Amphipathic Substrates Based on Crosslinker-Free Poly(ε-Caprolactone):Poly(2-Hydroxyethyl Methacrylate) Semi-Interpenetrated Networks Promote Serum Protein Adsorption

 ,
, 
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
3. Results
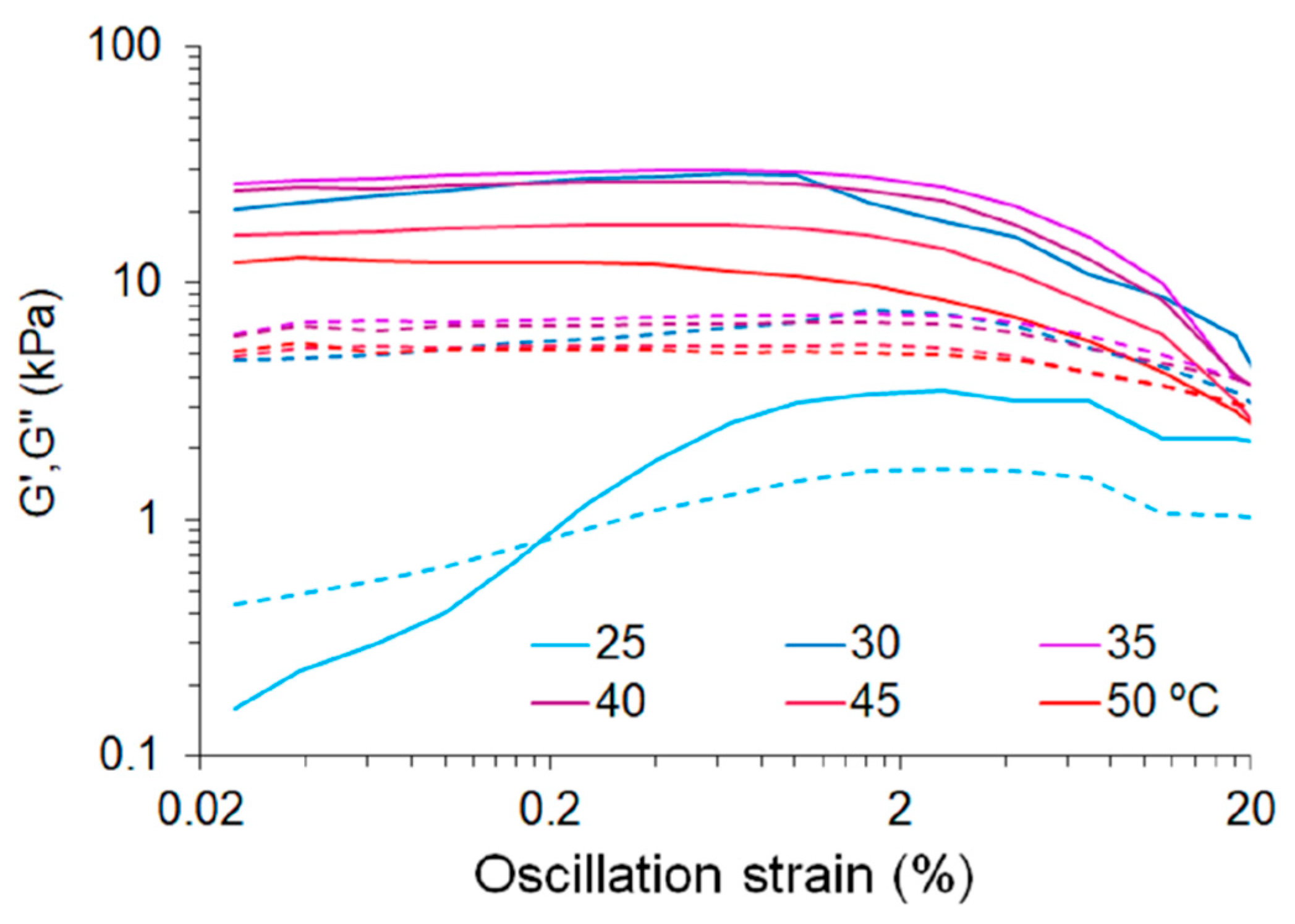
3.1. Production of Deployable Physically-Crosslinked PHEMA
3.2. Characterisation of APsIPN Substrates
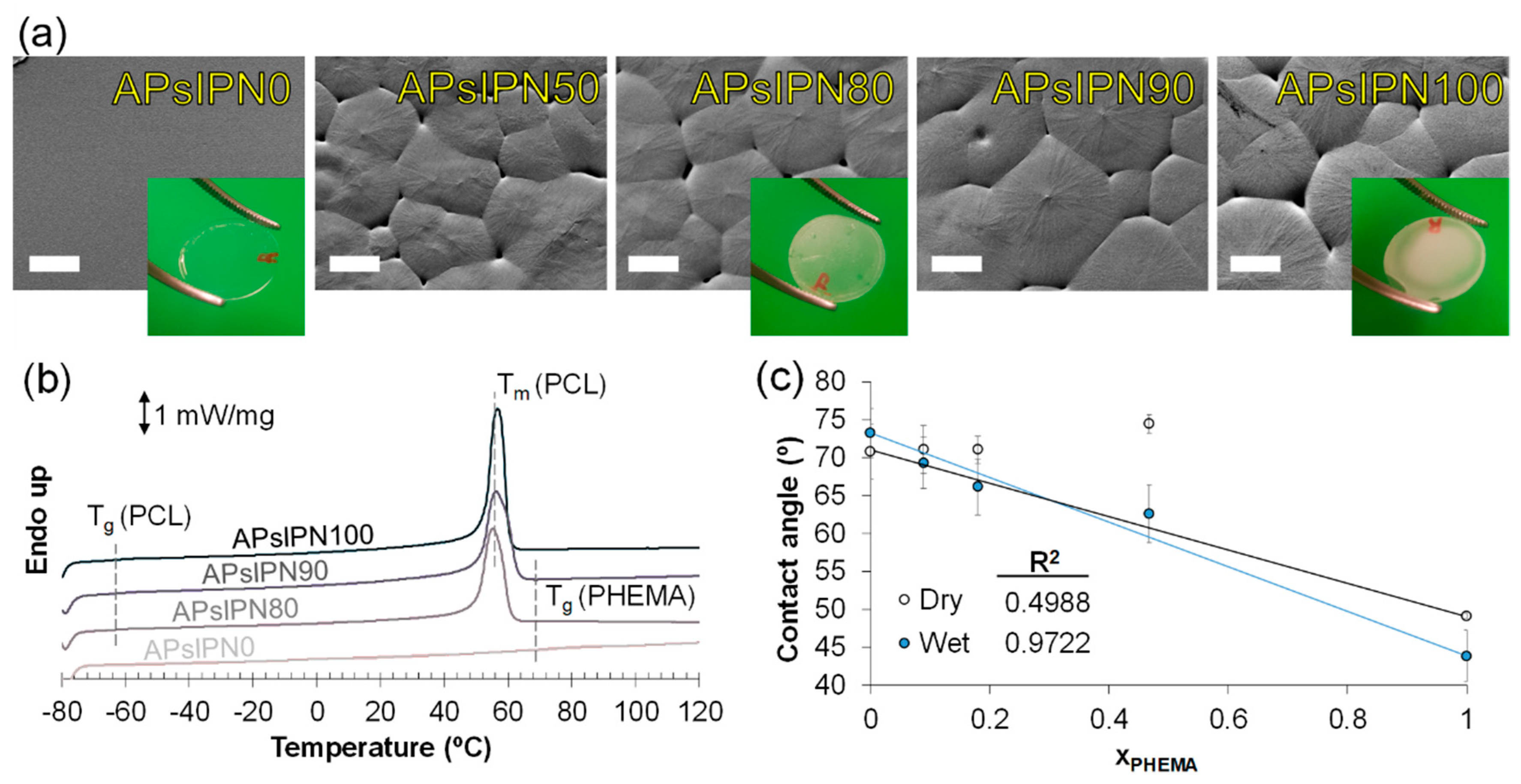
3.2.1. Microscopic Imaging and Thermophysical Assessment
3.2.2. Behaviour in Aqueous Solutions
3.3. Biomedical Approaches of APsIPN
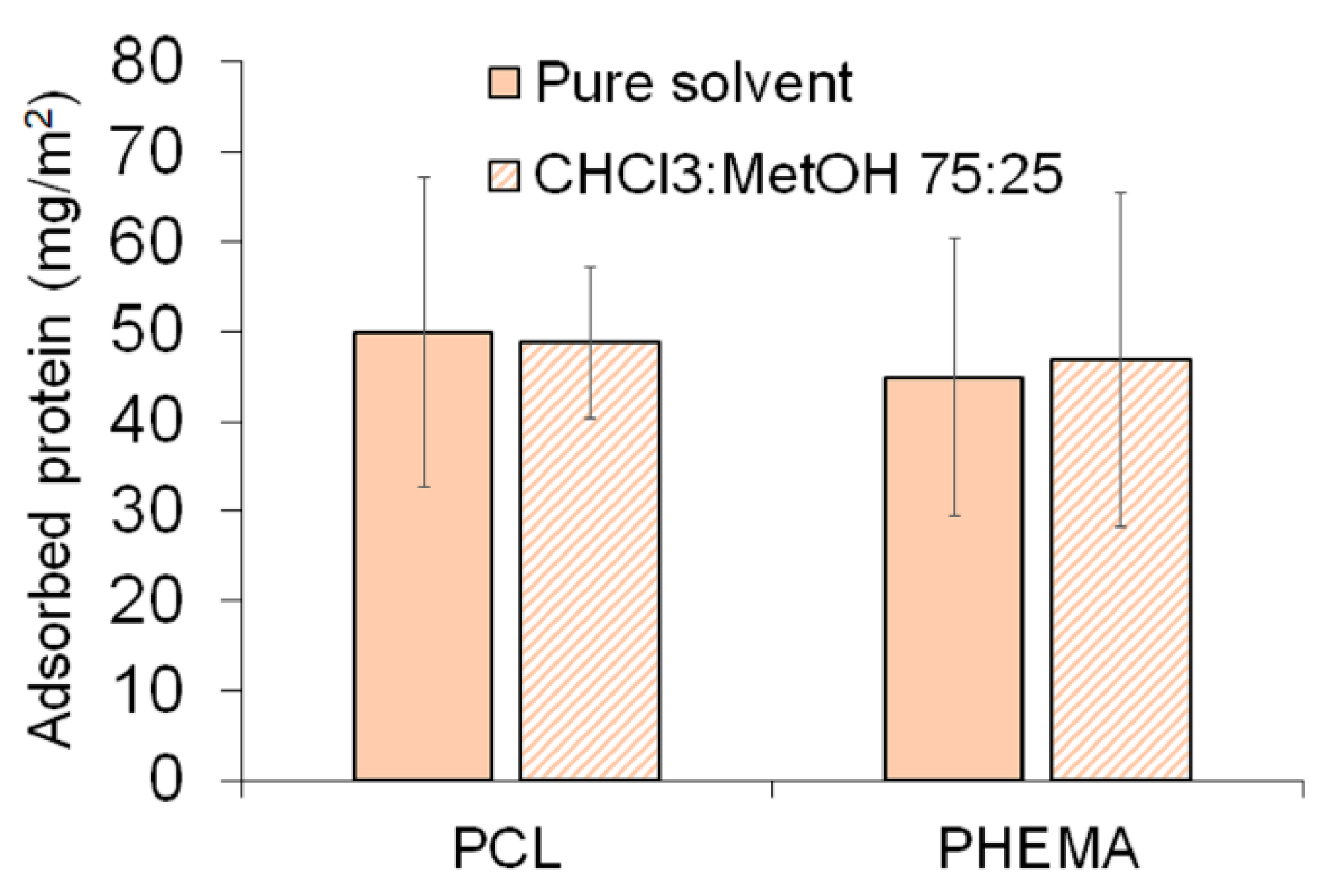
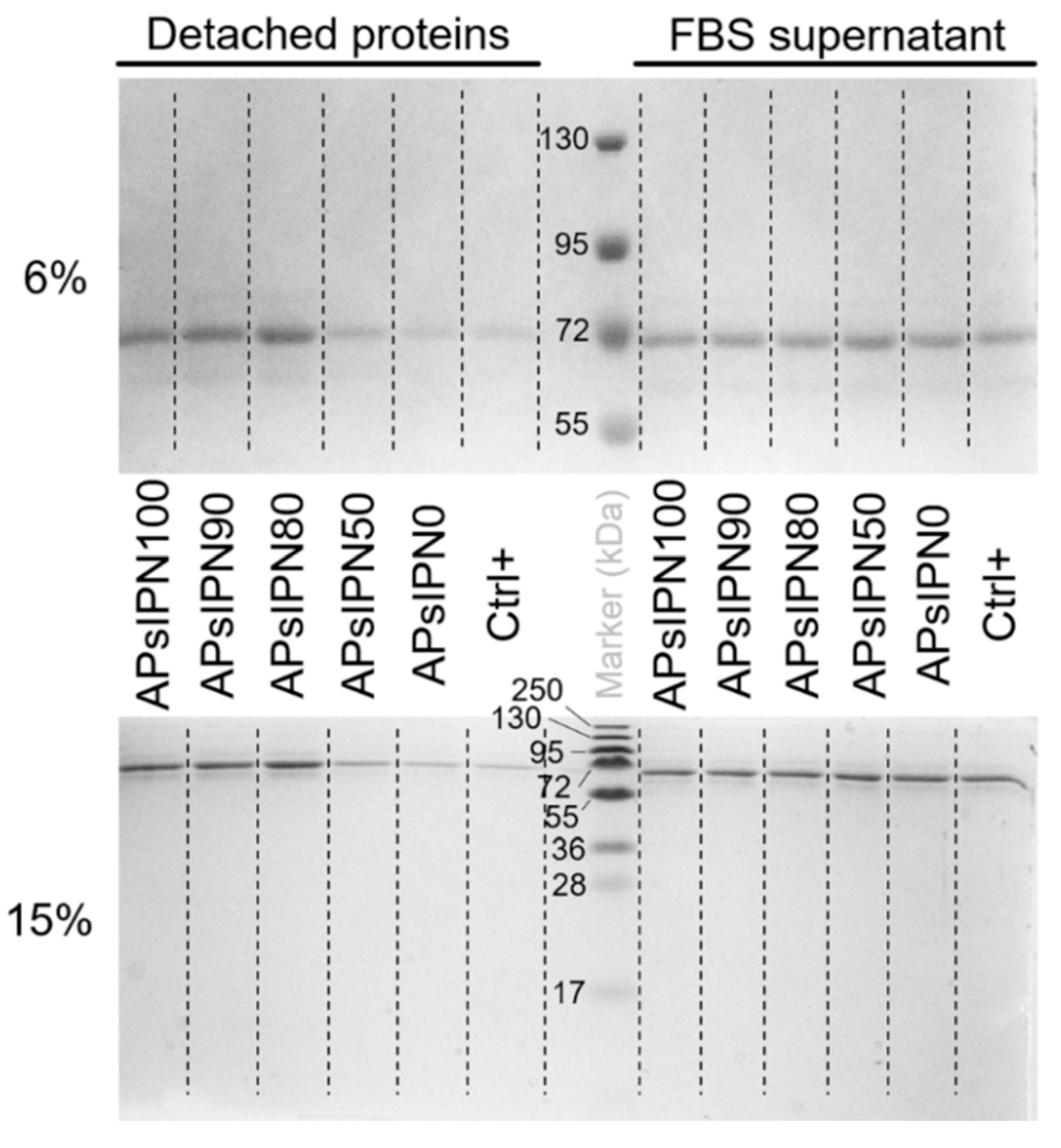
3.3.1. Serum Protein Adsorption on Amphiphilic Substrates
3.3.2. Electrospun Meshes Based on APsIPN
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




References
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Ratner, B.D.; Hoffman, A.S. Synthetic Hydrogels for Biomedical Applications. In Hydrogels for Medical and Related Applications; Andrade, J.D., Ed.; American Chemical Society: Washington, DC, USA, 1976; pp. 1–36. [Google Scholar] [CrossRef]
- Wheeler, J.C.; Woods, J.A.; Cox, M.J.; Cantrell, R.W.; Watkins, F.H.; Edlich, R.F. Evolution of hydrogel polymers as contact lenses, surface coatings, dressings, and drug delivery systems. J. Long. Term. Eff. Med. Implants. 1996, 6, 207–217. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10167362 (accessed on 3 December 2019).
- Atzet, S.; Curtin, S.; Trinh, P.; Bryant, S.; Ratner, B. Degradable poly(2-hydroxyethyl methacrylate)-co-polycaprolactone hydrogels for tissue engineering scaffolds. Biomacromolecules 2008, 9, 3370–3377. [Google Scholar] [CrossRef] [PubMed]
- Karabanova, L.V.; Mikhalovsky, S.V.; Lloyd, A.W. Gradient semi-interpenetrating polymer networks based on polyurethane and poly(2-hydroxyethyl methacrylate) for biomedical applications. J. Mater. Chem. 2012, 22, 7919–7928. [Google Scholar] [CrossRef]
- Rezaei, S.M.; Ishak, Z.A.M. Grafting of collagen onto interpenetrating polymer networks of poly(2-hydroxyethyl methacrylate) and poly(dimethyl siloxane) polymer films for biomedical applications. Express Polym. Lett. 2014, 8, 39–49. [Google Scholar] [CrossRef]
- Abbasi, F.; Mirzadeh, H.; Katbab, A.A. Sequential interpenetrating polymer networks of poly(2-hydroxyethyl methacrylate) and polydimethylsiloxane. J. Appl. Polym. Sci. 2002, 85, 1825–1831. [Google Scholar] [CrossRef]
- Kayaman-Apohan, N.; Baysal, B.M. Semi-Interpenetrating Hydrogel Networks of Poly(2-hydroxyethyl methacrylate) with Poly[(D,L-lactic acid)-co-(ɛ-caprolactam)]. Macromol. Chem. Phys. 2001, 202, 1182–1188. [Google Scholar] [CrossRef]
- Prashantha, K.; Pai, K.V.K.; Sherigara, B.S.; Prasannakumar, S. Interpenetrating polymer networks based on polyol modified castor oil polyurethane and poly(2-hydroxyethylmethacrylate): Synthesis, chemical, mechanical and thermal properties. Bull. Mater. Sci. 2001, 24, 535–538. [Google Scholar] [CrossRef]
- Krezović, B.D.; Dimitrijević, S.I.; Filipović, J.M.; Nikolić, R.R.; Tomić, S.L. Antimicrobial P(HEMA/IA)/PVP semi-interpenetrating network hydrogels. Polym. Bull. 2013, 70, 809–819. [Google Scholar] [CrossRef]
- Jones, D.S.; Andrews, G.P.; Caldwell, D.L.; Lorimer, C.; Gorman, S.P.; McCoy, C.P. Novel semi-interpenetrating hydrogel networks with enhanced mechanical properties and thermoresponsive engineered drug delivery, designed as bioactive endotracheal tube biomaterials. Eur. J. Pharm. Biopharm. 2012, 82, 563–571. [Google Scholar] [CrossRef]
- Lim, A.R.; Schueneman, G.T.; Novak, B.M. The T1ρ13C spin-lattice relaxation time of interpenetrating networks by solid state NMR. Solid State Commun. 1999, 109, 465–470. [Google Scholar] [CrossRef]
- Davis, P.A.; Nicolais, L.; Ambrosio, L.; Huang, S.J. Poly(2-hydroxyethyl methacrylate)/Poly(caprolactone) Semi-Interpenetrating Polymer Networks. J. Bioact. Compat. Polym. 1988, 3, 205–218. [Google Scholar] [CrossRef]
- Stamatopoulou, C.; Klonos, P.; Koutsoumpis, S.; Gun’Ko, V.; Pissis, P.; Karabanova, L. Hydrophilic nanocomposites based on polyurethane/poly(2-hydroxyethyl methacrylate) semi-IPNs and modified/unmodified nanosilica for biomedical applications. J. Polym. Sci. Part B Polym. Phys. 2014, 52, 397–408. [Google Scholar] [CrossRef]
- Davis, P.A.; Nicolais, L.; Ambrosio, L. Synthesis and characterization of semi-interpenetrating polymer networks of poly(2-hydroxyethyl methacrylate) and poly(caprolactone). In Proceedings of the ACS Division of Polymeric Material, Denver, CO, USA, 8 June 1987; Volume 56, pp. 536–540. [Google Scholar]
- Miyashita, Y.; Kobayashi, R.; Kimura, N.; Suzuki, H.; Nishio, Y. Transition behavior and phase structure of chitin/poly(2-hydroxyethyl methacrylate) composites synthesized by a solution coagulation/bulk polymerization method. Carbohydr. Polym. 1997, 34, 221–228. [Google Scholar] [CrossRef]
- Refojo, M.F.; Yasuda, H. Hydrogels from 2-hydroxyethyl methacrylate and propylene glycol monoacrylate. J. Appl. Polym. Sci. 1965, 9, 2425–2435. [Google Scholar] [CrossRef]
- Dušek, K.; Sedláček, B. Phase separation in poly(2-hydroxyethyl methacrylate) gels in the presence of water. Eur. Polym. J. 1971, 7, 1275–1285. [Google Scholar] [CrossRef]
- Yoon, S.C.; Jhon, M.S. Temperature effect on the permeation through poly(2-hydroxyethyl methacrylate) membrane. J. Appl. Polym. Sci. 1982, 27, 4661–4668. [Google Scholar] [CrossRef]
- Arica, M.Y.; Hasirci, V.; Alaeddinoǧlu, N.G. Covalent immobilization of α-amylase onto pHEMA microspheres: Preparation and application to fixed bed reactor. Biomaterials 1995, 16, 761–768. [Google Scholar] [CrossRef]
- Passos, M.F.; Dias, D.R.C.; Bastos, G.N.T.; Jardini, A.L.; Benatti, A.C.B.; Dias, C.G.B.T.; Filho, R.M. pHEMA hydrogels: Synthesis, kinetics and in vitro tests. J. Therm. Anal. Calorim. 2016, 125, 361–368. [Google Scholar] [CrossRef]
- Gregonis, D.E.; Russell, G.A.; Andrade, J.D.; DeVisser, A.C. Preparation and properties of stereoregular poly(hydroxyethyl methacrylate) polymers and hydrogels. Polymer 1978, 19, 1279–1284. [Google Scholar] [CrossRef]
- Paterson, S.M.; Brown, D.H.; Chirila, T.V.; Keen, I.; Whittaker, A.K.; Baker, M.V. The synthesis of water-soluble PHEMA via ARGET ATRP in protic media. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4084–4092. [Google Scholar] [CrossRef]
- Chirila, T.V.; Chen, Y.C.; Griffin, B.J.; Constable, I.J. Hydrophilic sponges based on 2-hydroxyethyl methacrylate. I. effect of monomer mixture composition on the pore size. Polym. Int. 1993, 32, 221–232. [Google Scholar] [CrossRef]
- Ratner, B.D.; Miller, I.F. Interaction of urea with poly(2-hydroxyethyl methacrylate) hydrogels. J. Polym. Sci. Part A-1 Polym. Chem. 1972, 10, 2425–2445. [Google Scholar] [CrossRef]
- Eschbach, F.O.; Huang, S.J. Hydrophilic-hydrophobic interpenetrating polymer networks and semi-interpenetrating polymer networks. In Interpenetrating Polymer Networks; Klempner, D., Sperling, L.H., Utracki, L.A., Eds.; Networks, American Chemical Society: New York, NY, USA, 1994; pp. 205–219. [Google Scholar]
- Shahrousvand, M.; Ghollasi, M.; Zarchi, A.A.K.; Salimi, A. Osteogenic differentiation of hMSCs on semi-interpenetrating polymer networks of polyurethane/poly(2-hydroxyethyl methacrylate)/cellulose nanowhisker scaffolds. Int. J. Biol. Macromol. 2019, 138, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Bartlett, P.D. The Kinetics of Decomposition of Benzoyl Peroxide in Solvents I. J. Am. Chem. Soc. 1946, 68, 1686–1692. [Google Scholar] [CrossRef]
- Crescenzi, V.; Manzini, G.; Calzolari, G.; Borri, C. Thermodynamics of fusion of poly-β-propiolactone and poly-ϵ-caprolactone. comparative analysis of the melting of aliphatic polylactone and polyester chains. Eur. Polym. J. 1972, 8, 449–463. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, D.; Somorjai, G.; Bertozzi, C.R. Probing the Surface Structural Rearrangement of Hydrogels by Sum-Frequency Generation Spectroscopy. J. Am. Chem. Soc. 1999, 121, 446–447. [Google Scholar] [CrossRef]
- Kim, S.H.; Opdahl, A.; Marmo, C.; Somorjai, G.A. AFM and SFG studies of pHEMA-based hydrogel contact lens surfaces in saline solution: Adhesion, friction, and the presence of non-crosslinked polymer chains at the surface. Biomaterials 2002, 23, 1657–1666. [Google Scholar] [CrossRef]
- Ramazani, S.; Karimi, M. Investigating the influence of temperature on electrospinning of polycaprolactone solutions. E-Polymers 2014, 14, 323–333. [Google Scholar] [CrossRef]
- Schoolaert, E.; Ryckx, P.; Geltmeyer, J.; Maji, S.; van Steenberge, P.H.M.; D’hooge, D.R.; Hoogenboom, R.; de Clerck, K. Waterborne Electrospinning of Poly(N-isopropylacrylamide) by Control of Environmental Parameters. ACS Appl. Mater. Interfaces 2017, 9, 24100–24110. [Google Scholar] [CrossRef]
- Pham, Q.P.; Sharma, U.; Mikos, A.G. Electrospun Poly(ε-caprolactone) Microfiber and Multilayer Nanofiber/Microfiber Scaffolds: Characterization of Scaffolds and Measurement of Cellular Infiltration. Biomacromolecules 2006, 7, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Wang, X.; Chen, W.; Meng, F.; Deng, C.; Liu, H.; Zhong, Z. Biodegradable poly(ε-caprolactone)-g-poly(2-hydroxyethyl methacrylate) graft copolymer micelles as superior nano-carriers for “smart” doxorubicin release. J. Mater. Chem. 2012, 22, 11730. [Google Scholar] [CrossRef]
- González-García, C.; Cantini, M.; Ballester-Beltrán, J.; Altankov, G.; Salmerón-Sánchez, M. The strength of the protein-material interaction determines cell fate. Acta Biomater. 2018, 77, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Rico, P.; Hernández, J.C.R.; Moratal, D.; Altankov, G.; Pradas, M.M.; Salmerón-Sánchez, M. Substrate-Induced Assembly of Fibronectin into Networks: Influence of Surface Chemistry and Effect on Osteoblast Adhesion. Tissue Eng. Part A 2009, 15, 3271–3281. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Solvent Fraction ( wt %) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 30 | 40 | 50 | 60 | 70 | 80 | 90 | |
| DMF | ISE | ISE | ISE | ISG | ISG | SG | SG | L |
| MetOH | ISE | ISE | ISE | ISG | ISG | SG | SG | L |
| Solvent | Time (h) | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 | 4 | 8 | 12 | 16 | 20 | 24 | 28 | |
| DMF | L | L | L | L | SG | SG | SG | ISG |
| MetOH | L | L | L | SG | SG | SG | SG | ISG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilariño-Feltrer, G.; Salgado-Gallegos, A.; de-la-Concepción-Ausina, J.; Rodríguez-Hernández, J.C.; Shahrousvand, M.; Vallés-Lluch, A. Amphipathic Substrates Based on Crosslinker-Free Poly(ε-Caprolactone):Poly(2-Hydroxyethyl Methacrylate) Semi-Interpenetrated Networks Promote Serum Protein Adsorption. Polymers 2020, 12, 1256. https://doi.org/10.3390/polym12061256
Vilariño-Feltrer G, Salgado-Gallegos A, de-la-Concepción-Ausina J, Rodríguez-Hernández JC, Shahrousvand M, Vallés-Lluch A. Amphipathic Substrates Based on Crosslinker-Free Poly(ε-Caprolactone):Poly(2-Hydroxyethyl Methacrylate) Semi-Interpenetrated Networks Promote Serum Protein Adsorption. Polymers. 2020; 12(6):1256. https://doi.org/10.3390/polym12061256
Chicago/Turabian StyleVilariño-Feltrer, Guillermo, Alfredo Salgado-Gallegos, Joan de-la-Concepción-Ausina, José Carlos Rodríguez-Hernández, Mohsen Shahrousvand, and Ana Vallés-Lluch. 2020. "Amphipathic Substrates Based on Crosslinker-Free Poly(ε-Caprolactone):Poly(2-Hydroxyethyl Methacrylate) Semi-Interpenetrated Networks Promote Serum Protein Adsorption" Polymers 12, no. 6: 1256. https://doi.org/10.3390/polym12061256
APA StyleVilariño-Feltrer, G., Salgado-Gallegos, A., de-la-Concepción-Ausina, J., Rodríguez-Hernández, J. C., Shahrousvand, M., & Vallés-Lluch, A. (2020). Amphipathic Substrates Based on Crosslinker-Free Poly(ε-Caprolactone):Poly(2-Hydroxyethyl Methacrylate) Semi-Interpenetrated Networks Promote Serum Protein Adsorption. Polymers, 12(6), 1256. https://doi.org/10.3390/polym12061256