Multiscale Experimental Evaluation of Agarose-Based Semi-Interpenetrating Polymer Network Hydrogels as Materials with Tunable Rheological and Transport Performance

 , , , and
, , , and
Abstract
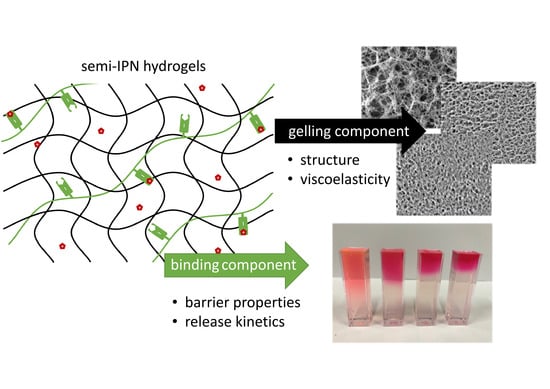
1. Introduction
2. Materials and Methods
2.1. Preparation of the Gels
2.2. Turbidimetry
2.3. Oscillatory Rheometry
2.4. Microrheometry
2.5. Macroscopic Diffusion Experiments
2.6. Fluorescence Correlation Spectroscopy
2.7. Scanning Electron Microscopy Imaging
3. Results
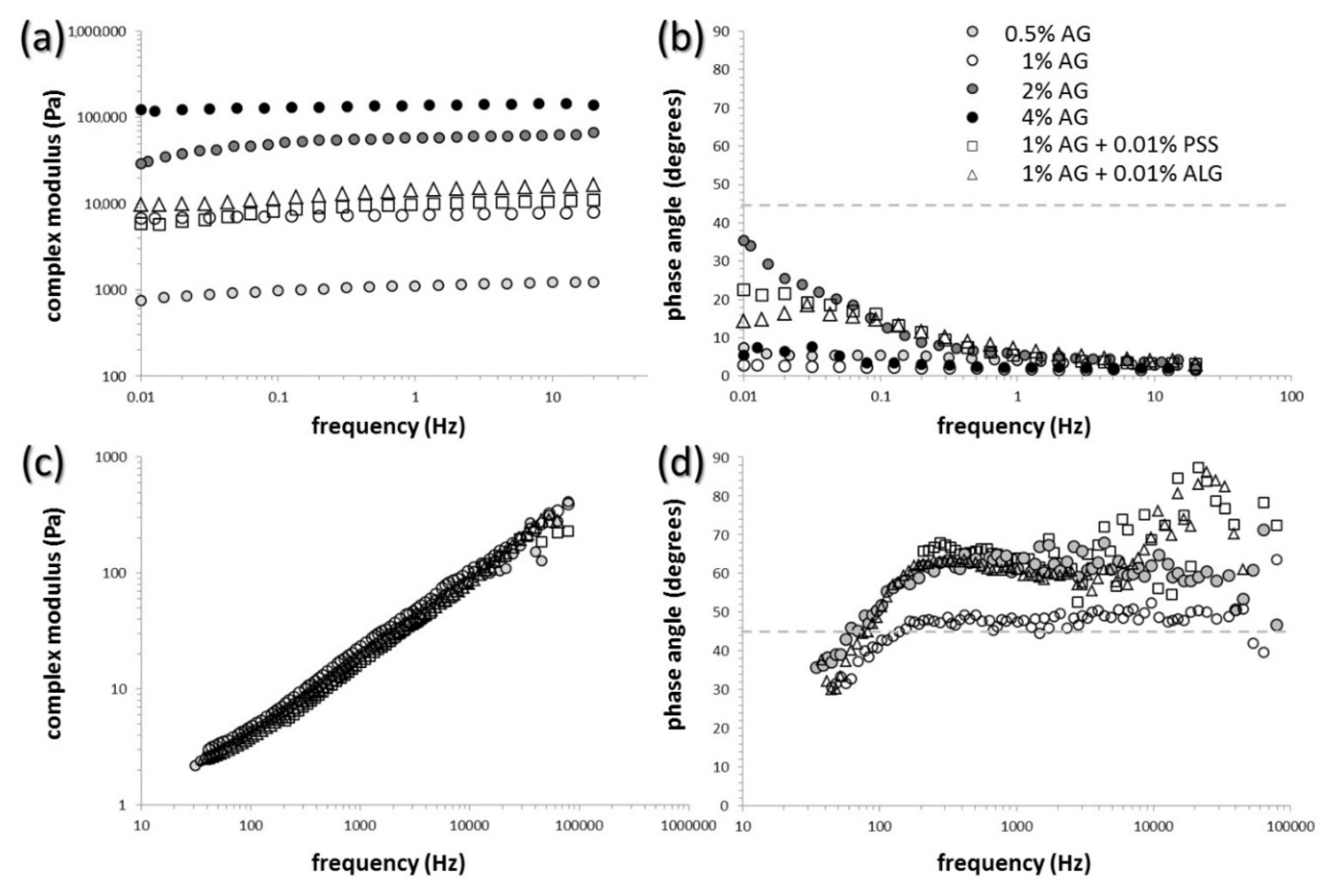
3.1. Mechanical Properties of the Gels
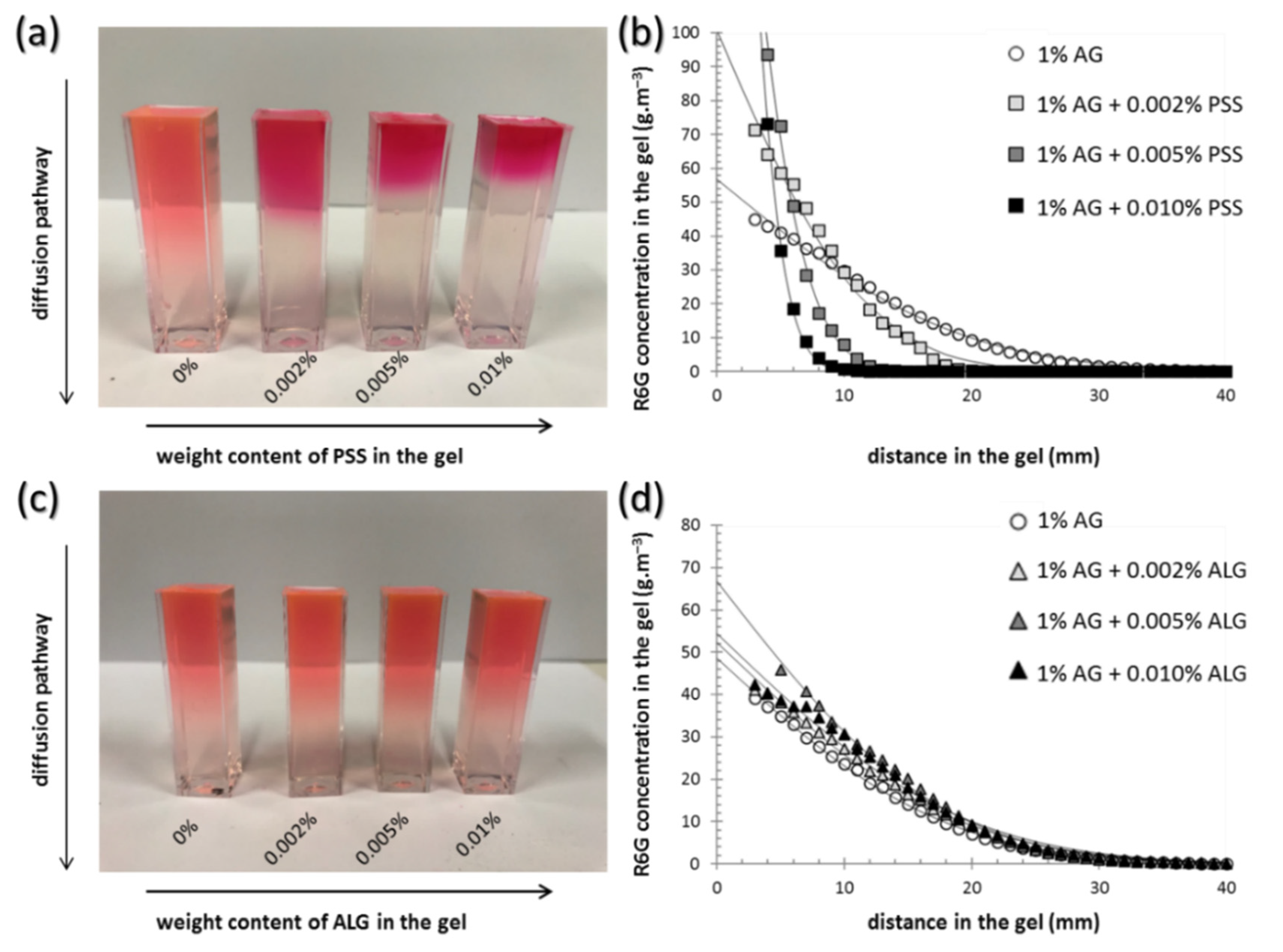
3.2. Transport Properties of the Gels
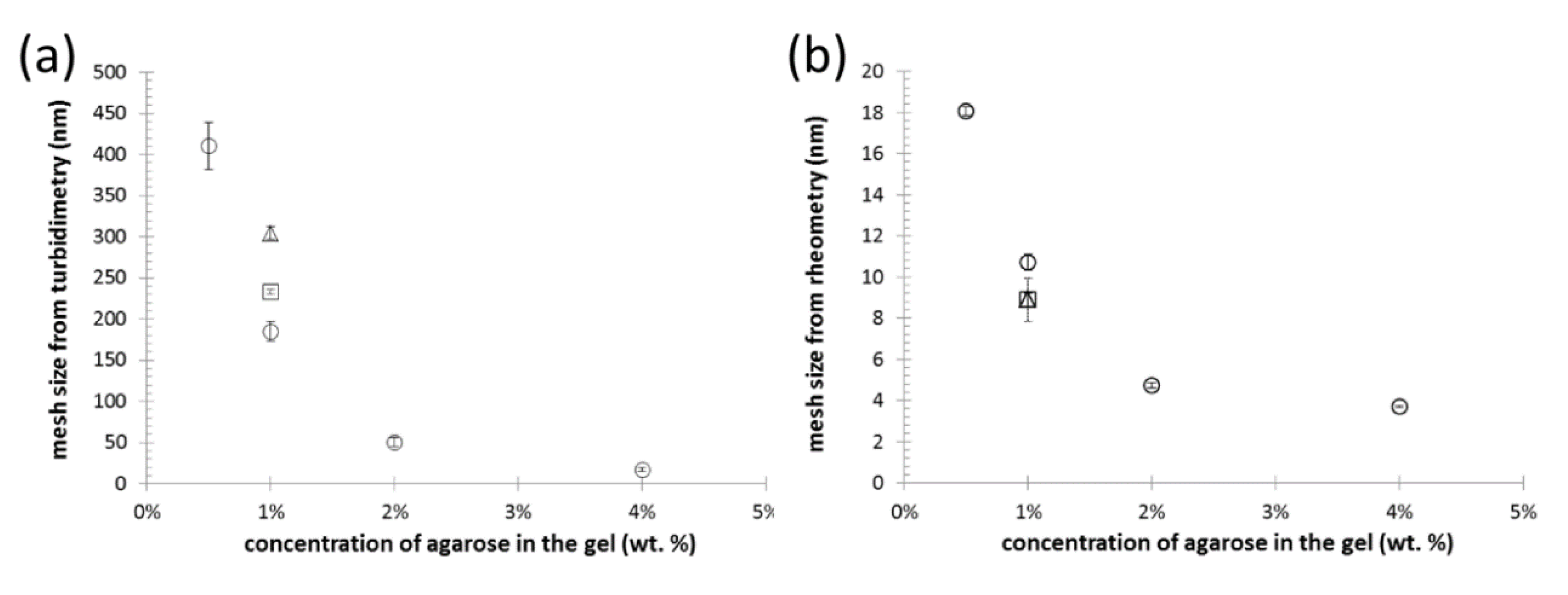
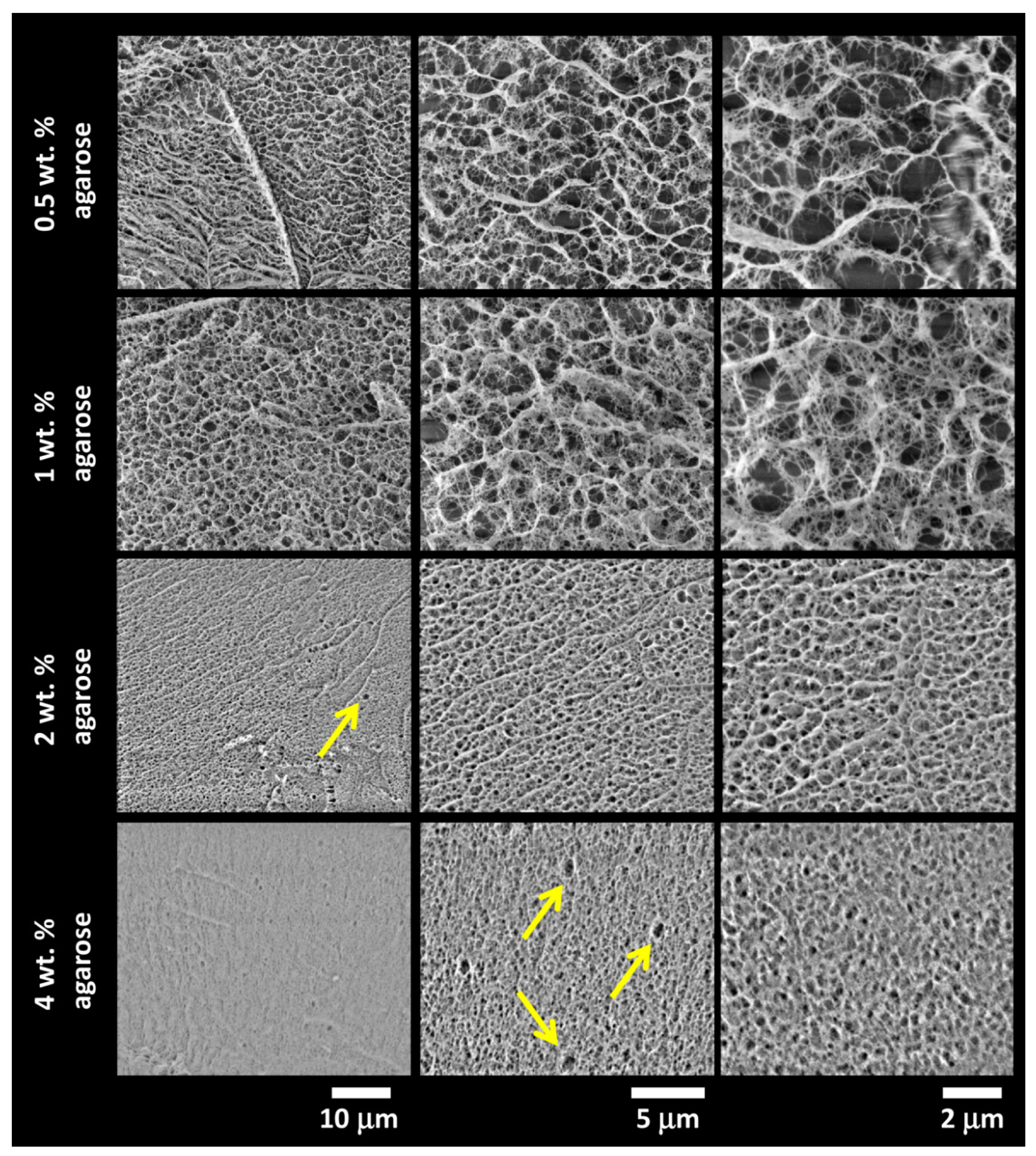
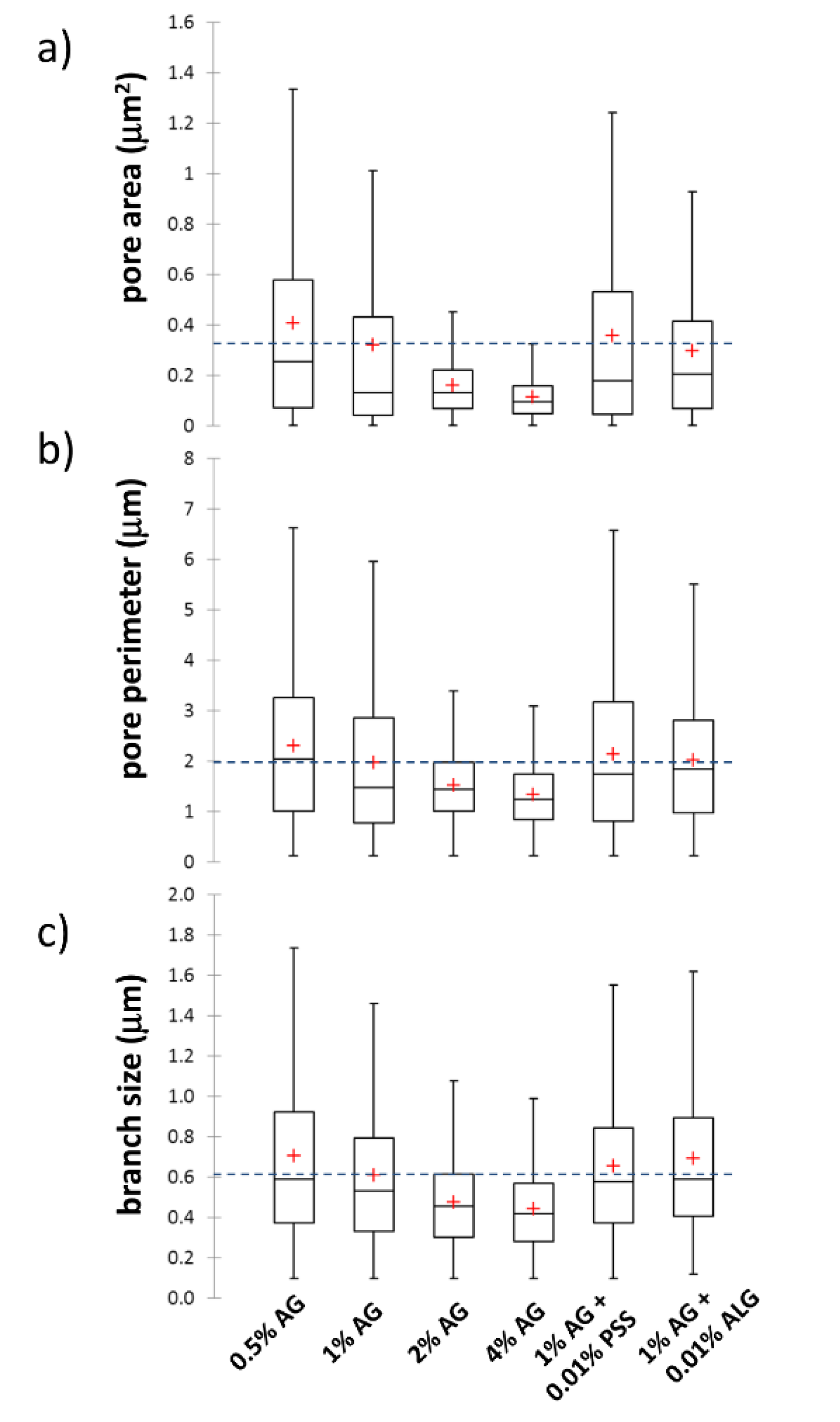
3.3. Characterization of the Internal Structure of the Gels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Kopeček, J. Hydrogel biomaterials: A smart future? Biomaterials 2007, 28, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Geckil, H.; Xu, F.; Zhang, X.; Moon, S.; Demirci, U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine 2010, 5, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Drumright, R.; Siegwart, D.J.; Matyjaszewski, K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008, 33, 448–477. [Google Scholar] [CrossRef]
- Vinogradov, S.V.; Bronich, T.K.; Kabanov, A.V. Nanosized cationic hydrogels for drug delivery: Preparation, properties and interactions with cells. Adv. Drug Deliver. Rev. 2002, 54, 135–147. [Google Scholar] [CrossRef]
- Tokarev, I.; Minko, S. Stimuli-responsive hydrogel thin films. Soft Matter 2009, 5, 511–524. [Google Scholar] [CrossRef]
- Ishihara, K.; Ueda, T.; Nakabayashi, N. Preparation of Phospholipid Polylners and Their Properties as Polymer Hydrogel Membranes. Polym. J. 1990, 22, 355–360. [Google Scholar] [CrossRef]
- Klouda, L.; Mikos, A.G. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [Google Scholar] [CrossRef]
- Klouda, L. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2015, 97, 338–349. [Google Scholar] [CrossRef]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliver. Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef]
- Miyata, T.; Asami, N.; Uragami, T. A reversibly antigen-responsive hydrogel. Nature 1999, 399, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Uragami, T.; Nakamae, K. Biomolecule-sensitive hydrogels. Adv. Drug Deliver. Rev. 2002, 54, 79–98. [Google Scholar] [CrossRef]
- Spiller, K.L.; Laurencin, S.J.; Charlton, D.; Maher, S.A.; Lowman, A.M. Superporous hydrogels for cartilage repair: Evaluation of the morphological and mechanical properties. Acta Biomater. 2008, 4, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Park, H.; Park, K. Synthesis of superporous hydrogels: Hydrogels with fast swelling and superabsorbent properties. J. Biomed. Mater. Res. 1999, 44, 53–62. [Google Scholar] [CrossRef]
- Petka, W.A.; Harden, J.L.; McGrath, K.P.; Wirtz, D.; Tirrell, D.A. Reversible Hydrogels from Self-Assembling Artificial Proteins. Science 1998, 281, 389–392. [Google Scholar] [CrossRef]
- Ehrick, J.D.; Deo, S.K.; Browning, T.W.; Bachas, L.G.; Madou, M.J.; Daunert, S. Genetically engineered protein in hydrogels tailors stimuli-responsive characteristics. Nat. Mater. 2005, 4, 298–302. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Yasuda, K.; Ping Gong, J.; Katsuyama, Y.; Nakayama, A.; Tanabe, Y.; Kondo, E.; Ueno, M.; Osada, Y. Biomechanical properties of high-toughness double network hydrogels. Biomaterials 2005, 26, 4468–4475. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in Regenerative Medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef]
- Annabi, N.; Tamayol, A.; Uquillas, J.A.; Akbari, M.; Bertassoni, L.E.; Cha, C.; Camci-Unal, G.; Dokmeci, M.R.; Peppas, N.A.; Khademhosseini, A. 25th Anniversary Article: Rational Design and Applications of Hydrogels in Regenerative Medicine. Adv. Mater. 2014, 26, 85–124. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, H.J.; Herber, S.; Olthuis, W.; Bergveld, P. Stimulus-sensitive hydrogels and their applications in chemical (micro)analysis. Analyst 2003, 128, 325–331. [Google Scholar] [CrossRef]
- Rubina, A.Y.; Pan’kov, S.V.; Dementieva, E.I.; Pen’kov, D.N.; Butygin, A.V.; Vasiliskov, V.A.; Chudinov, A.V.; Mikheikin, A.L.; Mikhailovich, V.M.; Mirzabekov, A.D. Hydrogel drop microchips with immobilized DNA: Properties and methods for large-scale production. Anal. Biochem. 2004, 325, 92–106. [Google Scholar] [CrossRef]
- Wang, K.L.; Burban, J.H.; Cussler, E.L. Hydrogels as separation agents. In Responsive Gels: Volume Transitions II.; Springer: Berlin/Heidelberg, Germany, 1993; pp. 67–79. [Google Scholar]
- Ozay, O.; Ekici, S.; Baran, Y.; Kubilay, S.; Aktas, N.; Sahiner, N. Utilization of magnetic hydrogels in the separation of toxic metal ions from aqueous environments. Desalination 2010, 260, 57–64. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Narayanaswamy, R.; Torchilin, V.P. Hydrogels and Their Applications in Targeted Drug Delivery. Molecules 2019, 24, 603. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 1–17. [Google Scholar] [CrossRef]
- Trongsatitkul, T.; Budhlall, B.M. Microgels or microcapsules? Role of morphology on the release kinetics of thermoresponsive PNIPAm-co-PEGMa hydrogels. Polym. Chem. 2013, 4, 1502–1516. [Google Scholar] [CrossRef]
- Perugini, P.; Genta, I.; Conti, B.; Modena, T.; Pavanetto, F. Long-term release of clodronate from biodegradable microspheres. AAPS PharmSciTech 2001, 2, 6–14. [Google Scholar] [CrossRef]
- Lee, B.H.; Lee, Y.M.; Sohn, Y.S.; Song, S.-C. A Thermosensitive Poly(organophosphazene) Gel. Macromolecules 2002, 35, 3876–3879. [Google Scholar] [CrossRef]
- Andrade-Vivero, P.; Fernandez-Gabriel, E.; Alvarez-Lorenzo, C.; Concheiro, A. Improving the Loading and Release of NSAIDs from pHEMA Hydrogels by Copolymerization with Functionalized Monomers. J. Pharm. Sci. 2007, 96, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Uchida, R.; Tanigawa, H.; Uno, K.; Murakami, A. Application of polymer gels containing side-chain phosphate groups to drug-delivery contact lenses. J. Appl. Polym. Sci. 2005, 98, 731–735. [Google Scholar] [CrossRef]
- Young, S.; Wong, M.; Tabata, Y.; Mikos, A.G. Gelatin as a delivery vehicle for the controlled release of bioactive molecules. J. Control. Release 2005, 109, 256–274. [Google Scholar] [CrossRef]
- Jenkins, A.D.; Kratochvíl, P.; Stepto, R.F.T.; Suter, U.W. Glossary of basic terms in polymer science (IUPAC Recommendations 1996). Pure Appl. Chem. 1996, 68, 2287–2311. [Google Scholar] [CrossRef]
- Zoratto, N.; Matricardi, P. Semi-IPNs and IPN-based hydrogels. In Polymeric Gels; Woodhead Publishing: Cambridge, UK, 2018; pp. 91–124. [Google Scholar]
- Aminabhavi, T.M.; Nadagouda, M.N.; More, U.A.; Joshi, S.D.; Kulkarni, V.H.; Noolvi, M.N.; Kulkarni, P.V. Controlled release of therapeutics using interpenetrating polymeric networks. Expert Opin. Drug Deliv. 2015, 12, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Rinaudo, M. Main properties and current applications of some polysaccharides as biomaterials. Polym. Int. 2008, 57, 397–430. [Google Scholar] [CrossRef]
- Zarrintaj, P.; Manouchehri, S.; Ahmadi, Z.; Saeb, M.R.; Urbanska, A.M.; Kaplan, D.L.; Mozafari, M. Agarose-based biomaterials for tissue engineering. Carbohydr. Polym. 2018, 187, 66–84. [Google Scholar] [CrossRef]
- Wang, N.; Wu, X.S. Preparation and Characterization of Agarose Hydrogel Nanoparticles for Protein and Peptide Drug Delivery. Pharm. Dev. Technol. 1997, 2, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Xu, J.; Weng, L.; Dai, H.; Zhang, X.; Zhang, L. Protein diffusion in agarose hydrogel in situ measured by improved refractive index method. J. Control. Release 2006, 115, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Marras-Marquez, T.; Peña, J.; Veiga-Ochoa, M.D. Agarose drug delivery systems upgraded by surfactants inclusion: Critical role of the pore architecture. Carbohydr. Polym. 2014, 103, 359–368. [Google Scholar] [CrossRef]
- Meilander, N.J.; Yu, X.; Ziats, N.P.; Bellamkonda, R.V. Lipid-based microtubular drug delivery vehicles. J. Control. Release 2001, 71, 141–152. [Google Scholar] [CrossRef]
- Narahashi, T.; Yamada, M.; Frazier, D.T. Cationic Forms of Local Anaesthetics block Action Potentials from Inside the Nerve Membrane. Nature 1969, 223, 748–749. [Google Scholar] [CrossRef]
- Gunasekaran, P.; Rajasekaran, G.; Han, E.H.; Chung, Y.-H.; Choi, Y.-J.; Yang, Y.J.; Lee, J.E.; Kim, H.N.; Lee, K.; Kim, J.-S.; et al. Cationic Amphipathic Triazines with Potent Anti-bacterial, Anti-inflammatory and Anti-atopic Dermatitis Properties. Sci. Rep. 2019, 9, 1292. [Google Scholar] [CrossRef]
- Broderick, E.; Lyons, H.; Pembroke, T.; Byrne, H.; Murray, B.; Hall, M. The characterisation of a novel, covalently modified, amphiphilic alginate derivative, which retains gelling and non-toxic properties. J. Colloid Interface Sci. 2006, 298, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Aymard, P.; Martin, D.R.; Plucknett, K.; Foster, T.J.; Clark, A.H.; Norton, I.T. Influence of thermal history on the structural and mechanical properties of agarose gels. Biopolymers 2001, 59, 131–144. [Google Scholar] [CrossRef]
- Lapasin, R.; Pricl, S. Rheology of Industrial Polysaccharides: Theory and Applications; Springer: Boston, MA, USA, 1995; ISBN 978-1-4613-5915-9. [Google Scholar]
- Pescosolido, L.; Feruglio, L.; Farra, R.; Fiorentino, S.; Colombo, I.; Coviello, T.; Matricardi, P.; Hennink, W.E.; Vermonden, T.; Grassi, M. Mesh size distribution determination of interpenetrating polymer network hydrogels. Soft Matter 2012, 8, 7708–7715. [Google Scholar] [CrossRef]
- Draper, N.R.; Smith, H. Applied Regression Analysis; John Wiley & Sons, Inc: New York, NY, USA, 1996. [Google Scholar]
- Sedláček, P.; Smilek, J.; Klučáková, M. How the interactions with humic acids affect the mobility of ionic dyes in hydrogels—2. Non-stationary diffusion experiments. React. Funct. Polym. 2014, 75, 41–50. [Google Scholar] [CrossRef]
- Smilek, J.; Sedláček, P.; Kalina, M.; Klučáková, M. On the role of humic acids’ carboxyl groups in the binding of charged organic compounds. Chemosphere 2015, 138, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Sedláček, P.; Smilek, J.; Laštůvková, M.; Kalina, M.; Klučáková, M. Hydrogels: Invaluable experimental tool for demonstrating diffusion phenomena in physical chemistry laboratory courses. J. Mater. Educ. 2017, 39, 59–90. [Google Scholar]
- Crank, J. The Mathematics of Diffusion; Oxford University Press Inc: New York, NY, USA, 1979. [Google Scholar]
- ImageJ Website. Available online: https://imagej.nih.gov/ij/ (accessed on 20 August 2020).
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Normand, V.; Lootens, D.L.; Amici, E.; Plucknett, K.P.; Aymard, P. New Insight into Agarose Gel Mechanical Properties. Biomacromolecules 2000, 1, 730–738. [Google Scholar] [CrossRef]
- Grillet, A.M.; Wyatt, N.B.; Gloe, L.M. Polymer Gel Rheology and Adhesion. In Rheology; InTech: Rijeka, Croatia, 2012; pp. 59–80. [Google Scholar]
- Weng, L.; Chen, X.; Chen, W. Rheological Characterization of in Situ Crosslinkable Hydrogels Formulated from Oxidized Dextran and N -Carboxyethyl Chitosan. Biomacromolecules 2007, 8, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Trenkmann, I.; Bok, S.; Korampally, V.R.; Gangopadhyay, S.; Graaf, H.; von Borczyskowski, C. Counting single Rhodamine 6G dye molecules in organosilicate nanoparticles. Chem. Phys. 2012, 406, 41–46. [Google Scholar] [CrossRef][Green Version]
- Penzkofer, A.; Leupacher, W. Fluorescence behaviour of highly concentrated rhodamine 6G solutions. J. Lumin. 1987, 37, 61–72. [Google Scholar] [CrossRef]
- Doty, P.; Steiner, R.F. Light Scattering and Spectrophotometry of Colloidal Solutions. J. Chem. Phys. 1950, 18, 1211–1220. [Google Scholar] [CrossRef]
- Horne, D.S. Determination of the fractal dimension using turbidimetric techniques. Application to aggregating protein systems. Faraday Discuss. Chem. Soc. 1987, 83, 259–280. [Google Scholar] [CrossRef]
- Camerini-Otero, R.D.; Day, L.A. The wavelength dependence of the turbidity of solutions of macromolecules. Biopolymers 1978, 17, 2241–2249. [Google Scholar] [CrossRef]
- Leone, M.; Sciortino, F.; Migliore, M.; Fornili, S.L.; Vittorelli, M.B.P. Order parameters of gels and gelation kinetics of aqueous agarose systems: Relation to the spinodal decomposition of the sol. Biopolymers 1987, 26, 743–761. [Google Scholar] [CrossRef]
- Aymard, P.; Williams, M.A.K.; Clark, A.H.; Norton, I.T. A Turbidimetric Study of Phase Separating Biopolymer Mixtures during Thermal Ramping. Langmuir 2000, 16, 7383–7391. [Google Scholar] [CrossRef]
- Singh, T.R.R. Hydrogels; CRC Press: Boca Raton, FL, USA, 2018; ISBN 9781315152226. [Google Scholar]
- Stellwagen, N.C. Effect of the electric field on the apparent mobility of large DNA fragments in agarose gels. Biopolymers 1985, 24, 2243–2255. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, New York, NY, USA, 1953. [Google Scholar]
- Larson, R.G. The Structure and Rheology of Complex Fluids; Oxford University Press Inc.: New York, NY, USA, 1999. [Google Scholar]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for Biomedical Applications: Their Characteristics and the Mechanisms behind Them. Gels 2017, 3, 6. [Google Scholar] [CrossRef]
- Calhoun, M.A.; Bentil, S.A.; Elliott, E.; Otero, J.J.; Winter, J.O.; Dupaix, R.B. Beyond linear elastic modulus: Viscoelastic models for brain and brain mimetic hydrogels. ACS Biomater. Sci. Eng. 2019, 5, 3964–3973. [Google Scholar] [CrossRef]
- Smilek, J.; Jarábková, S.; Velcer, T.; Pekař, M. Compositional and Temperature Effects on the Rheological Properties of Polyelectrolyte–Surfactant Hydrogels. Polymers 2019, 11, 927. [Google Scholar] [CrossRef]
- Galway, M.E.; Heckman, J.W., Jr.; Hyde, G.J.; Fowke, L.C. Advances in high-pressure and plunge-freeze fixation. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 1995; Volume 49, pp. 3–19. [Google Scholar] [CrossRef]
- Hrubanova, K.; Nebesarova, J.; Ruzicka, F.; Krzyzanek, V. The innovation of cryo-SEM freeze-fracturing methodology demonstrated on high pressure frozen biofilm. Micron 2018, 110, 28–35. [Google Scholar] [CrossRef]
- Paradossi, G.; Cavalieri, F.; Chiessi, E.; Spagnoli, C.; Cowman, M.K. Poly (vinyl alcohol) as versatile biomaterial for potential biomedical applications. J. Mater. Sci. Mater. 2003, 14, 687–691. [Google Scholar] [CrossRef]
- Holly, F.J.; Refojo, M.F. Wettability of hydrogels I. Poly(2-hydroxyethyl methacrylate). J. Biomed. Mater. Res. 1975, 9, 315–326. [Google Scholar] [CrossRef]
- Zhu, J. Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials 2010, 31, 4639–4656. [Google Scholar] [CrossRef]
- Osmałek, T.; Froelich, A.; Tasarek, S. Application of gellan gum in pharmacy and medicine. Int. J. Pharm. 2014, 466, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Park, K.; Shalaby, W.S.W. Biodegradable Hydrogels for Drug Delivery; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Xiong, X.Y.; Tam, K.C.; Gan, L.H. Polymeric Nanostructures for Drug Delivery Applications Based on Pluronic Copolymer Systems. J. Nanosci. Nanotechno. 2006, 6, 2638–2650. [Google Scholar] [CrossRef] [PubMed]
- Amiji, M.; Tailor, R.; Ly, M.-K.; Goreham, J. Gelatin-Poly(Ethylene Oxide) Semi-interpenetrating Polymer Network with pH-Sensitive Swelling and Enzyme-Degradable Properties for Oral Drug Delivery. Drug Dev. Ind. Pharm. 1997, 23, 575–582. [Google Scholar] [CrossRef]
- Elisseeff, J.; McIntosh, W.; Anseth, K.; Riley, S.; Ragan, P.; Langer, R. Photoencapsulation of chondrocytes in poly(ethylene oxide)-based semi-interpenetrating networks. J. Biomed. Mater. Res. 2000, 51, 164–171. [Google Scholar] [CrossRef]
- Sedláček, P.; Smilek, J.; Klučáková, M. How the interactions with humic acids affect the mobility of ionic dyes in hydrogels—Results from diffusion cells. React. Funct. Polym. 2013, 73, 1500–1509. [Google Scholar] [CrossRef]
- Larrañeta, E.; Stewart, S.; Ervine, M.; Al-Kasasbeh, R.; Donnelly, R. Hydrogels for Hydrophobic Drug Delivery. Classification, Synthesis and Applications. J. Funct. Biomater. 2018, 9, 13. [Google Scholar] [CrossRef]
- Venerová, T.; Pekař, M. Rheological properties of gels formed by physical interactions between hyaluronan and cationic surfactants. Carbohydr. Polym. 2017, 170, 176–181. [Google Scholar] [CrossRef]
- Enev, V.; Sedláček, P.; Jarábková, S.; Velcer, T.; Pekař, M. ATR-FTIR spectroscopy and thermogravimetry characterization of water in polyelectrolyte-surfactant hydrogels. Colloids Surf. A 2019, 575, 1–9. [Google Scholar] [CrossRef]
- Moschakis, T. Microrheology and particle tracking in food gels and emulsions. Curr. Opin. Colloid Interface Sci. 2013, 18, 311–323. [Google Scholar] [CrossRef]
- Rathgeber, S.; Beauvisage, H.-J.; Chevreau, H.; Willenbacher, N.; Oelschlaeger, C. Microrheology with Fluorescence Correlation Spectroscopy. Langmuir 2009, 25, 6368–6376. [Google Scholar] [CrossRef]
- García-Aparicio, C.; Quijada-Garrido, I.; Garrido, L. Diffusion of small molecules in a chitosan/water gel determined by proton localized NMR spectroscopy. J. Colloid Interface Sci. 2012, 368, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Severs, N.; Shotton, D. Rapid Freezing of Biological Specimens for Freeze Fracture and Deep Etching. Cell Biol. 2006, 3, 249–255. [Google Scholar]
- Landry, M.R. Thermoporometry by differential scanning calorimetry: Experimental considerations and applications. Thermochim. Acta 2005, 433, 27–50. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight Content of PSS in the Gel | Macroscopic Diffusivity Deff (µm2·s–1) | FCS Diffusivity * Ds (µm2·s–1) | Partition Coefficient ε (-) | Fluorescence Lifetime τ (ns) |
|---|---|---|---|---|
| 0% | 389 ± 7 | 343 ± 7 | 5.3 ± 2.0 | 4.21 ± 0.06 |
| 0.002% | 158 ± 27 | 196 ± 27 | 10.2 ± 2.6 | 4.21 ± 0.09 |
| 0.005% | 52 ± 25 | 148 ± 25 | 31.0 ± 10.1 | 4.27 ± 0.07 |
| 0.010% | 27 ± 22 | 106 ± 22 | 64.8 ± 36.7 | 4.30 ± 0.06 |
| Weight Content of ALG in the Gel | Macroscopic Diffusivity Deff (µm2·s–1) | FCS Diffusivity Ds (µm2·s–1) | Partition Coefficient ε (-) | Fluorescence Lifetime τ (ns) |
|---|---|---|---|---|
| 0% | 333 ± 33 | 385 ± 15 | 7.0 ± 1.6 | 3.70 ± 0.02 |
| 0.002% | 379 ± 27 | 309 ± 21 | 6.4 ± 1.2 | 3.73 ± 0.03 |
| 0.005% | 379 ± 46 | 304 ± 48 | 6.4 ± 1.0 | 3.69 ± 0.01 |
| 0.010% | 394 ± 42 | 298 ± 21 | 6.1 ± 1.0 | 3.64 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trudicova, M.; Smilek, J.; Kalina, M.; Smilkova, M.; Adamkova, K.; Hrubanova, K.; Krzyzanek, V.; Sedlacek, P. Multiscale Experimental Evaluation of Agarose-Based Semi-Interpenetrating Polymer Network Hydrogels as Materials with Tunable Rheological and Transport Performance. Polymers 2020, 12, 2561. https://doi.org/10.3390/polym12112561
Trudicova M, Smilek J, Kalina M, Smilkova M, Adamkova K, Hrubanova K, Krzyzanek V, Sedlacek P. Multiscale Experimental Evaluation of Agarose-Based Semi-Interpenetrating Polymer Network Hydrogels as Materials with Tunable Rheological and Transport Performance. Polymers. 2020; 12(11):2561. https://doi.org/10.3390/polym12112561
Chicago/Turabian StyleTrudicova, Monika, Jiri Smilek, Michal Kalina, Marcela Smilkova, Katerina Adamkova, Kamila Hrubanova, Vladislav Krzyzanek, and Petr Sedlacek. 2020. "Multiscale Experimental Evaluation of Agarose-Based Semi-Interpenetrating Polymer Network Hydrogels as Materials with Tunable Rheological and Transport Performance" Polymers 12, no. 11: 2561. https://doi.org/10.3390/polym12112561
APA StyleTrudicova, M., Smilek, J., Kalina, M., Smilkova, M., Adamkova, K., Hrubanova, K., Krzyzanek, V., & Sedlacek, P. (2020). Multiscale Experimental Evaluation of Agarose-Based Semi-Interpenetrating Polymer Network Hydrogels as Materials with Tunable Rheological and Transport Performance. Polymers, 12(11), 2561. https://doi.org/10.3390/polym12112561