1. Introduction
Polymer brushes are layers of polymer chains with the end link firmly (almost irreversibly) connected to the surface. Recently, the anniversary of their appearance in polymer science was celebrated [
1,
2]. They are widely used for various modifications of the properties of surfaces [
3,
4,
5]. One of the areas of application of polymer brushes is a stabilization of colloidal and nanoparticles in solutions and filler nanoparticles in polymer nanocomposites. First of all, decorating of the particle by the brush has to provide a possibility to include particles into the matrix, even by the mutual incompatibility of them [
3,
4,
5,
6]. On the other hand, the distribution of such particles in the medium depends on the interaction between grafted brushes. If the brushes repel each other, this helps to avoids segregation of polymer modified particles and ensures a more uniform distribution of them in the matrix. The structure of the brush and its interaction with other brushes depends on the molecular characteristics of grafted chains such as the chemical structure and the molecular weight, charging, the grafting density, the matrix properties as well as on the geometry of the surface. In the present work, we will limit ourselves to considering planar polymer brushes formed by neutral homopolymers.
There are a lot of works where so-called conventional brushes were studied theoretically. The structure of such brushes depends on the solvent strength and is determined by a compromise between the tendency to decrease the brush density due to volume interactions and the need for this to stretch the grafted chains, losing conformational entropy. In a good or θ-solvent (Flory–Huggins parameter χ ≤ 0.5) the brush is “wet”, it contains a large amount of solvent, becoming “dry” only at extremely high grafting density. The main feature of the structure is the strong extension of the grafted chains and a wide distribution in the degree of extension. The brush is not uniform in the density, which is maximum at the grafting surface and drops to zero towards the periphery (see, for example, the review [
7]). The opposing brushes are virtually mutually impenetrable and repel each other when approaching due to the volume interactions.
The substitution of the low molecular solvent by the high molecular melt changes both the structure of conventional brushes and the interaction between them. Such a situation, in particular, arises in nanocomposites when nanofillers decorated by the brush are immersed into the polymer melt consisting of flexible chains chemically identical to the grafted ones. The chemical identity of the grafted chains in the brush and the macromolecules of the polymer melt (χ = 0) is a sufficient (although not necessary) condition to overcome incompatibility between the filler and the melt. Under these conditions, free polymer chains of the melt screen the volume interactions of the monomer units of grafted macromolecules. As a result, the brush becomes denser than in the solution, and the stretching degree of grafted chains decreases. With sparse grafting, the chains do not stretch, so the brush is a system of overlapping Gaussian coils and contains free polymer (“wet” brush). At large grafting densities, the stretched chains form an almost dry brush without melt chains (except for the peripheral part) [
2,
8,
9].
The theoretical study of the problem of the mutual interaction of such brushes in a polymer matrix has a long history [
8,
9,
10,
11,
12,
13,
14]. The main result is the existence of the weak short-range attraction between them. This attraction is connected with the tendency of grafted chains of the brush to mix more easily with grafted chains from the other brush than with the free polymer chains in the melt. The above-mentioned conclusions concerning the structure and interaction of conventional brushes in different media were confirmed in a large number of experimental and computer simulation studies (see [
7] for a review).
Recently, Glova et al. [
15,
16] have found a new “unusual structure” of a brush in a chemically equivalent polymer environment. By using the fully atomistic molecular dynamics (MD), they have studied the structure of the brush consisting of the lactic acid oligomers (OLA) covalently grafted to the cellulose nanocrystals (CNC) immersed into the polylactic acid (PLA) matrix. Polylactide-based bionanocomposites are a promising class of materials [
12,
17]. It turned out that the grafted chains separate in two populations: the extended chains with the ends on the brush periphery and backfolded ones («hairpins») with the ends near the grafting surface. The analysis has shown that such an unusual behavior is caused by the interaction of atomic partial charges in the brush. By exclusion of electrostatic interactions, the brush structure becomes similar to that for conventional brushes.
Theoretical studies [
18] made by using the lattice Scheutjens–Fleer self-consistent field (SF-SCF) method have shown that such an unusual structure arises due to the presence of longitudinal dipole moments in grafted macromolecules. These dipole moments are connected with the distribution of partial charges and are directed along the monomers of the grafted chains. When all chains of the brush are grafted onto the surface via the same end, the longitudinal dipoles of all stretched chains are directed in the same way. This causes the appearance of the population of backfolded chains.
Note that the longitudinal component of the dipole moment can arise in macromolecules with three or more different bonds in monomer units of the main chain (polyesters, in particular PLA, or polypeptides, for example). For reasons of symmetry polar macromolecules with two different bonds in the monomer unit such as, for example, polyvinyl chloride, cannot have a longitudinal component of the dipole moment [
19]. Following the classification of Stockmayer [
17], in what follows, we will call macromolecules with longitudinal dipole moments macromolecules of type A. We will also call the brushes formed by A-type chains, which are grafted with the same ends A-type brushes.
The obvious question arises how this unusual structure will affect the mutual interactions of brushes in polymer medium. The answer to this question is one of the goals of the present work. It should be mentioned the theoretical works where dipolar brushes in polar solvent [
20,
21,
22,
23] were considered. In contrast to our case the directions of dipoles in the grafted chains were not correlated with the directions of monomer units. The dipole moments of macromolecules in this case do not contain longitudinal components and are directed perpendicular to the chain length vectors (contain only a transverse components). Such macromolecules and brushes made of such grafted macromolecules are not type A systems, which are considered in our work. Kumar et al. [
20], Mahalik et al. [
21], Budkov et al. [
22], Gordievskaya et al. [
23], show that the effect of dipole-dipole interaction between transverse dipoles can be described by the introduction of the macroscopic effective Flory–Huggins-like parameter which depends on the polymer concentration. These interactions can lead to the collapse of the dipolar flat brushes and to an attraction between two opposite brushes at intermediate separation distances [
24]. To avoid misunderstandings, A-type brushes were not considered in these works.
Another goal of the present work is to study the influence of the limited flexibility of the grafted chains and free chains in the polymer matrix on the internal structure and interaction of brushes. The formulation of this problem is motivated by the results of further atomistic MD modeling [
25], which demonstrated the difference in the structure of brushes formed on the surface of CNCs particles by grafted OLA and oligohydroxybutyrate (OHB) in melts of PLA and polyhydroxybutyrate (PHB) chains, respectively. The fraction of “hairpins” in the OLA brush was larger than that in the OHB brush at the same grafting densities. The authors have concluded that this difference is caused by the lower flexibility of PLA macromolecules as compared to PHB ones.
It should be noted that the effect of limited flexibility of macromolecules on the structure and mutual interaction of brushes in the polymer melt has not been studied even for conventional brushes. For conventional brushes in solution, such an analysis was carried out already at the earliest stage in the development of theoretical studies of brushes [
10,
13,
14].
All the studies in the present work were performed by using the numerical lattice SF-SCF method, which has proven effective for many polymer systems, including polymer brushes.
The outline of the paper is the following one. In
Section 2, the model of the brush and method are described. The diagram of states of a brush formed by macromolecules of limited flexibility in a polymer melt and the results for a single planar dipolar brush immersed into the melt will be presented in
Section 3.1. The effect of such parameters as the polymerization degree
N, grafting density
σ, parameter τ of the dipole-dipole interactions, and the Kuhn segment length
p on the brush structure will be studied. In
Section 3.2, the effect of the above-mentioned parameters on the mutual interaction of two such brushes will be studied.
Section 4 concludes the paper.
2. Model and Method
In present work, a coarse-grained model of the planar polymer brushes consisting of polymer chains pinned by one end to a flat impenetrable surface was used. It is assumed that all monomer units in chain are identical and have the same linear size a. The value of a is used as the unit length.
The grafting density of the chains is characterized by the dimensionless ratio σ = a2/s, where s is an average surface area per one grafted macromolecule.
To investigate the properties of dipolar brushes, the Scheutjens–Fleer self-consistent field (SF-SCF) method [
26,
27] was used. In the SF-SCF method, all the various interactions between the particles of the system are taken into account not explicitly but through a mean effective field acting on the given particle. This reduces the problem of the determination of properties of the system consisting of many polymer chains to the problem of the behavior of a single chain in the effective field. This problem is quite easy to solve with a relatively low computational cost. The disadvantage of the mean field approximation as compared with more detailed methods (Monte-Carlo, molecular or stochastic dynamics) is its inability to take into account the correlations in the system. However, this disadvantage weakly affects the statistical properties of the brushes. Advantages of SF-SCF method are high computational efficiency combined with the accuracy of the results reproducibility.
We use the lattice numerical SF-SCF method. The space is taken to be composed of cubic lattice sites with characteristic length a and the volume per cell a3. Due to the symmetry of the system all brush characteristics change only along z axis directed normally to the grafting surface. The space is divided into layers parallel to the surface which are numbered from z = 0 to zmax. Layer z = 0 corresponds to the grafting surface, the first segments of all grafted chains are located in the layer z = 1. When two opposite brushes are considered, the opposite grafting surface is located in the layer (z = zmax + 1) and the first segments of chains belonging to another brush are in the layer z = zmax. The distance between the two grafting surfaces is D = zmax·a. The chain grafting is supposed to be sufficiently dense, so the chains overlap strongly and form a relatively uniform brush. Under these conditions, the polymer volume fraction ϕ and the exchange chemical potential u remain constant within the same layer and have one nonzero gradient in z-direction.
The grafted chains are modeled as lattice chains with restricted flexibility in an external effective field
u(
z), which describes the effect of the inter- and intramolecular interactions. Two adjacent monomer units of the chain occupy neighboring lattice sites. However, the long range correlations are ignored. Hence, the overlap of two monomer units on the same lattice site is, in principle, allowed, but is prevented effectively through the incompressibility condition, which is to be fulfilled in each layer:
Hereinafter, the subscripts indicate that the parameter belongs to the polymer chain of the type 1, type 2 or to the solvent (s) molecule, respectively.
The chain stiffness is characterized by the Kuhn segment length
p, which obeys the equation [
28]:
where
γ =
π −
θ is the supplementary angle to the valence angle
θ.
In this model, the correlation between two consecutive chain links is given by the potential:
where
K is a coefficient of the bonds correlation.
On the cubic lattice, two consecutive bonds can have three relative orientations: (1) straight conformation, where a bond makes an angle
θ =
π with the preceding one (that corresponds to
γ = 0 and
Ua(s) = 0); (2) perpendicular kink (
γ =
π/2 and
Ua(p) =
K)); (3) backfold “fracture” (
γ =
π and
Ua(b) = 2
K). The weighting factor
λs of the straight conformation and the weighting factor
λb of the backfold conformation are related to the weighting factor
λp of the perpendicular kink as
The sum of the weighting factors in the case of cubic lattice obeys the normalization condition:
The average cosine of the angle
γ can be calculated by the equation:
According to the above Equations (2) and (4)–(7), weight coefficient
λp can be found by the formulae:
where
Thus, using expressions (4), (5), (8) and (9), the weighting factors (λb, λp, λs) for different orientations of adjacent segments along the chain are uniquely determined through the length Kuhn segment, p. Note that the given model does not prohibit the reverse movement along the chain and allows the overlap of segments, but such overlapping is prevented through the incompressibility condition.
The grafted chains are immersed in the melt of exactly the same chains. The free energy per unit area of the grafting surface is calculated as the energy of dipole-dipole interactions
Fd−d and the sum of the negative logarithms of the partition functions
QX of all components (
X = 1,2,
s) in a constant effective external fields
minus the work of these fields
(according to the Legendre transform):
where
σX is the
X-chain grafting density.
The main goal of SCF calculations is to determine the system characteristics which correspond to the minimum of the free energy under the incompressibility condition. This goal is achieved through the optimization of the functional:
where
α(
z) is Lagrange field (set of Lagrange multipliers). The Lagrange field
α(
z) corresponding to the minimum of functional
(Equation (11)) is calculated during the iterative procedure of gradient descent:
where
η < 1/2 is the convergence step size, which corresponds to the highest convergence rate.
Minimization of
with respect to the volume fractions
ϕX(
z) allows to calculate the potential fields
uX(
z):
Minimization of
with respect to the potential fields
uX(
z) provides a way to calculate the volume fraction distributions
ϕX(
z):
For the calculation of the energy of dipole-dipole interactions
Fd−d we apply a mean-field approach used in [
18]:
Here
τ is the dimensionless parameter of dipole-dipole interactions (which is proportional to the dipole moment µ of the monomer unit),
s1(
z) is the first order parameter of dipoles in
z-th layer. Based on the assumption that the dipoles are directed along the bonds, the order parameter has the form:
where
ϕ(
z,
d = 1) and
ϕ(
z,
d = −1) are volume fractions of segments oriented in different directions
d relative to the grafting surface. There are three possible directions d of monomer unit in each lattice layer: from layer
z to layer
z − 1 (
d = −1), within layer
z (
d = 0), or from layer
z to layer
z + 1 (
d = 1). The potential of dipole-dipole interactions acts on the monomer depending on its orientation in space:
The value of
τ can be expressed as:
where
is the relative dielectric constant of the polymer melt and
is the vacuum electric permittivity.
The analytical solution of Equations (13) and (14) for the case of polymer molecules is rather difficult. They are solved by using the iterative procedure which also takes into account the linking of monomer units in chains.
The relative preference of any monomer unit to be in layer z with respect to the bulk melt is determined by the statistical Boltzmann weight:
The chain conformation is considered as a set of trajectories of particle diffusion from fixed to free end (forward propagate) and vice versa from free to fixed end (back propagate). Both processes are characterized by the probability density matrices of monomer units. These matrices are called forward
Gf and back
Gb propagators. Propagation matrices also differ in directions (
d = −1, 0, 1). The way to “fill” these matrices is as follows: first, initial conditions are set up. For instance, for a polymer grafted to a surface (
z = 0), the initial conditions are as follows
which means that the first monomer unit (
s = 1) of the chain is fixed in layer
z = 1, and the last segment (
s =
N) can be in all accessible layers. In the case of a solvent molecule, which is also a chain of length
N, both end segments are free:
Each next step of the propagators
Gf(
z,
s,
d) and
Gb(
z,
s,
d) is computed during iterations respect with
s and
z. This procedure is described in detail in [
29,
30].
The statistical weight
q(
z,
s,
d) of the chain conformations in which the
s-th segment with orientation
d is in the
z-th layer can be represented as a composition of two propagators (the composition law) [
27]:
The total partition function of chain is equal to
The product of propagators gives the volume fraction profile:
where
C is a normalization constant. For the free chain in melt
C = 1/
N. For the polymer grafted in the first layer, this constant is equal to
that ensures the normalization of the volume fraction profile:
The volume fraction profile can also be split into three components depending on the segment orientation:
Under described approach, chain conformations are considered using a second-order Markov model. This implementation of Markov formalism for chain stiffness was proposed in pioneering works [
29,
30,
31,
32].
To undertake the above calculations, the special software was developed.
4. Conclusions
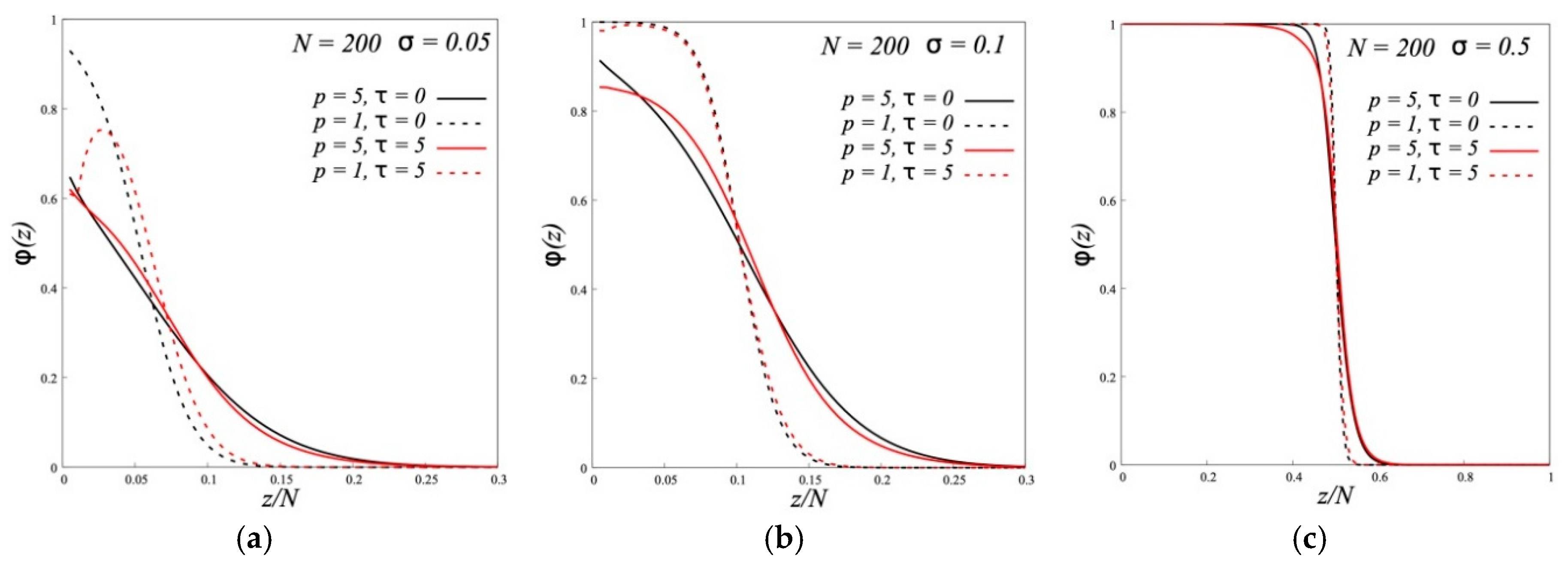
By using the numerical lattice SF-SCF method, we have studied the effect of the increase in the Kuhn length of grafted chains on the structure and mutual interaction of two opposing planar non-polar and A-type dipolar brushes. Brushes are immersed in the solvent consisting of chains similar to the grafted ones. The degrees of polymerization as well as the parameters of flexibility and dipole-dipole interactions are the same for free chains in solution and chains in a brush.
It is shown that for brushes made of semi-rigid chains, as for brushes made of flexible chains, the modes of wet and dry brush are possible at low and high grafting densities, respectively. Reducing the chain flexibility leads to the loosening of the brush and to the increase of its thickness in the case of wet brush and to more gentle decline in brush density at the brush periphery, which contains free polymer, in the case of “dry” brush.
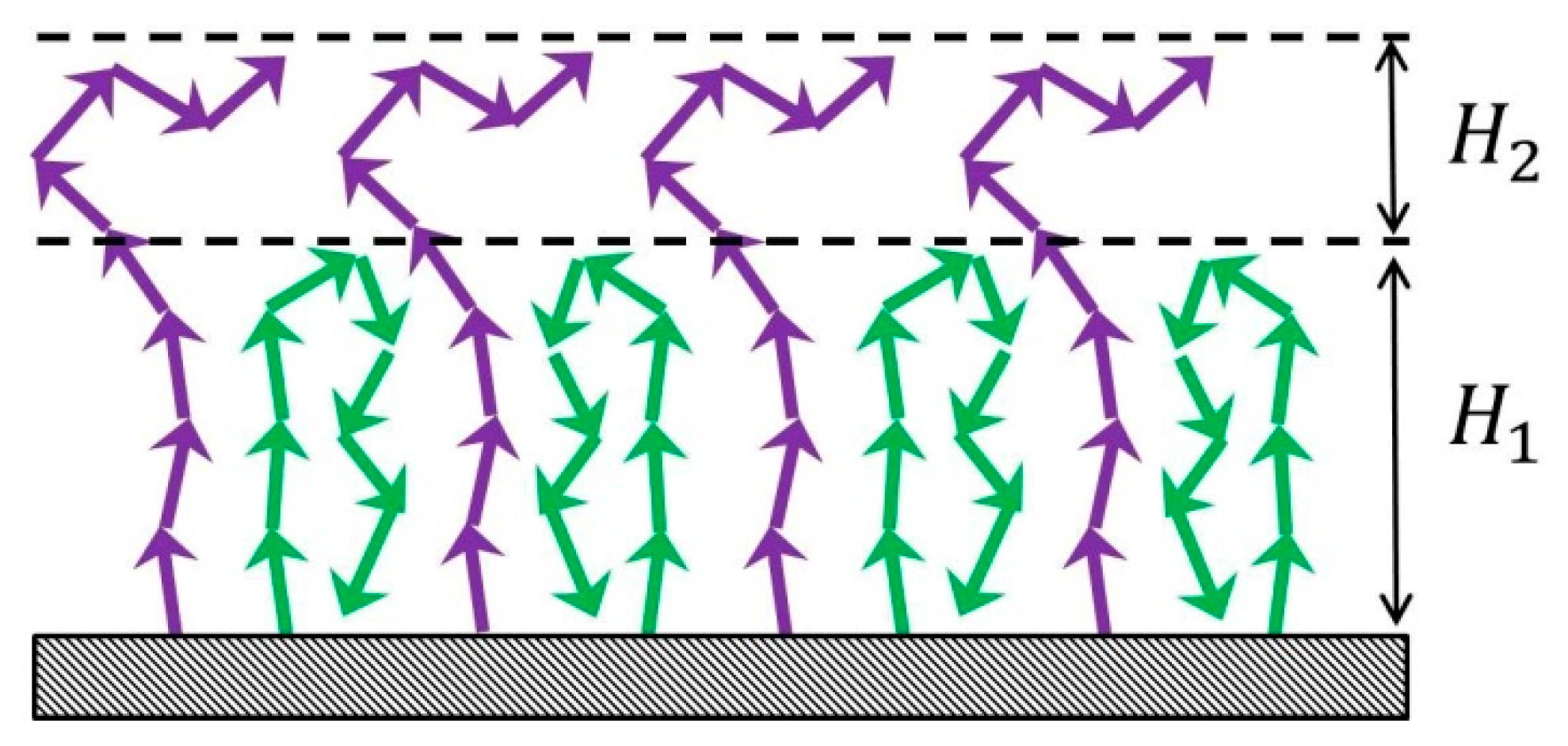
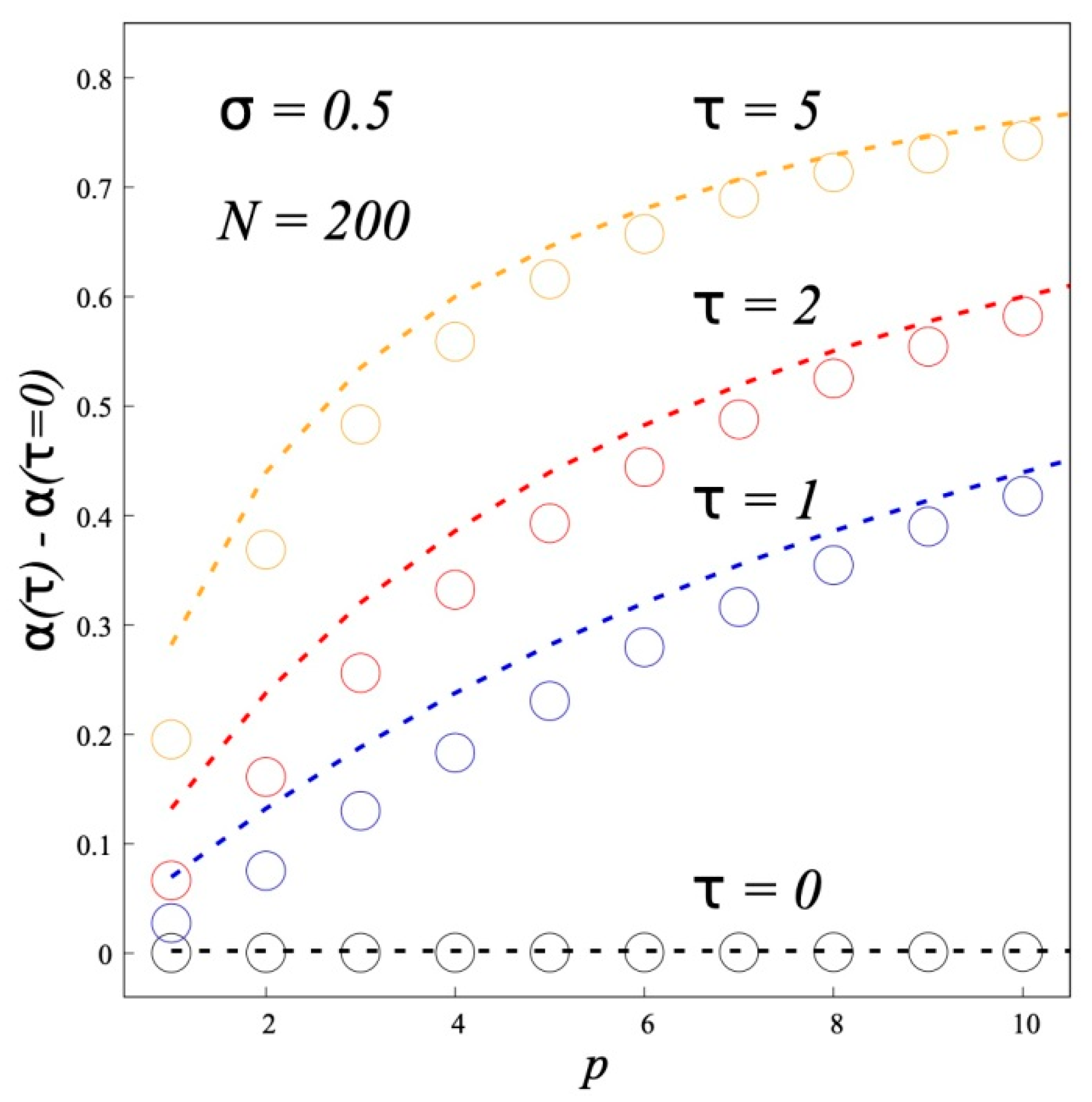
For unusual structure of A-type dipolar brushes the increase of the chain rigidity enhances the separation of grafted chains in a brush into two populations: backfolded chains with terminal monomers near the grafting surface and chains with the ends at the brush periphery. The fraction of backfolded chains grows by increase of the Kuhn segment length.
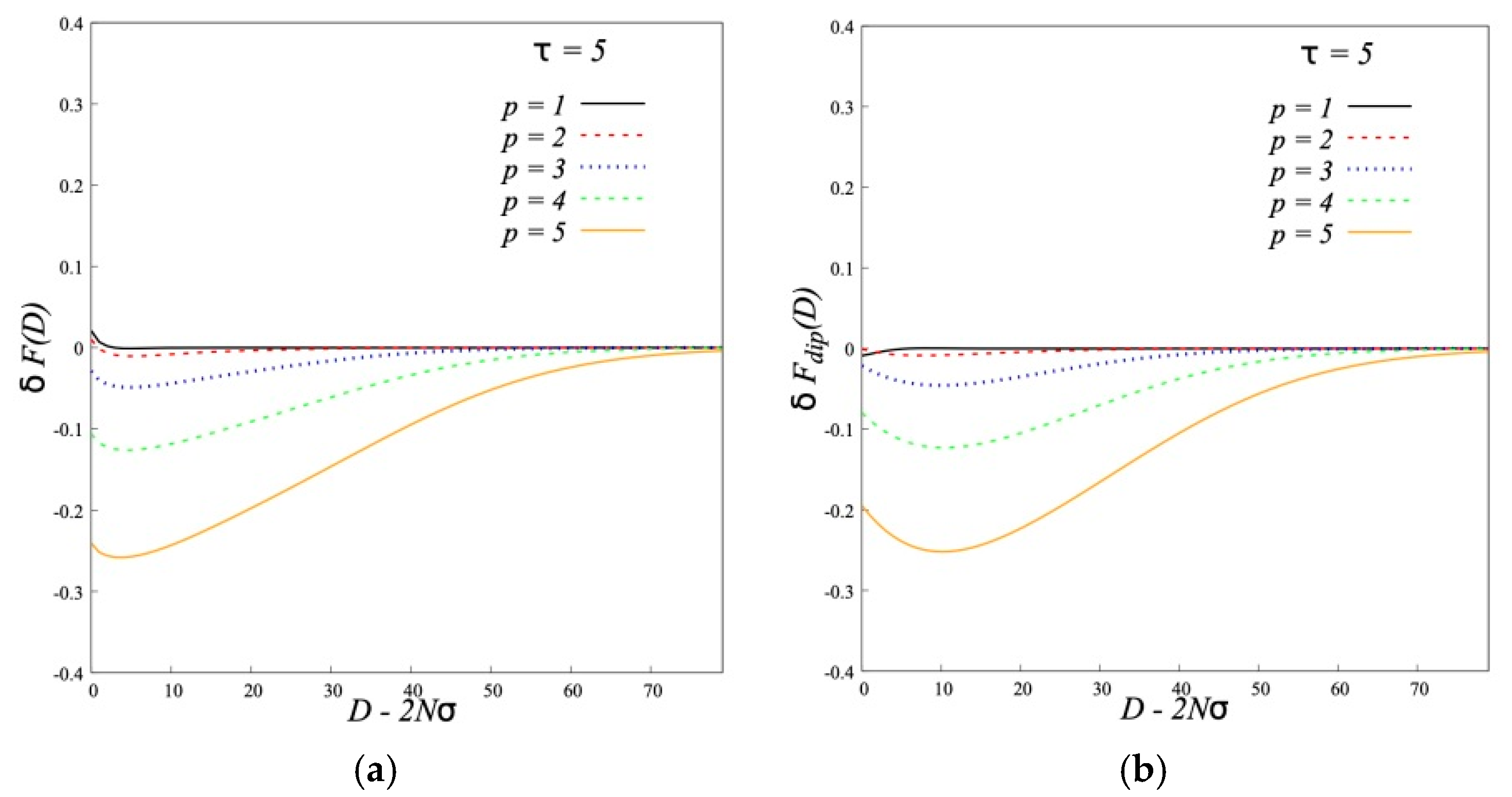
As to the interaction of opposite A-type dipolar brushes our main result is that the brushes from semi-rigid chains are attracted to each other at short distances. The attraction becomes more pronounced and begins at larger distances for more rigid chains at the same brush characteristics (polymerization degree, grafting density and dipole moments of monomer units). This effect of the chain rigidity is opposite to that for conventional brushes without dipoles in the chains. For such brushes the growth of the chain rigidity leads to the enhanced repulsion between them.
It is shown that the strong attraction of A-type dipolar brushes is energetically favorable and is connected with the dipole-dipole interactions. Chains of opposing brushes with oppositely directed dipoles penetrate deeply into each other upon contact. The decrease of fraction of «hairpins» shows that some backfolded chains unfold and penetrate into the opposite brush.
Note that the deep interpenetration of opposing brushes predicted in this work for unusual brushes was previously discussed for brushes with mesogenic groups in the backbone of semi-rigid grafted chains, undergoing a phase transition to a liquid-crystalline (LC) state [
30,
34]. The brushes were placed in a low molecular weight solvent and the transition was initiated by an increase in the orientation-dependent interaction of mesogenic groups. This transition occurs through the micro (nano)phase separated state of a brush (vertical phase segregation).
In this state, the grafted chains are divided into two populations [
35]: a set of folded chains that form a dense internal LC microphase, and a set of more extended chains that form a microphase containing a solvent at the periphery of the brush. Upon contact of the brushes, the chains stretch, penetrating into the counter brush and forming a single LC phase. Brushes attract each other and even stick together. This is somewhat similar to what happens when A-type bipolar brushes made of semi-rigid chains approach each other, although the common property of the considered systems is only orientation-dependent interaction in the brushes.
All obtained results should be taken into account using the grafting of polymer chains on the surface of nanoparticles to prevent their aggregation in the polymer matrix. It is possible to obtain repulsive brushes from the A-type macromolecules by the alternative grafting of them at different ends. In this case the longitudinal dipole moments of any pair of differently grafted chains are directed antiparallel. As it was shown [
18] by SF-SCF method, and confirmed for OLA brush by fully atomistic MD simulations the structure of such a brush is similar to the structure of conventional brushes without dipoles. As shown in this paper, conventional brushes from semi-rigid chains repel each other. As it was shown in [
10], repulsive brushes can be obtained also in the case of chemical non-identity of the grafted chains in the brush and polymer macromolecules under the additional condition of attraction between them (the Flory interaction parameter χ < 0).



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}