Effect of Poly(vinyl alcohol) on Nanoencapsulation of Budesonide in Chitosan Nanoparticles via Ionic Gelation and Its Improved Bioavailability

 ,
, 

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Molecular Weight Determination
2.3. Nanoparticles’ Preparation
2.4. Nanoparticle Characterization
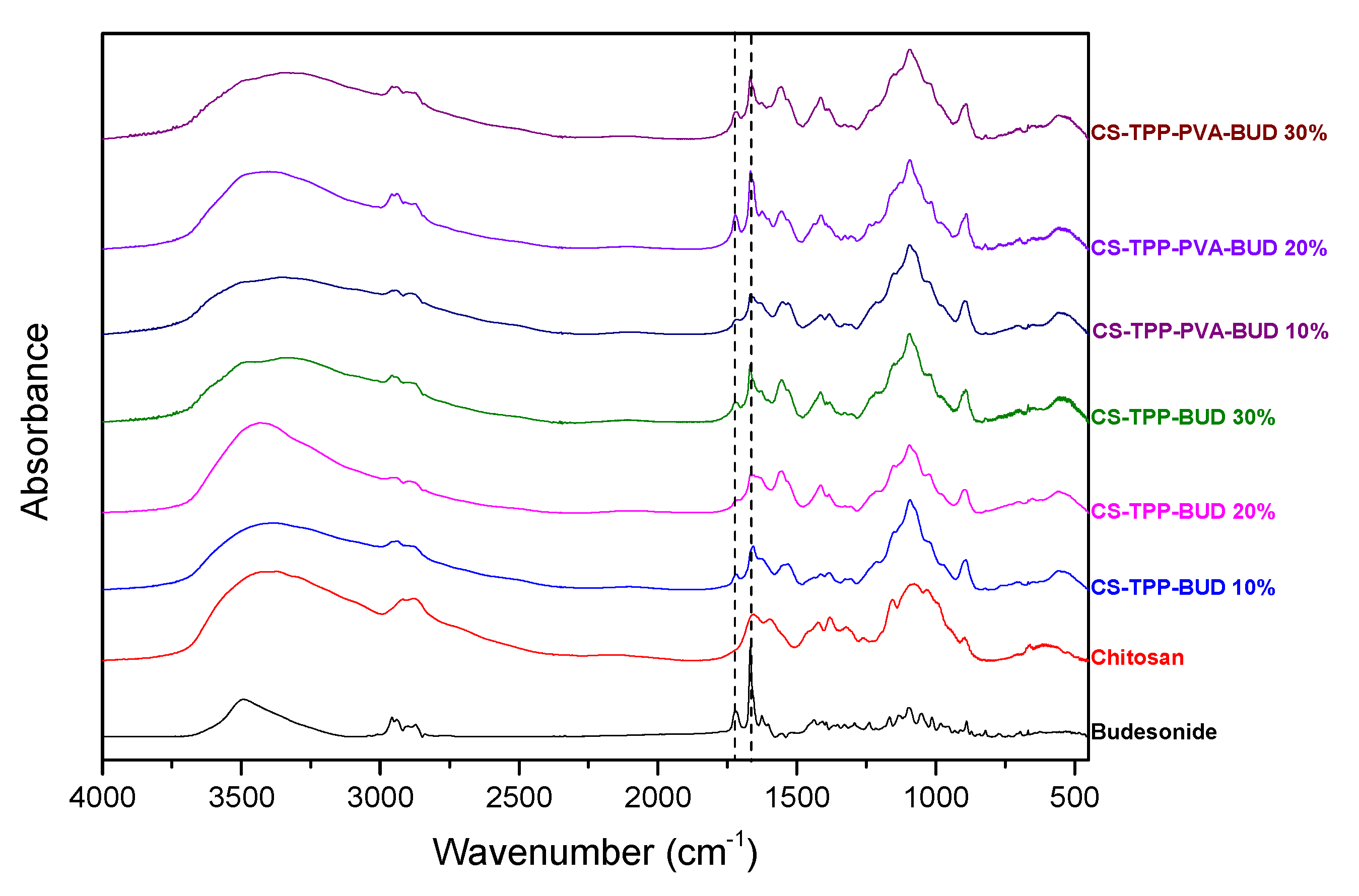
2.4.1. Fourier-Transformed Infrared Spectroscopy (FTIR)
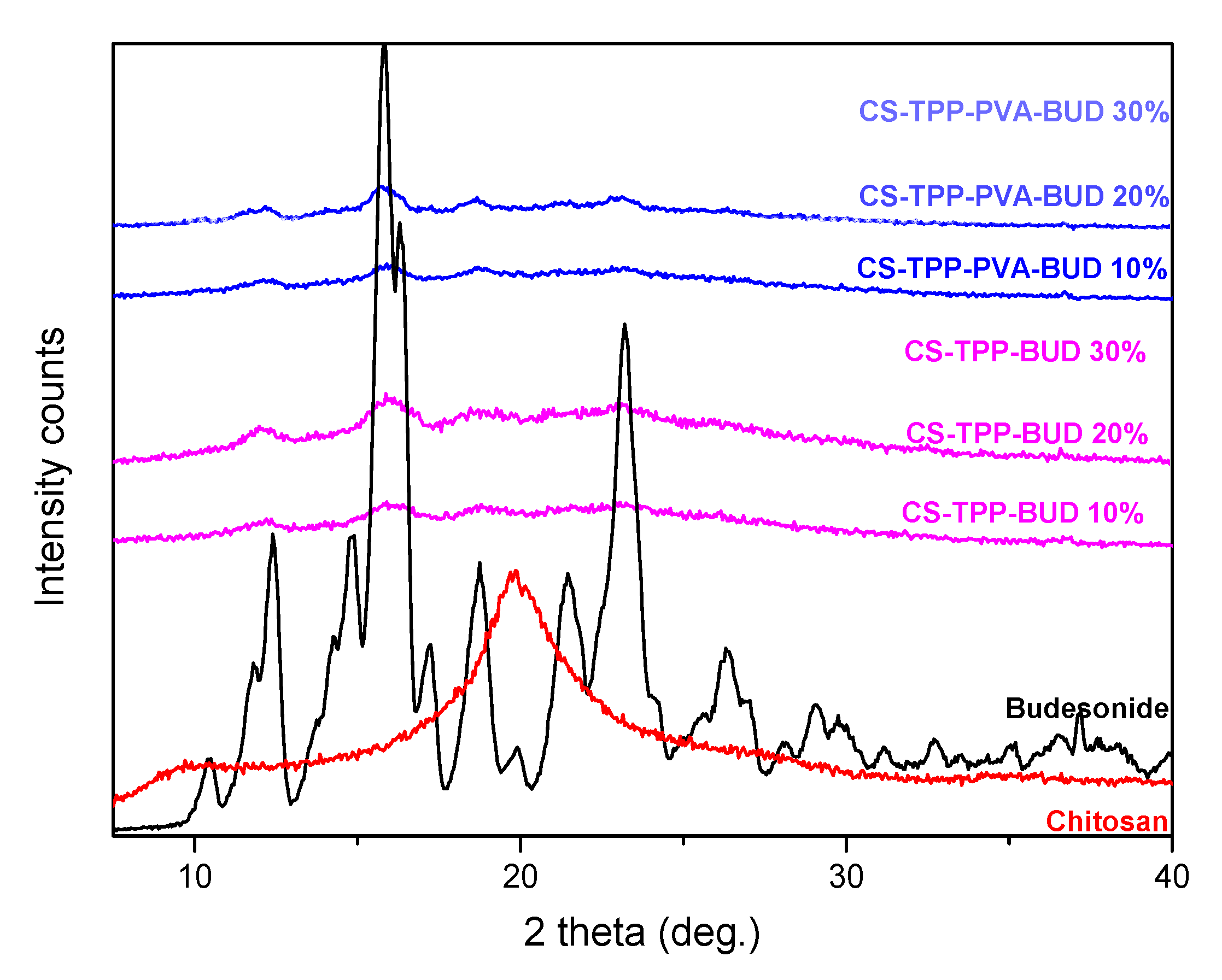
2.4.2. Wide-Angle X-ray Scattering (XRD)
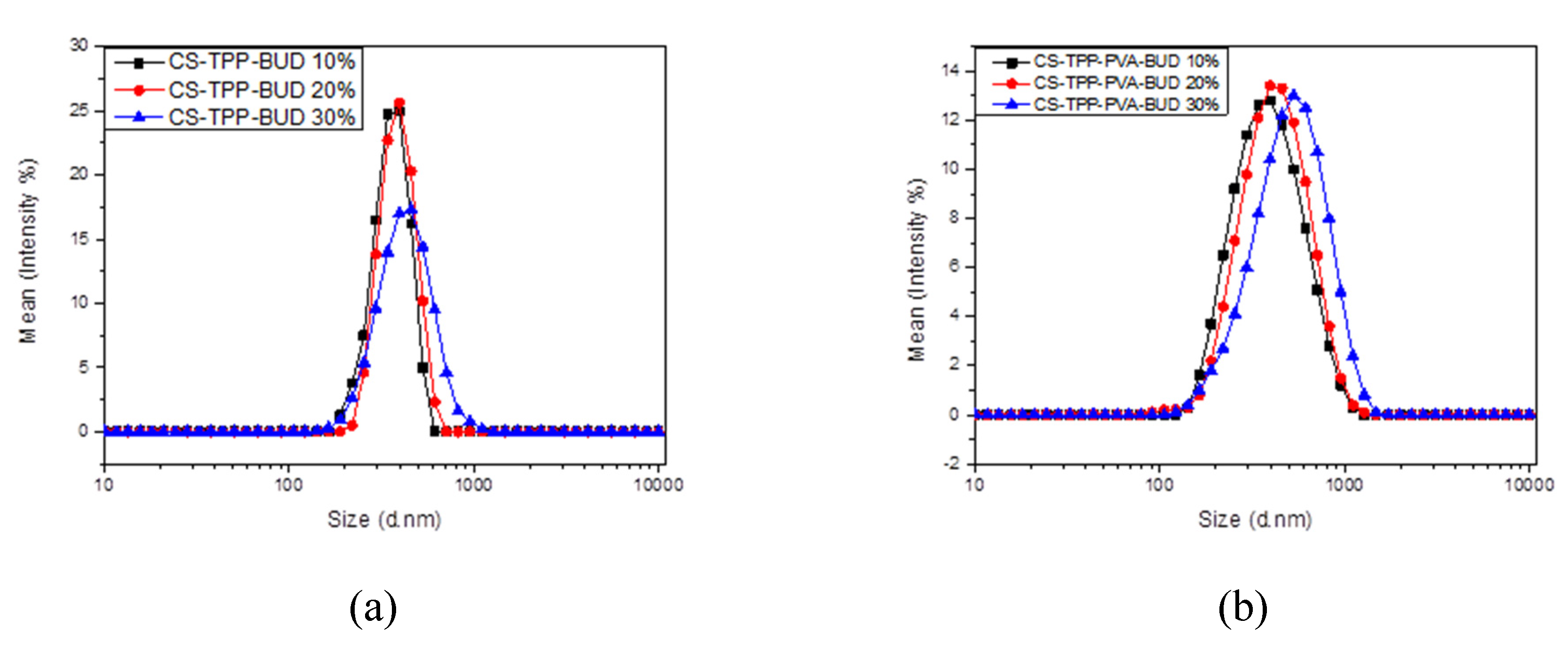
2.4.3. Dynamic Light Scattering (DLS)
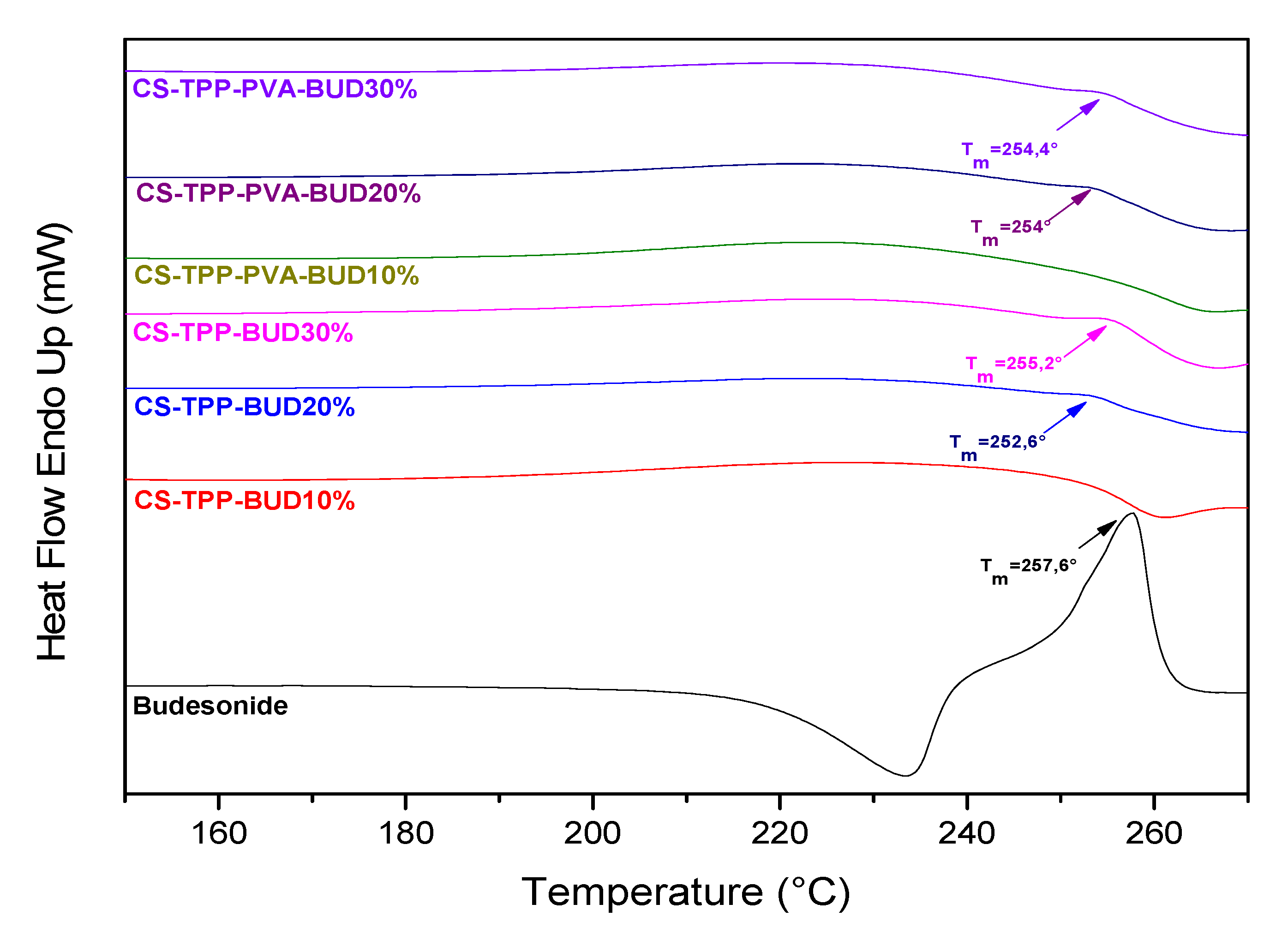
2.4.4. Differential Scanning Calorimetry (DSC)
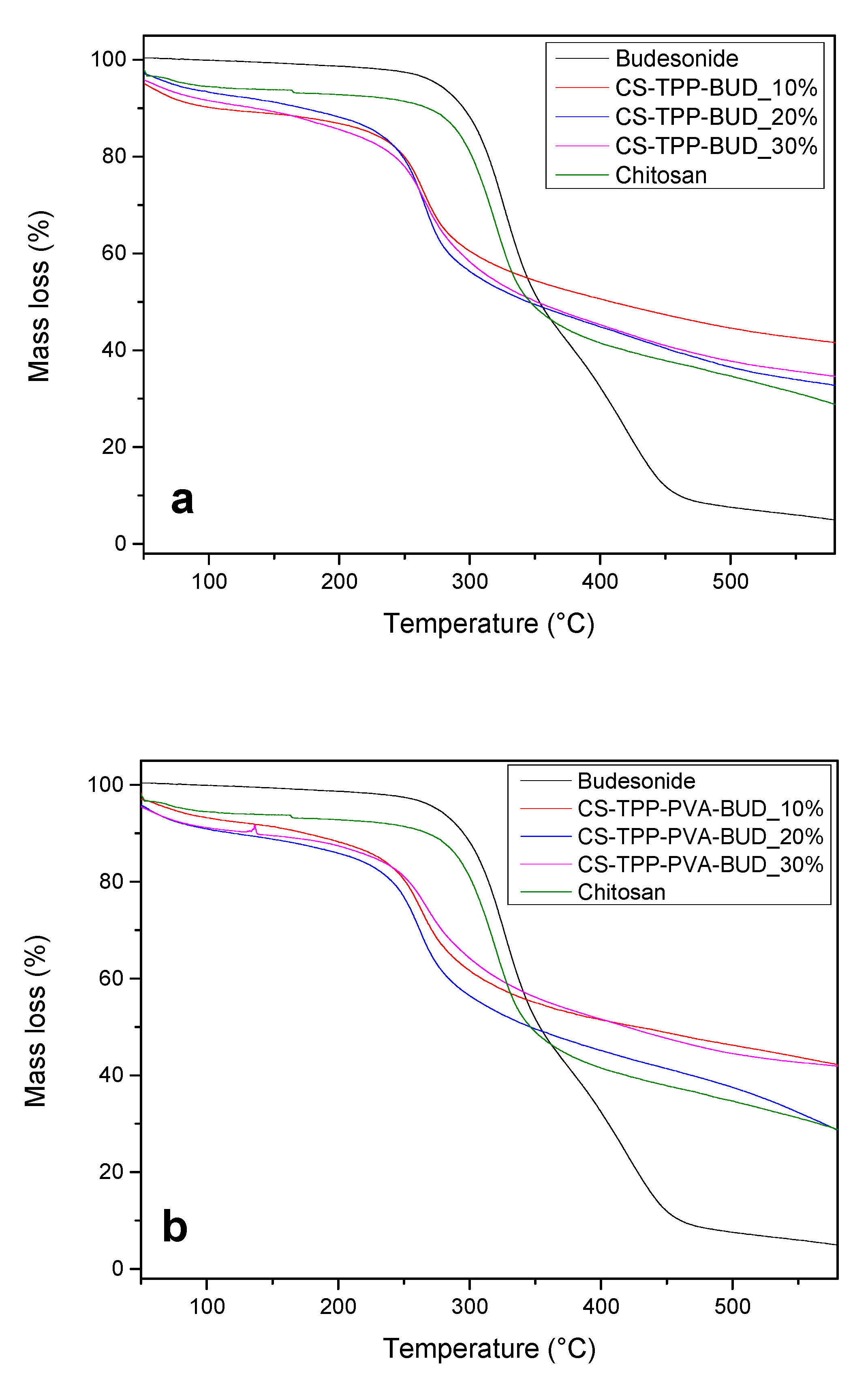
2.4.5. Thermogravimetric Analysis (TGA)
2.4.6. Scanning electron Microscopy (SEM)
2.4.7. High-Pressure Liquid Chromatography (HPLC), Quantitative Analysis and Drug Loading
2.4.8. Yield, Encapsulation Efficiency and Drug Loading
2.4.9. In vitro Dissolution Studies
2.5. Technical Details on the Computation
2.5.1. Vibrational Analysis
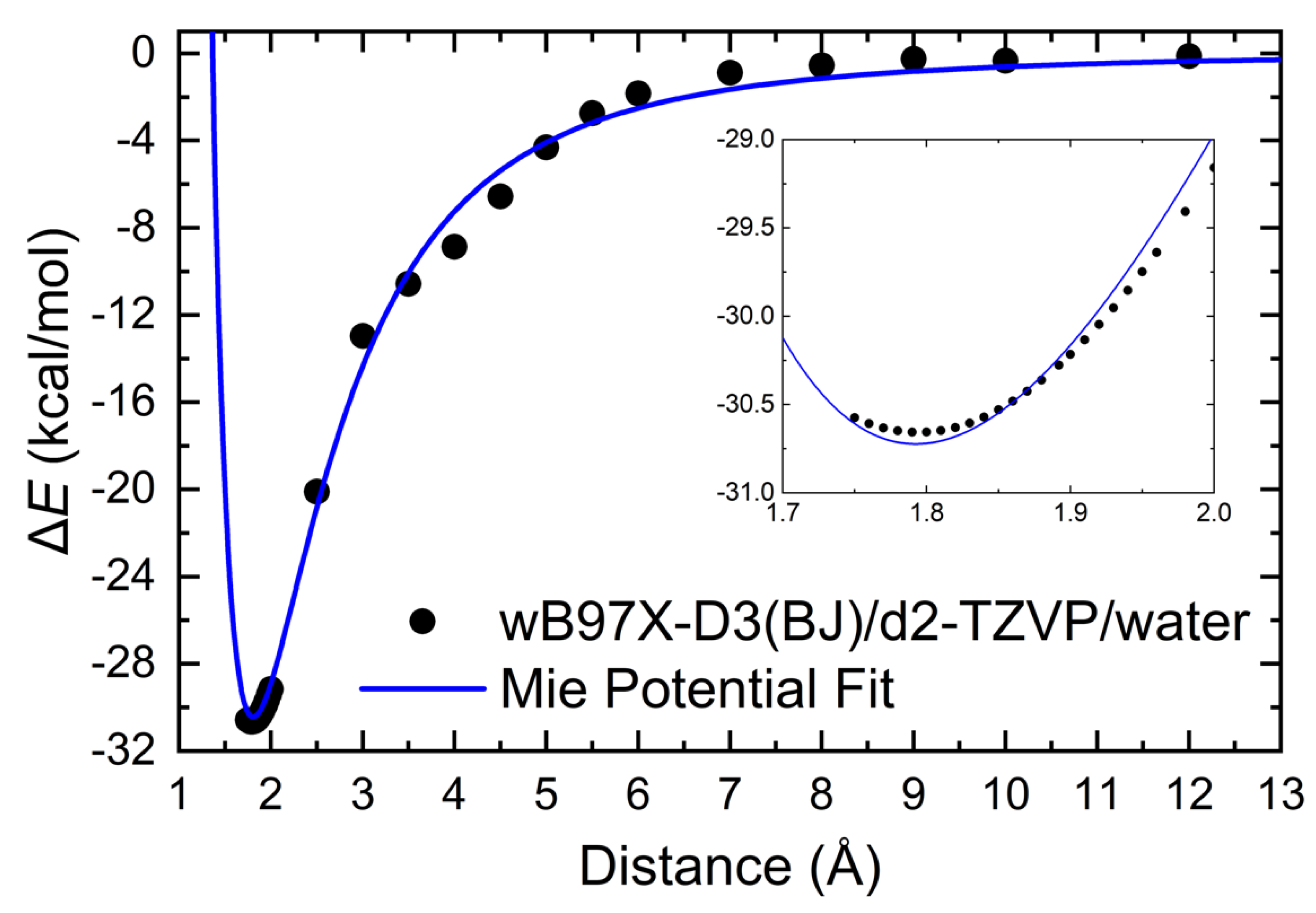
2.5.2. Interaction Energy
3. Results and Discussion
3.1. Characterization of Budesonide-Loaded Nanoparticles
3.2. Computational Study
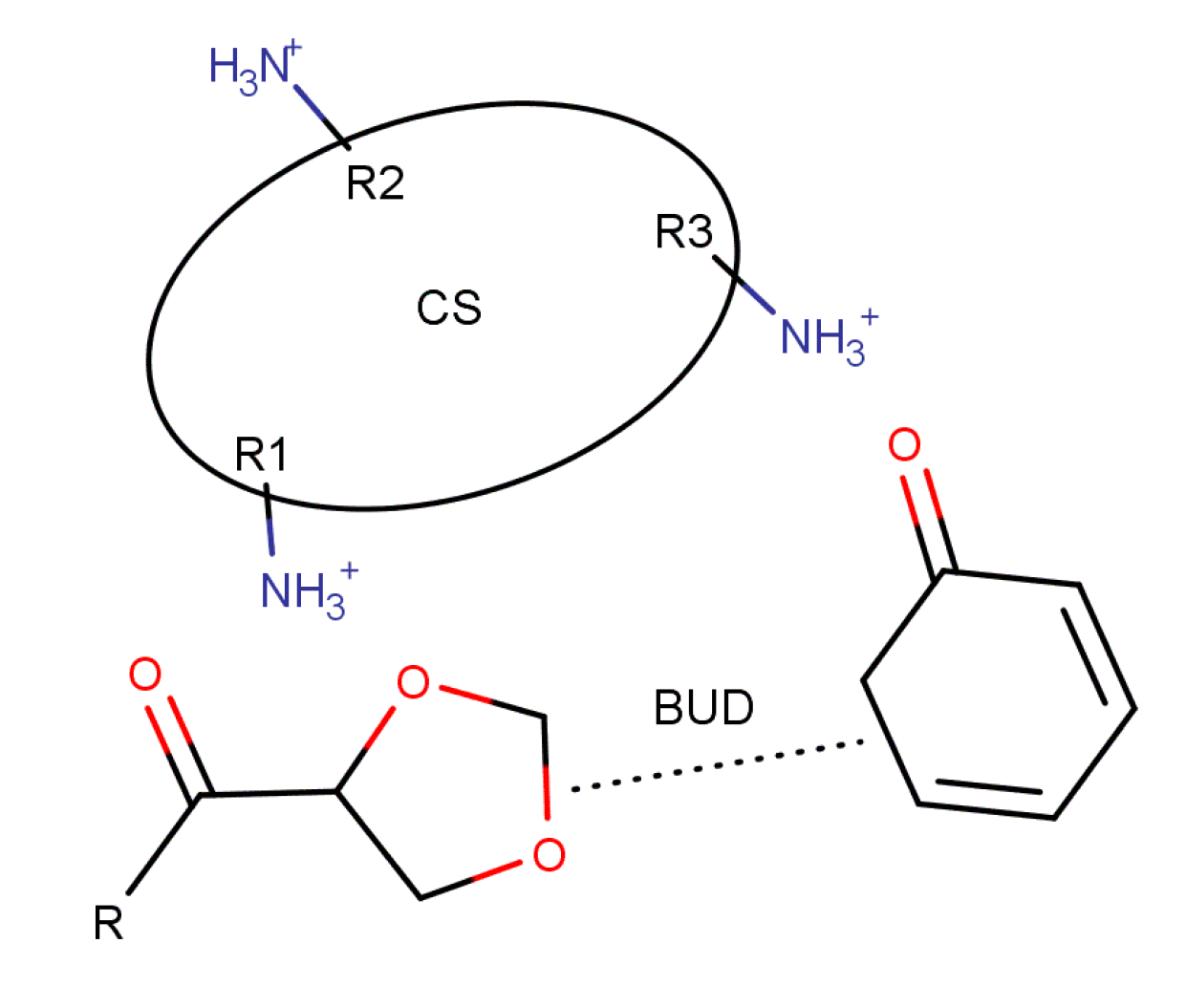
3.2.1. Intermolecular Interactions
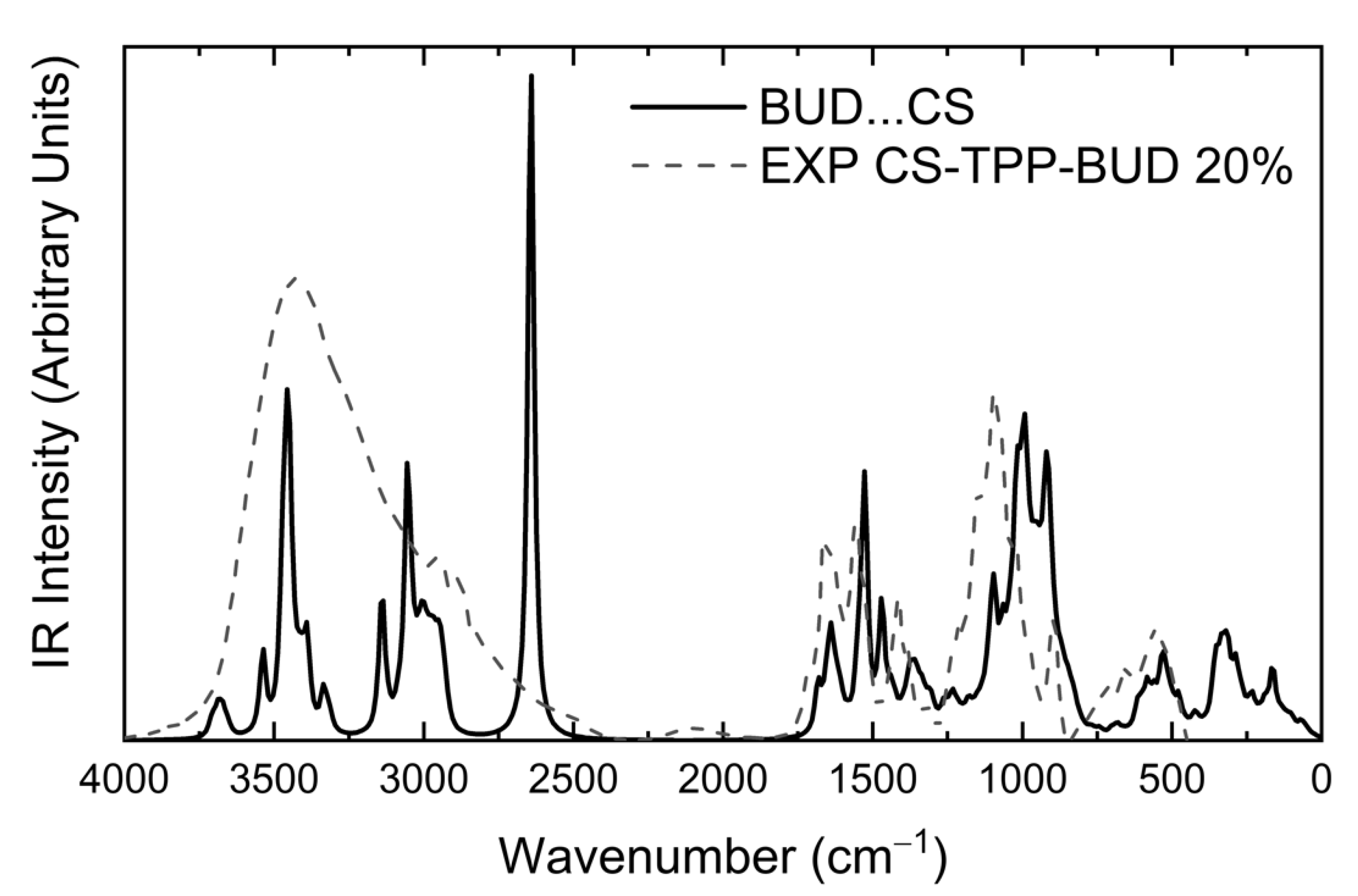
3.2.2. Infrared Spectra
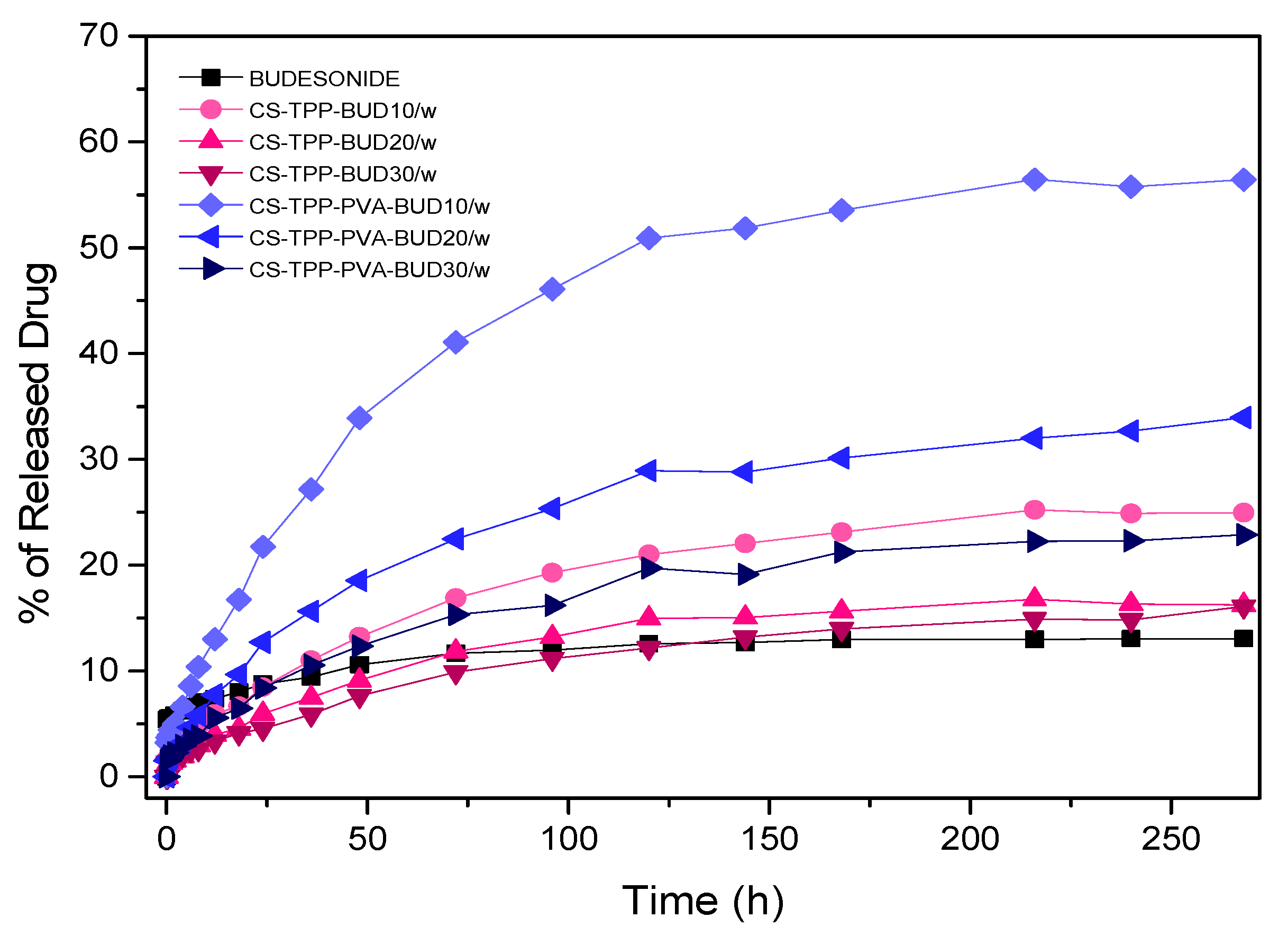
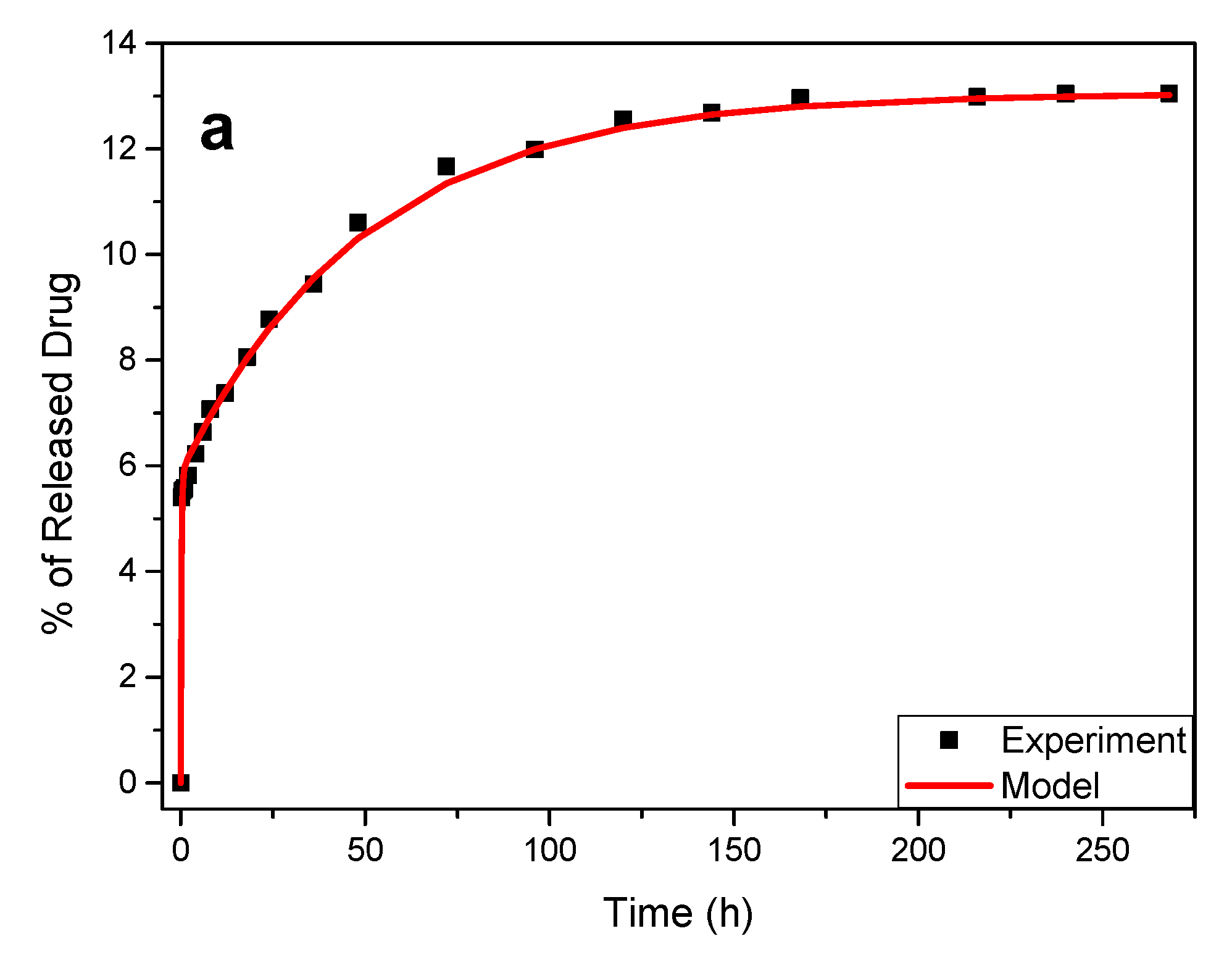
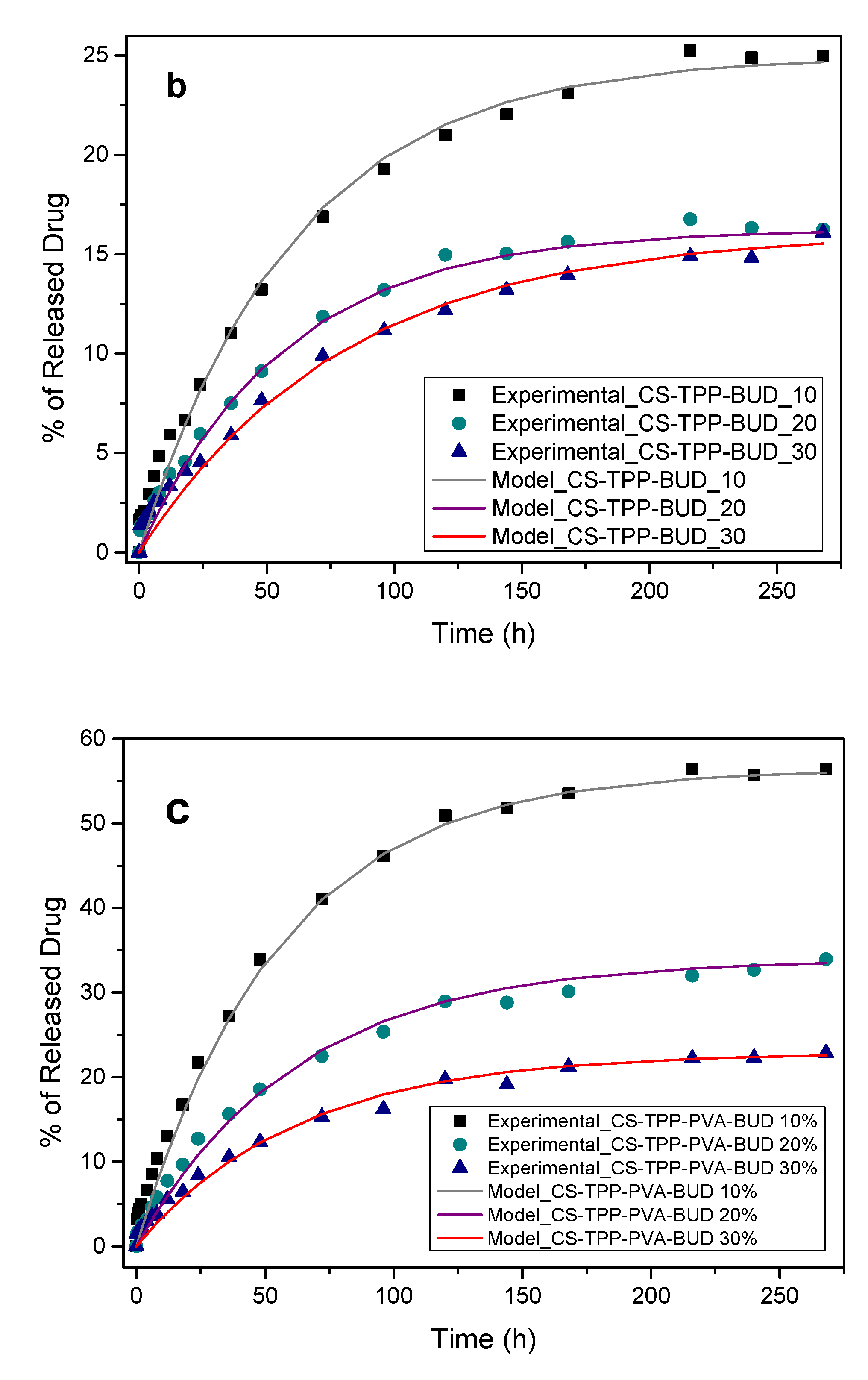
3.3. Drug Release Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Coster, D.; Jones, M. Tailoring of corticosteroids in COPD management. Curr. Respir. Care Rep. 2014, 3, 121–132. [Google Scholar] [CrossRef]
- Celli, B.R. Pharmacological Therapy of COPD: Reasons for Optimism. Chest 2018, 154, 1404–1415. [Google Scholar] [CrossRef]
- Onoue, S.; Misaka, S.; Kawabata, Y.; Yamada, S. New treatments for chronic obstructive pulmonary disease and viable formulation/device options for inhalation therapy. Expert Opin. Drug Deliv. 2009, 6, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Rogliani, P.; Calzetta, L.; Coppola, A.; Cavalli, F.; Ora, J.; Puxeddu, E.; Matera, M.G.; Cazzola, M. Optimizing drug delivery in COPD: The role of inhaler devices. Respir. Med. 2017, 124, 6–14. [Google Scholar] [CrossRef]
- Usmani, O.S. Treating the small airways. Respiration 2012, 84, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.; Saad, N.; Suissa, S. Inhaled corticosteroids in COPD: The clinical evidence. Eur. Respir. J. 2015, 45, 525–537. [Google Scholar] [CrossRef]
- Choulis, N.H. Chapter 14—Dermatological Drugs, Topical Agents, and Cosmetics. In A Worldwide Yearly Survey of New Data in Adverse Drug Reactions; Ray, S.D.B.T.-S.E., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 36, pp. 203–231. ISBN 0378-6080. [Google Scholar]
- Odonnell, S.; Omorain, C.A. Therapeutic benefits of budesonide in gastroenterology. Ther. Adv. Chronic Dis. 2010, 1, 177–186. [Google Scholar] [CrossRef]
- Ali, H.; Weigmann, B.; Collnot, E.M.; Khan, S.A.; Windbergs, M.; Lehr, C.M. Budesonide loaded PLGA nanoparticles for targeting the inflamed intestinal mucosa—Pharmaceutical characterization and fluorescence imaging. Pharm. Res. 2016, 33, 1085–1092. [Google Scholar] [CrossRef]
- Buhecha, M.D.; Lansley, A.B.; Somavarapu, S.; Pannala, A.S. Development and characterization of PLA nanoparticles for pulmonary drug delivery: Co-encapsulation of theophylline and budesonide, a hydrophilic and lipophilic drug. J. Drug Deliv. Sci. Technol. 2019, 53, 101128. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Zhang, X.; Jiaqi, L.; Fan, L.; Beck-Broichsitter, M.; Zhang, X.; Muenster, U.; Wang, X.; Zhao, J.; et al. Sustained therapeutic efficacy of budesonide-loaded chitosan swellable microparticles after lung delivery: Influence of in vitro release, treatment interval and dose. J. Control. Release 2018, 283, 163–174. [Google Scholar] [CrossRef]
- Campos, J.C.; Ferreira, D.C.; Lima, S.; Reis, S.; Costa, P.J. Swellable polymeric particles for the local delivery of budesonide in oral mucositis. Int. J. Pharm. 2019, 566, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Bodas, D.S.; Ige, P.P. Central composite rotatable design for optimization of budesonide-loaded cross-linked chitosan–dextran sulfate nanodispersion: Characterization, in vitro diffusion and aerodynamic study. Drug Dev. Ind. Pharm. 2019, 45, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Qelliny, M.R.; Aly, U.F.; Elgarhy, O.H.; Khaled, K.A. Budesonide-Loaded Eudragit S 100 Nanocapsules for the Treatment of Acetic Acid-Induced Colitis in Animal Model. AAPS PharmSciTech 2019, 20, 237. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, M.; Tian, F.; Cun, D.; Rantanen, J.; Yang, M. Budesonide nanocrystal-loaded hyaluronic acid microparticles for inhalation: In vitro and in vivo evaluation. Carbohydr. Polym. 2018, 181, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Prosapio, V.; De Marco, I.; Reverchon, E. PVP/corticosteroid microspheres produced by supercritical antisolvent coprecipitation. Chem. Eng. J. 2016, 292, 264–275. [Google Scholar] [CrossRef]
- Lu, W.; Rades, T.; Rantanen, J.; Yang, M. Inhalable co-amorphous budesonide-arginine dry powders prepared by spray drying. Int. J. Pharm. 2019, 565, 1–8. [Google Scholar] [CrossRef]
- Kaialy, W.; Nokhodchi, A. The use of freeze-dried mannitol to enhance the in vitro aerosolization behaviour of budesonide from the Aerolizer®. Powder Technol. 2016, 288, 291–302. [Google Scholar] [CrossRef]
- Nagavarma, B.V.N.; Yadav, H.K.S.; Ayaz, A.; Vasudha, L.S.; Shivakumar, H.G. Different techniques for preparation of polymeric nanoparticles—A review. Asian J. Pharm. Clin. Res. 2012, 5, 16–23. [Google Scholar]
- Sung, J.C.; Pulliam, B.L.; Edwards, D.A. Nanoparticles for drug delivery to the lungs. Trends Biotechnol. 2007, 25, 563–570. [Google Scholar] [CrossRef]
- Yhee, J.; Im, J.; Nho, R. Advanced Therapeutic Strategies for Chronic Lung Disease Using Nanoparticle-Based Drug Delivery. J. Clin. Med. 2016, 5, 82. [Google Scholar] [CrossRef]
- Younes, I.; Rinaudo, M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Koukaras, E.N.; Papadimitriou, S.A.; Bikiaris, D.N.; Froudakis, G.E. Insight on the formation of chitosan nanoparticles through ionotropic gelation with tripolyphosphate. Mol. Pharm. 2012, 9, 2856–2862. [Google Scholar] [CrossRef] [PubMed]
- Koutroumanis, K.P.; Avgoustakis, K.; Bikiaris, D. Synthesis of cross-linked N-(2-carboxybenzyl)chitosan pH sensitive polyelectrolyte and its use for drug controlled delivery. Carbohydr. Polym. 2010, 82, 181–188. [Google Scholar] [CrossRef]
- Filippousi, M.; Siafaka, P.I.; Amanatiadou, E.P.; Nanaki, S.G.; Nerantzaki, M.; Bikiaris, D.N.; Vizirianakis, I.S.; Van Tendeloo, G. Modified chitosan coated mesoporous strontium hydroxyapatite nanorods as drug carriers. J. Mater. Chem. B 2015, 3, 5991–6000. [Google Scholar] [CrossRef]
- Janes, K.A.; Alonso, M.J. Depolymerized chitosan nanoparticles for protein delivery: Preparation and characterization. J. Appl. Polym. Sci. 2003, 88, 2769–2776. [Google Scholar] [CrossRef]
- Siafaka, P.I.; Titopoulou, A.; Koukaras, E.N.; Kostoglou, M.; Koutris, E.; Karavas, E.; Bikiaris, D.N. Chitosan derivatives as effective nanocarriers for ocular release of timolol drug. Int. J. Pharm. 2015, 495, 249–264. [Google Scholar] [CrossRef]
- Ahirrao, S.P.; Gide, P.S.; Shrivastav, B.; Sharma, P. Ionotropic Gelation: A Promising Cross Linking Technique for Hydrogels. J. Pharm. Nanotechnol. 2013, 2, 1–6. [Google Scholar]
- Lavertu, M.; Xia, Z.; Serreqi, A.N.; Berrada, M.; Rodrigues, A.; Wang, D.; Buschmann, M.D.; Gupta, A. A validated 1H NMR method for the determination of the degree of deacetylation of chitosan. J. Pharm. Biomed. Anal. 2003, 32, 1149–1158. [Google Scholar] [CrossRef]
- Kasaai, M.R.; Arul, J.; Charlet, G. Fragmentation of chitosan by acids. Sci. World J. 2013. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Bikiaris, D.; Avgoustakis, K.; Karavas, E.; Georgarakis, M. Chitosan nanoparticles loaded with dorzolamide and pramipexole. Carbohydr. Polym. 2008, 73, 44–54. [Google Scholar] [CrossRef]
- Koukaras, E.N.; Papadimitriou, S.A.; Bikiaris, D.N.; Froudakis, G.E. Properties and energetics for design and characterization of chitosan nanoparticles used for drug encapsulation. RSC Adv. 2014, 4, 12653–12661. [Google Scholar] [CrossRef]
- Leng, D.; Thanki, K.; Foged, C.; Yang, M. Formulating Inhalable Dry Powders Using Two-Fluid and Three-Fluid Nozzle Spray Drying. Pharm. Res. 2018, 35, 247. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Sirianni, D.A.; Alenaizan, A.; Cheney, D.L.; Sherrill, C.D. Assessment of Density Functional Methods for Geometry Optimization of Bimolecular van der Waals Complexes. J. Chem. Theory Comput. 2018, 14, 3004–3013. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to Isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: Theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Chipman, D.M. Comparison of solvent reaction field representations. Theor. Chem. Acc. 2002, 107, 80–89. [Google Scholar] [CrossRef]
- Mardirossian, N.; Ruiz Pestana, L.; Womack, J.C.; Skylaris, C.K.; Head-Gordon, T.; Head-Gordon, M. On the Use of the rVV10 Nonlocal Correlation Functional in the B97M-V Density Functional: Defining B97M-rV and Related Functionals. J. Phys. Chem. Lett. 2017, 8, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Gräfenstein, J.; Izotov, D.; Cremer, D. Avoiding singularity problems associated with meta-GGA (generalized gradient approximation) exchange and correlation functionals containing the kinetic energy density. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Nanaki, S.; Tseklima, M.; Christodoulou, E.; Triantafyllidis, K.; Kostoglou, M.; Bikiaris, D.N. Thiolated chitosan masked polymeric microspheres with incorporated mesocellular silica foam (MCF) for intranasal delivery of paliperidone. Polymers (Basel) 2017, 9, 617. [Google Scholar] [CrossRef]
- Ntohogian, S.; Gavriliadou, V.; Christodoulou, E.; Nanaki, S.; Lykidou, S.; Naidis, P.; Mischopoulou, L.; Barmpalexis, P.; Nikolaidis, N.; Bikiaris, D. Chitosan Nanoparticles with Encapsulated Natural and UF-Purified Annatto and Saffron for the Preparation of UV Protective Cosmetic Emulsions. Molecules 2018, 23, 2107. [Google Scholar] [CrossRef]
- Muralidharan, P.; Malapit, M.; Mallory, E.; Hayes, D.; Mansour, H.M. Inhalable nanoparticulate powders for respiratory delivery. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1189–1199. [Google Scholar] [CrossRef]
- Papadimitriou, S.A.; Achilias, D.S.; Bikiaris, D.N. Chitosan-g-PEG nanoparticles ionically crosslinked with poly(glutamic acid) and tripolyphosphate as protein delivery systems. Int. J. Pharm. 2012, 430, 318–327. [Google Scholar] [CrossRef]
- Gagnadoux, F.; Hureaux, J.; Vecellio, L.; Urban, T.; Le Pape, A.; Valo, I.; Montharu, J.; Leblond, V.; Boisdron-Celle, M.; Lerondel, S.; et al. Aerosolized chemotherapy. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. Review article DLS and zeta potential—What they are and what they are not ? J. Control. Release 2016, 235, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Abouelhag, H.A.; Sivakumar, S.M.; Bagul, U.S.; Eltyep, E.M. preparation and physical characterization of cisplatin chitosan nanoparticles by zeta nanosizer “prime step for formulation and development”. Int. J. Pharm. Sci. Res. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Tiǧli Aydin, R.S.; Pulat, M. 5-fluorouracil encapsulated chitosan nanoparticles for pH-stimulated drug delivery: Evaluation of controlled release kinetics. J. Nanomater. 2012, 2012. [Google Scholar] [CrossRef]
- Sahib, M.N.; Darwis, Y.; Peh, K.K.; Abdulameer, S.A.; Tan, Y.T.F. Rehydrated sterically stabilized phospholipid nanomicelles of budesonide for nebulization: Physicochemical characterization and in vitro, in vivo evaluations. Int. J. Nanomed. 2011, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, P.Y.; Li, Y.L.; Fan, L.; Lin, J.; Hu, Q.L. Modification of chitosan membrane with poly(vinyl alcohol) and biocompatibility evaluation. Int. J. Biol. Macromol. 2012, 50, 658–663. [Google Scholar] [CrossRef]
- Naveen Kumar, H.M.P.; Prabhakar, M.N.; Venkata Prasad, C.; Madhusudhan Rao, K.; Ashok Kumar Reddy, T.V.; Chowdoji Rao, K.; Subha, M.C.S. Compatibility studies of chitosan/PVA blend in 2% aqueous acetic acid solution at 30 °C. Carbohydr. Polym. 2010, 82, 251–255. [Google Scholar] [CrossRef]
- Shetta, A.; Kegere, J.; Mamdouh, W. Comparative study of encapsulated peppermint and green tea essential oils in chitosan nanoparticles: Encapsulation, thermal stability, in-vitro release, antioxidant and antibacterial activities. Int. J. Biol. Macromol. 2019, 126, 731–742. [Google Scholar] [CrossRef]
- Kahdestani, S.A.; Shahriari, M.H.; Abdouss, M. Synthesis and characterization of chitosan nanoparticles containing teicoplanin using sol–gel. Polym. Bull. 2020. [Google Scholar] [CrossRef]
- Ong, S.G.M.; Ming, L.C.; Lee, K.S.; Yuen, K.H. Influence of the encapsulation efficiency and size of liposome on the oral bioavailability of griseofulvin-loaded liposomes. Pharmaceutics 2016, 8, 25. [Google Scholar] [CrossRef]
- Kanaze, F.I.; Kokkalou, E.; Niopas, I.; Georgarakis, M.; Stergiou, A.; Bikiaris, D. Dissolution enhancement of flavonoids by solid dispersion in PVP and PEG matrixes: A comparative study. J. Appl. Polym. Sci. 2006, 102, 460–471. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.; Kanaze, F.I.; Karavas, E.; Stergiou, A.; Georgarakis, E. Tailoring the release rates of fluconazole using solid dispersions in polymer blends. Drug Dev. Ind. Pharm. 2008, 34, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, G.Z.; Bikiaris, D.; Karavas, E.; Politis, S.; Docoslis, A.; Park, Y.; Stergiou, A.; Georgarakis, E. Effect of physical state and particle size distribution on dissolution enhancement of nimodipine/PEG solid dispersions prepared by melt mixing and solvent evaporation. AAPS J. 2006, 8, 623–631. [Google Scholar] [CrossRef]
- Karavas, E.; Georgarakis, E.; Sigalas, M.P.; Avgoustakis, K.; Bikiaris, D. Investigation of the release mechanism of a sparingly water-soluble drug from solid dispersions in hydrophilic carriers based on physical state of drug, particle size distribution and drug-polymer interactions. Eur. J. Pharm. Biopharm. 2007, 66, 334–347. [Google Scholar] [CrossRef]
- Bikiaris, D.N. Solid dispersions, Part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef]
- Bikiaris, D.N. Solid dispersions, Part II: New strategies in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1663–1680. [Google Scholar] [CrossRef]
- Karavas, E.; Georgarakis, E.; Bikiaris, D.; Thomas, T.; Catsos, V.; Xenakis, A. Hydrophilic matrices as carriers in felodipine solid dispersion systems. Prog. Colloid Polym. Sci. 2001, 118, 149. [Google Scholar] [CrossRef]
- Hattori, Y.; Haruna, Y.; Otsuka, M. Dissolution process analysis using model-free Noyes-Whitney integral equation. Colloids Surf. B Biointerfaces 2013, 102, 227–231. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion; Oxford University press: Oxford, UK, 1975. [Google Scholar]
- Tien, C. Adsorption Calculations and Modeling; Butterworth-Heinemann: Boston, MA, USA, 1994. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Z-Average (d.nm) | PdI | Zeta Potential (mV) |
|---|---|---|---|
| CS-TPP-BUD 10% | 363 | 0.52 | +36.3 |
| CS-TPP-BUD 20% | 394 | 0.60 | +37.6 |
| CS-TPP-BUD 30% | 443 | 0.53 | +46.9 |
| CS-TPP-PVA-BUD 10% | 416 | 0.26 | +38.6 |
| CS-TPP-PVA-BUD 20% | 448 | 0.35 | +43.9 |
| CS-TPP-PVA-BUD 30% | 543 | 0.45 | +48.7 |
| Sample | Yield (%) | Drug Loading (%) | EE (%) |
|---|---|---|---|
| CS-TPP-BUD 10% | 38.1 | 15.4 | 64.5 |
| CS-TPP-BUD 20% | 41.8 | 19.5 | 48.8 |
| CS-TPP-BUD 30% | 44.2 | 15.6 | 29.8 |
| CS-TPP-PVA-BUD 10% | 38.2 | 17.7 | 74.5 |
| CS-TPP-PVA-BUD 20% | 45.1 | 20.2 | 54.5 |
| CS-TPP-PVA-BUD 30% | 49.3 | 21.2 | 36.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michailidou, G.; Ainali, N.M.; Xanthopoulou, E.; Nanaki, S.; Kostoglou, M.; Koukaras, E.N.; Bikiaris, D.N. Effect of Poly(vinyl alcohol) on Nanoencapsulation of Budesonide in Chitosan Nanoparticles via Ionic Gelation and Its Improved Bioavailability. Polymers 2020, 12, 1101. https://doi.org/10.3390/polym12051101
Michailidou G, Ainali NM, Xanthopoulou E, Nanaki S, Kostoglou M, Koukaras EN, Bikiaris DN. Effect of Poly(vinyl alcohol) on Nanoencapsulation of Budesonide in Chitosan Nanoparticles via Ionic Gelation and Its Improved Bioavailability. Polymers. 2020; 12(5):1101. https://doi.org/10.3390/polym12051101
Chicago/Turabian StyleMichailidou, Georgia, Nina Maria Ainali, Eleftheria Xanthopoulou, Stavroula Nanaki, Margaritis Kostoglou, Emmanuel N. Koukaras, and Dimitrios N. Bikiaris. 2020. "Effect of Poly(vinyl alcohol) on Nanoencapsulation of Budesonide in Chitosan Nanoparticles via Ionic Gelation and Its Improved Bioavailability" Polymers 12, no. 5: 1101. https://doi.org/10.3390/polym12051101
APA StyleMichailidou, G., Ainali, N. M., Xanthopoulou, E., Nanaki, S., Kostoglou, M., Koukaras, E. N., & Bikiaris, D. N. (2020). Effect of Poly(vinyl alcohol) on Nanoencapsulation of Budesonide in Chitosan Nanoparticles via Ionic Gelation and Its Improved Bioavailability. Polymers, 12(5), 1101. https://doi.org/10.3390/polym12051101