholar] [CrossRef] [PubMed]
- Dahm, C.E.; Peters, D.G. Catalytic reduction of Iodoethane and 2-Iodopropane at Carbon Electrodes Coated with Anodically Polymerized Films of Nickel(II) Salen. Anal. Chem. 1994, 66, 3117–3123. [Google Scholar] [CrossRef]
- Konev, A.S.; Kayumov, M.Y.; Karushev, M.P.; Novoselova, Y.V.; Lukyanov, D.A.; Alekseeva, E.V.; Levin, O.V. Polymeric Metal Salen-Type Complexes as Catalysts for Photoelectrocatalytic Hydrogen Peroxide Production. ChemElectroChem 2018, 5, 3138–3142. [Google Scholar] [CrossRef]
- Novozhilova, M.; Anischenko, D.; Chepurnaya, I.; Dmitrieva, E.; Malev, V.; Timonov, A.; Karushev, M. Metal-centered redox activity in a polymeric Cobalt(II) complex of a sterically hindered salen type ligand. Electrochim. Acta 2020, 353, 136496. [Google Scholar] [CrossRef]
- Novozhilova, M.V.; Smirnova, E.A.; Polozhentseva, J.A.; Danilova, J.A.; Chepurnaya, I.A.; Karushev, M.P.; Malev, V.V.; Timonov, A.M. Multielectron redox processes in polymeric cobalt complexes with N2O2 Schiff base ligands. Electrochim. Acta 2018, 282, 105–115. [Google Scholar] [CrossRef]
- Kingsborough, R.P.; Swager, T.M. Electroactivity Enhancement by Redox Matching in Cobalt Salen-Based Conducting Polymers. Adv. Mater. 1998, 10, 1100–1104. [Google Scholar] [CrossRef]
- Kingsborough, R.P.; Swager, T.M. Electrocatalytic Conducting Polymers: Oxygen Reduction by a Polythiophene−Cobalt Salen Hybrid. Chem. Mater. 2000, 12, 872–874. [Google Scholar] [CrossRef]
- Dmitrieva, E.; Rosenkranz, M.; Danilova, J.S.; Smirnova, E.A.; Karushev, M.P.; Chepurnaya, I.A.; Timonov, A.M. Radical formation in polymeric nickel complexes with N2O2 Schiff base ligands: An in situ ESR and UV–vis–NIR spectroelectrochemical study. Electrochim. Acta 2018, 283, 1742–1752. [Google Scholar] [CrossRef]
- Łępicka, K.; Pieta, P.; Shkurenko, A.; Borowicz, P.; Majewska, M.; Rosenkranz, M.; Avdoshenko, S.; Popov, A.A.; Kutner, W. Spectroelectrochemical Approaches to Mechanistic Aspects of Charge Transport in meso-Nickel(II) Schiff Base Electrochromic Polymer. J. Phys. Chem. C 2017, 121, 16710–16720. [Google Scholar] [CrossRef]
- Martins, M.; Boas, M.V.; de Castro, B.; Hillman, A.R.; Freire, C. Spectroelectrochemical characterisation of copper salen-based polymer-modified electrodes. Electrochimica Acta 2005, 51, 304–314. [Google Scholar] [CrossRef]
- Polozhentseva, Y.A.; Novozhilova, M.V.; Bykov, V.A.; Karushev, M.P. Modification of Porous Carbon Material with Polymeric Cobalt Complex with a Schiff Base of Salen-Type for Electrodes of Electrochemical Supercapacitors. Tech. Phys. Lett. 2020, 46, 913–915. [Google Scholar] [CrossRef]
- Shioya, T.; Swager, T.M. A reversible resistivity-based nitric oxide sensorThis work was made possible by a Postdoctoral Fellowship from the Japan Society for the Promotion of Science to T. Shioya and the generous financial support of the Office of Naval Research. Chem. Commun. 2002, 1364–1365. [Google Scholar] [CrossRef]
- Swager, T.M. 50th Anniversary Perspective: Conducting/Semiconducting Conjugated Polymers. A Personal Perspective on the Past and the Future. Macromolecules 2017, 50, 4867–4886. [Google Scholar] [CrossRef]
- Holliday, B.J.; Swager, T.M. Conducting metallopolymers: The roles of molecular architecture and redox matching. Chem. Commun. 2005, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Understanding the Effect of Metal Centers on Charge Transport and Delocalization in Conducting Metallopolymers. Macromolecules 2017, 50, 872–883. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Effect of conjugation length and metal-backbone interactions on charge transport properties of conducting metallopolymers. Polym. Chem. 2017, 8, 4359–4367. [Google Scholar] [CrossRef]
- Green, M.; Tasker, P.A. NN′-ethylenebis-(o-aminobenzylideneiminato)cobalt(II) and its derivatives. Part I. Physical properties and shape. J. Chem. Soc. A 1970, 3105–3108. [Google Scholar] [CrossRef]
- Higson, B.; McKenzie, E. The redox properties of some planar Schiff compounds of cobalt, nickel and copper. Inorg. Nucl. Chem. Lett. 1970, 6, 209–213. [Google Scholar] [CrossRef]
- Higson, B.M.; McKenzie, E.D. The structure, redox properties, and reactions of some planar [MIIN4] chelate compounds of cobalt, nickel, and copper, and their oxidised products, including paramagnetic cobalt(III) species. J. Chem. Soc. Dalton Trans. 1972, 269–280. [Google Scholar] [CrossRef]
- Karlsson, R.; Engelhardt, L.M.; Green, M. Crystal structure of [NN′-ethylenebis-(2-amino-5-chlorobenzylideneiminato)]cobalt(II). J. Chem. Soc. Dalton Trans. 1972, 2463–2465. [Google Scholar] [CrossRef]
- Kochem, A.; Gellon, G.; Jarjayes, O.; Philouze, C.; Leconte, N.; Van Gastel, M.; Bill, E.; Thomas, F. A singlet ground state for a cobalt(ii)–anilinosalen radical complex. Chem. Commun. 2014, 50, 4924–4926. [Google Scholar] [CrossRef] [PubMed]
- Kochem, A.; Thomas, F.; Jarjayes, O.; Gellon, G.; Philouze, C.; Weyhermüller, T.; Neese, F.; Van Gastel, M. Structural and Spectroscopic Investigation of an Anilinosalen Cobalt Complex with Relevance to Hydrogen Production. Inorg. Chem. 2013, 52, 14428–14438. [Google Scholar] [CrossRef] [PubMed]
- Shagisultanova, G.A.; Ardasheva, L.P. Electrochemical Polymerization of Ni(II) and Pd(II) Complexes with 1,2-Bis(o-aminobenzylidene)ethylenediamine. Russ. J. Coord. Chem. 2004, 30, 94–99. [Google Scholar] [CrossRef]
- Karushev, M.; Smirnova, E.; Chepurnaya, I. Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States. Molecules 2021, 26, 2646. [Google Scholar] [CrossRef]
- Inzelt, G. Methods of Investigation. Underpotential Depos. 2012, 83–147. [Google Scholar] [CrossRef]
- Casado, N.; Mecerreyes, D. Chapter 1. Introduction to Redox Polymers: Classification, Characterization Methods and Main Applications. Polymer Chem. Series 2020, 1–26. [Google Scholar] [CrossRef]
- Salinas, G.; Frontana-Uribe, B.A. Analysis of Conjugated Polymers Conductivity by in situ Electrochemical-Conductance Method. ChemElectroChem 2019, 6, 4105–4117. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung Dünner Schichten und zur Mikrowägung. Eur. Phys. J. A 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Zotti, G.; Zecchin, S.; Schiavon, G.; Groenendaal, L. “Bert” Conductive and Magnetic Properties of 3,4-Dimethoxy- and 3,4-Ethylenedioxy-Capped Polypyrrole and Polythiophene. Chem. Mater. 2000, 12, 2996–3005. [Google Scholar] [CrossRef]
- Yurchenko, O.; Heinze, J.; Ludwigs, S. Electrochemically Induced Formation of Independent Conductivity Regimes in Polymeric Tetraphenylbenzidine Systems. ChemPhysChem 2010, 11, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Paynter, R.; Dao, L.H. Synthesis and characterization of poly(diarylamines): A new class of electrochromic conducting polymers. Macromolecules 1990, 23, 3598–3605. [Google Scholar] [CrossRef]
- Malacrida, C.; Lu, Y.; Dirnberger, K.; Gámez-Valenzuela, S.; Delgado, M.C.R.; Ludwigs, S. Towards highly conducting bicarbazole redox polymer films with plateau-like conductivities. J. Mater. Chem. C 2020, 8, 15393–15405. [Google Scholar] [CrossRef]
- Kochem, A.; Kanso, H.; Baptiste, B.; Arora, H.; Philouze, C.; Jarjayes, O.; Vezin, H.; Luneau, D.; Orio, M.; Thomas, F. Ligand Contributions to the Electronic Structures of the Oxidized Cobalt(II) salen Complexes. Inorg. Chem. 2012, 51, 10557–10571. [Google Scholar] [CrossRef] [PubMed]
- Otteny, F.; Perner, V.; Wassy, D.; Kolek, M.; Bieker, P.; Winter, M.; Esser, B. Poly(vinylphenoxazine) as Fast-Charging Cathode Material for Organic Batteries. ACS Sustain. Chem. Eng. 2019, 8, 238–247. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Range | Charge Carrier Molar Mass, g mol−1 |
|---|---|
| −0.5 to −0.15 V | 25 |
| −0.15 to +0.2 V | 94 |
| +0.35 to +0.95 V | 185 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karushev, M. A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex. Polymers 2021, 13, 1667. https://doi.org/10.3390/polym13101667
Karushev M. A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex. Polymers. 2021; 13(10):1667. https://doi.org/10.3390/polym13101667
Chicago/Turabian StyleKarushev, Mikhail. 2021. "A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex" Polymers 13, no. 10: 1667. https://doi.org/10.3390/polym13101667
APA StyleKarushev, M. (2021). A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex. Polymers, 13(10), 1667. https://doi.org/10.3390/polym13101667