Pyrene-Functionalized Polyacetylenes: Synthesis and Photoluminescence Property

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
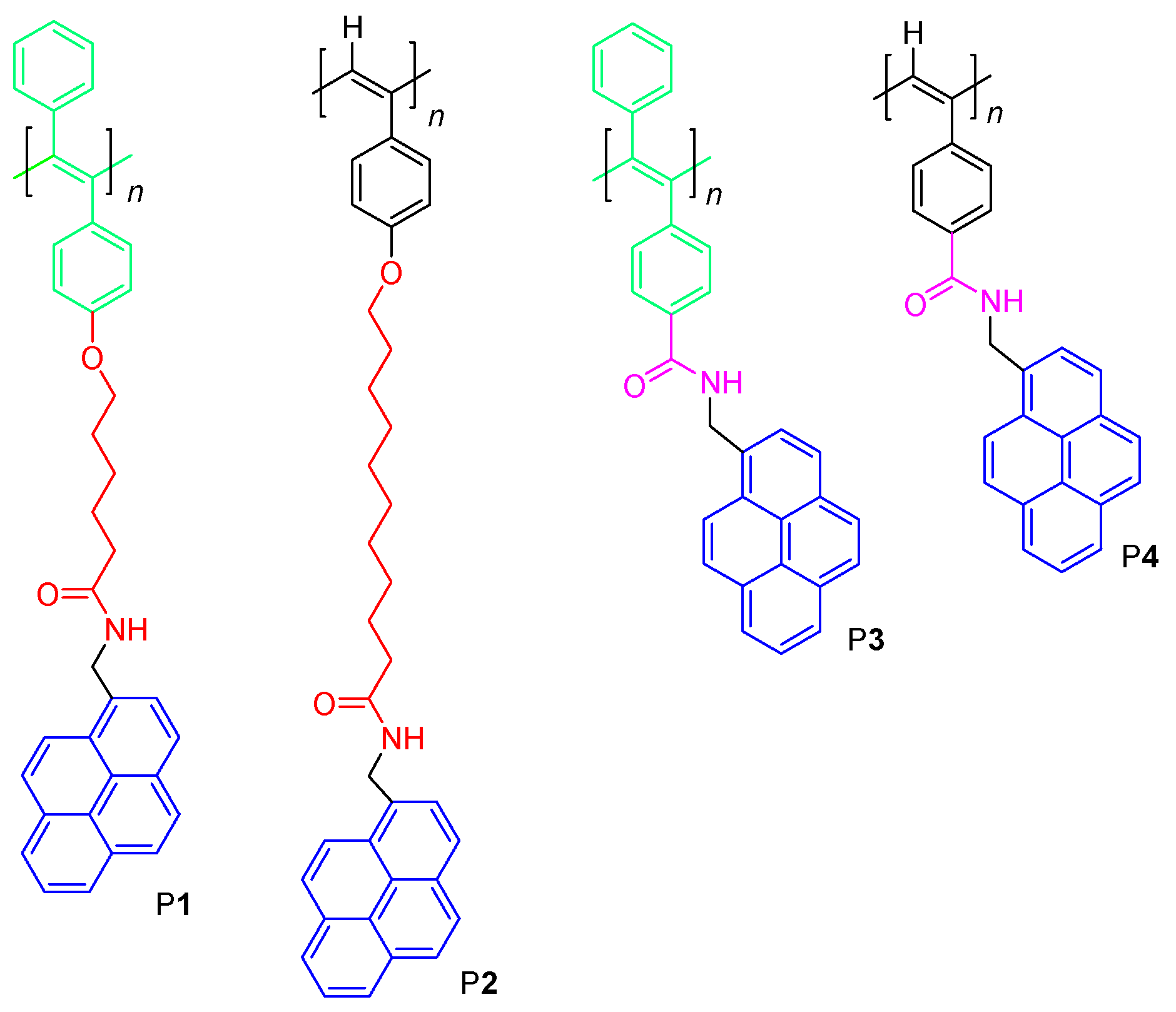
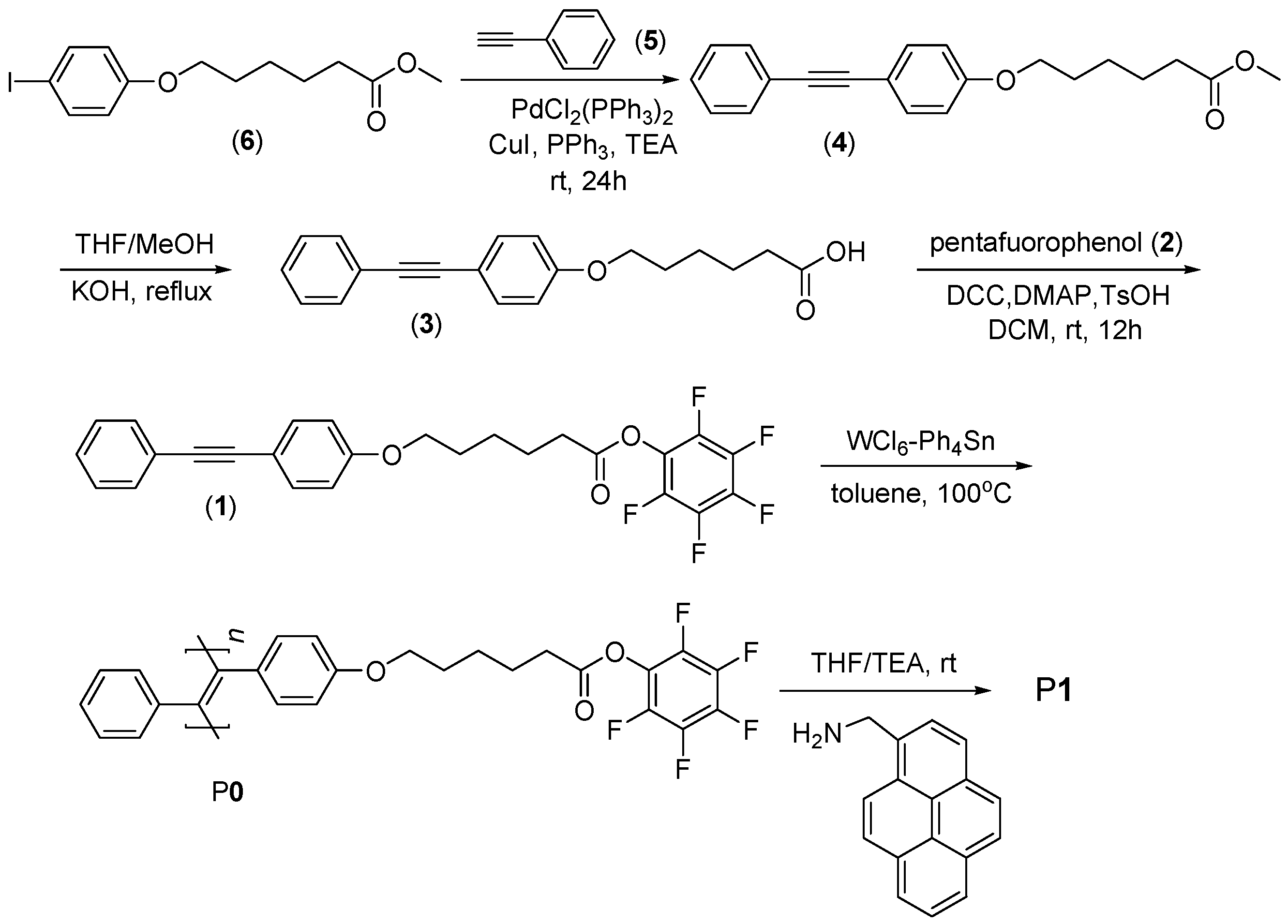
3.1. Polymer Synthesis
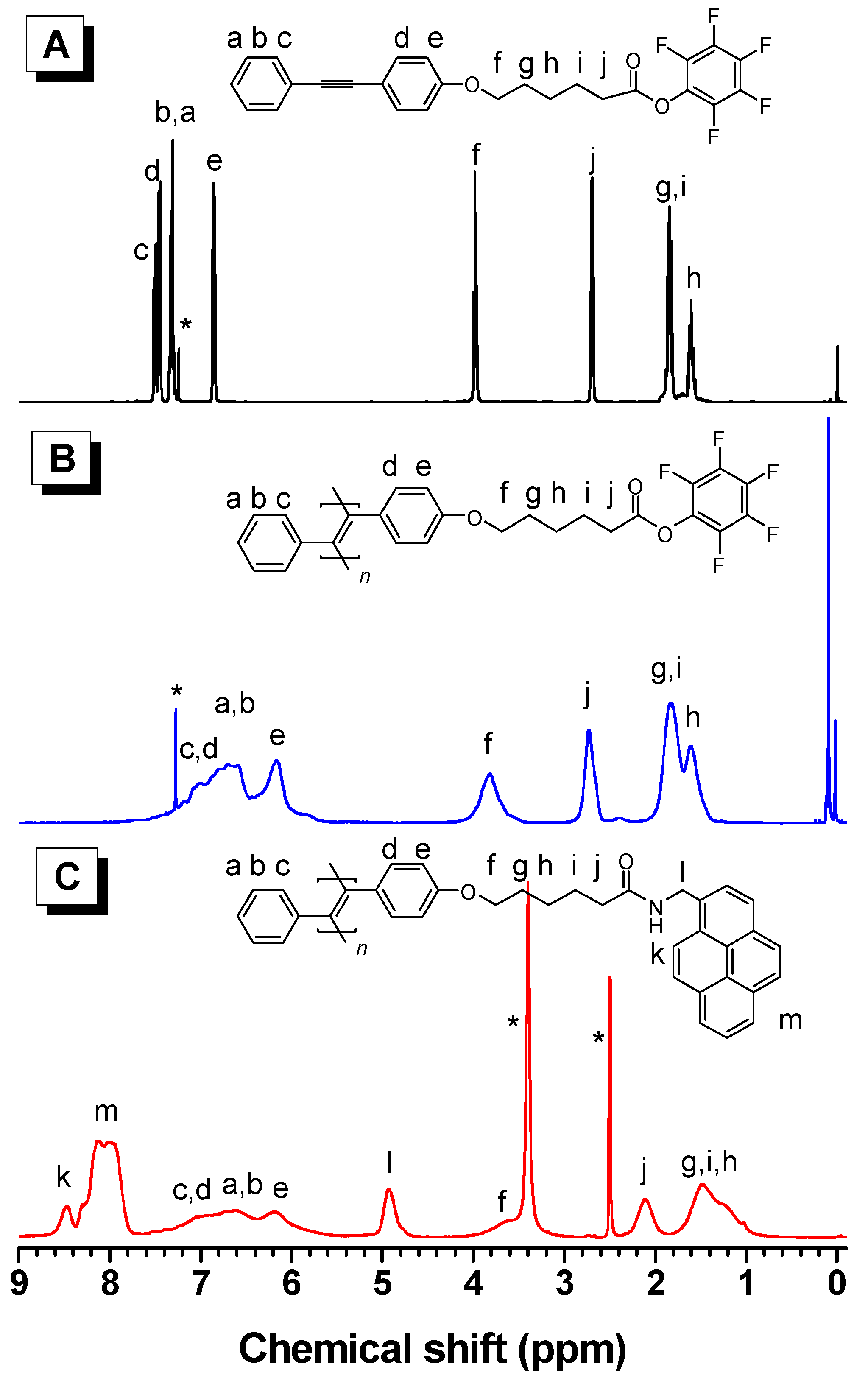
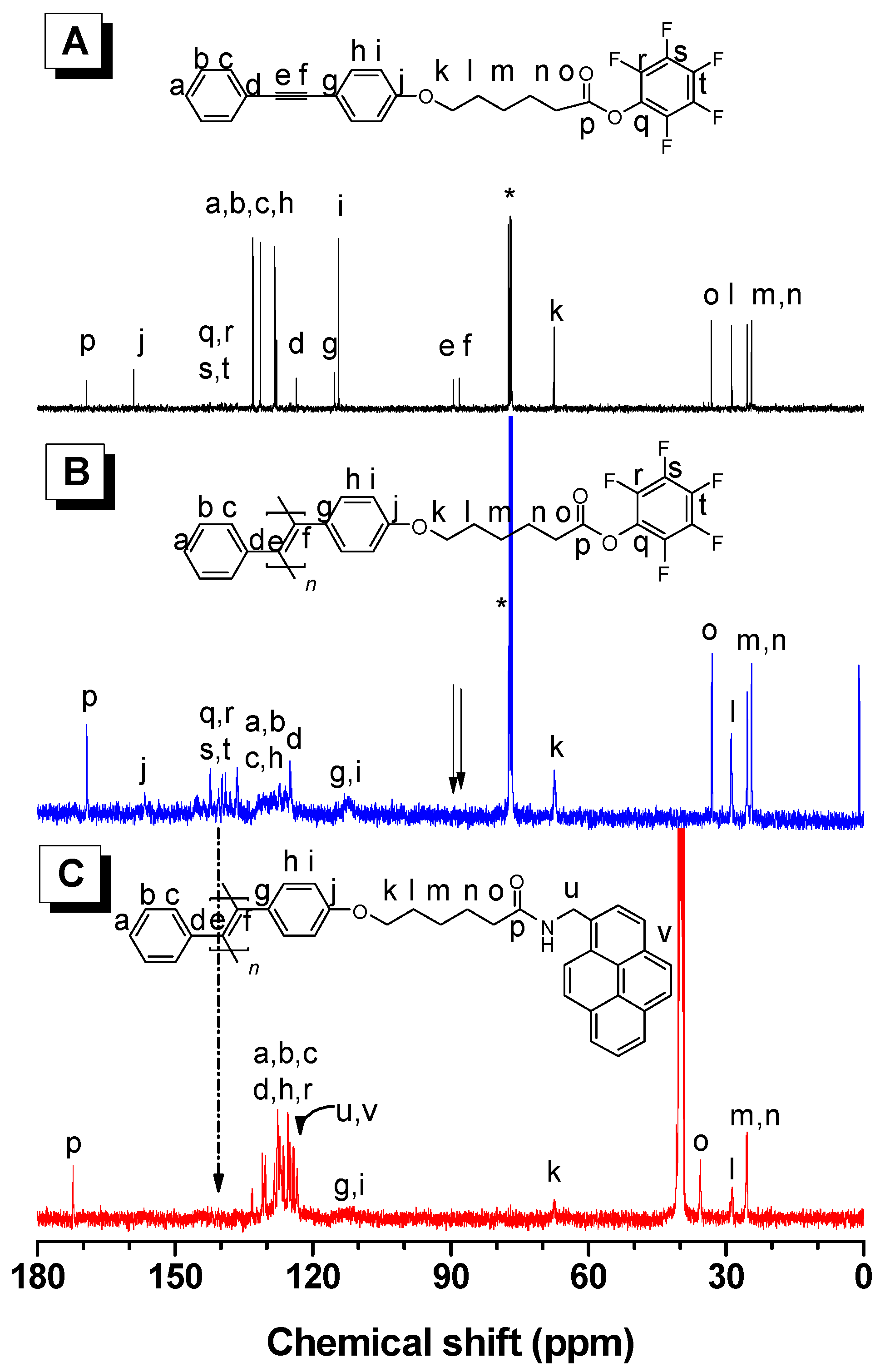
3.2. Structure Characterization
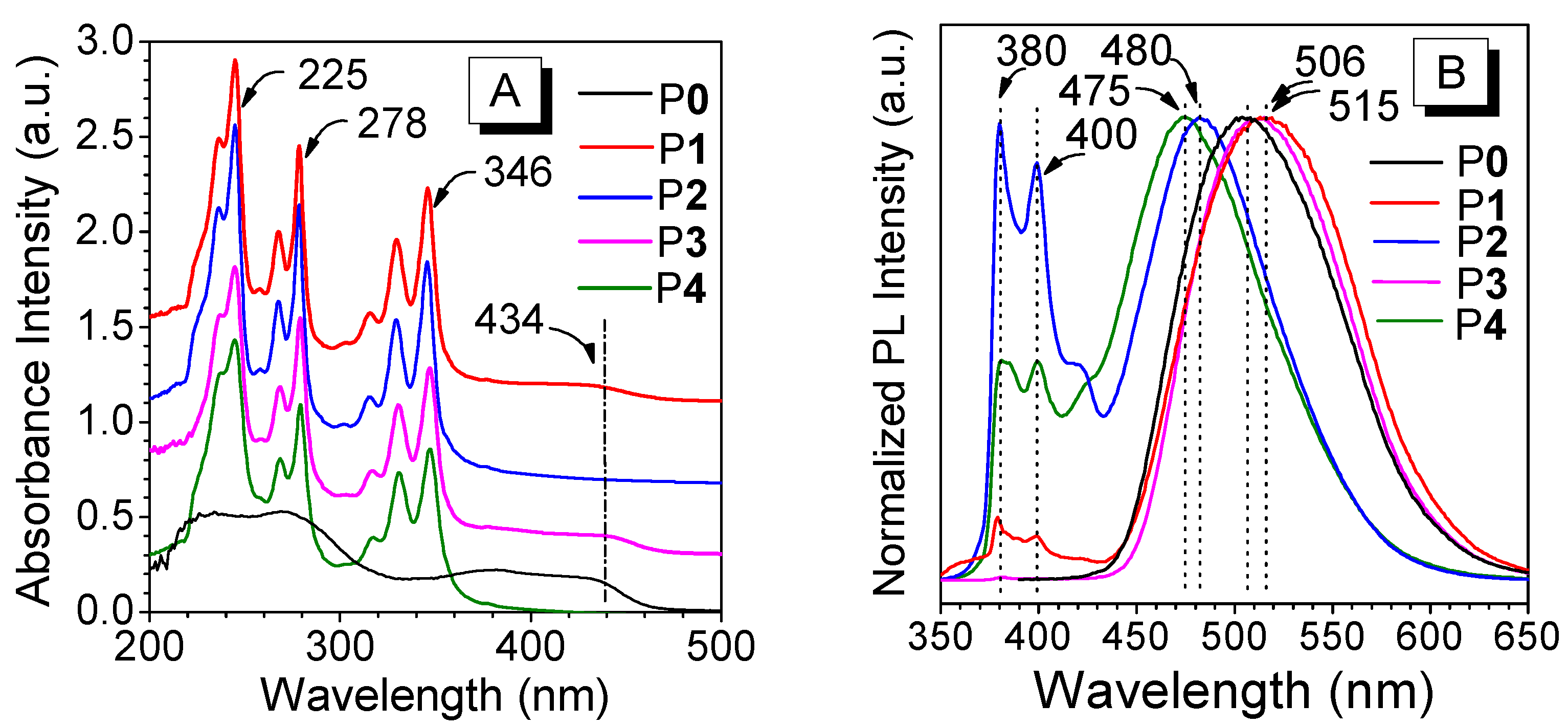
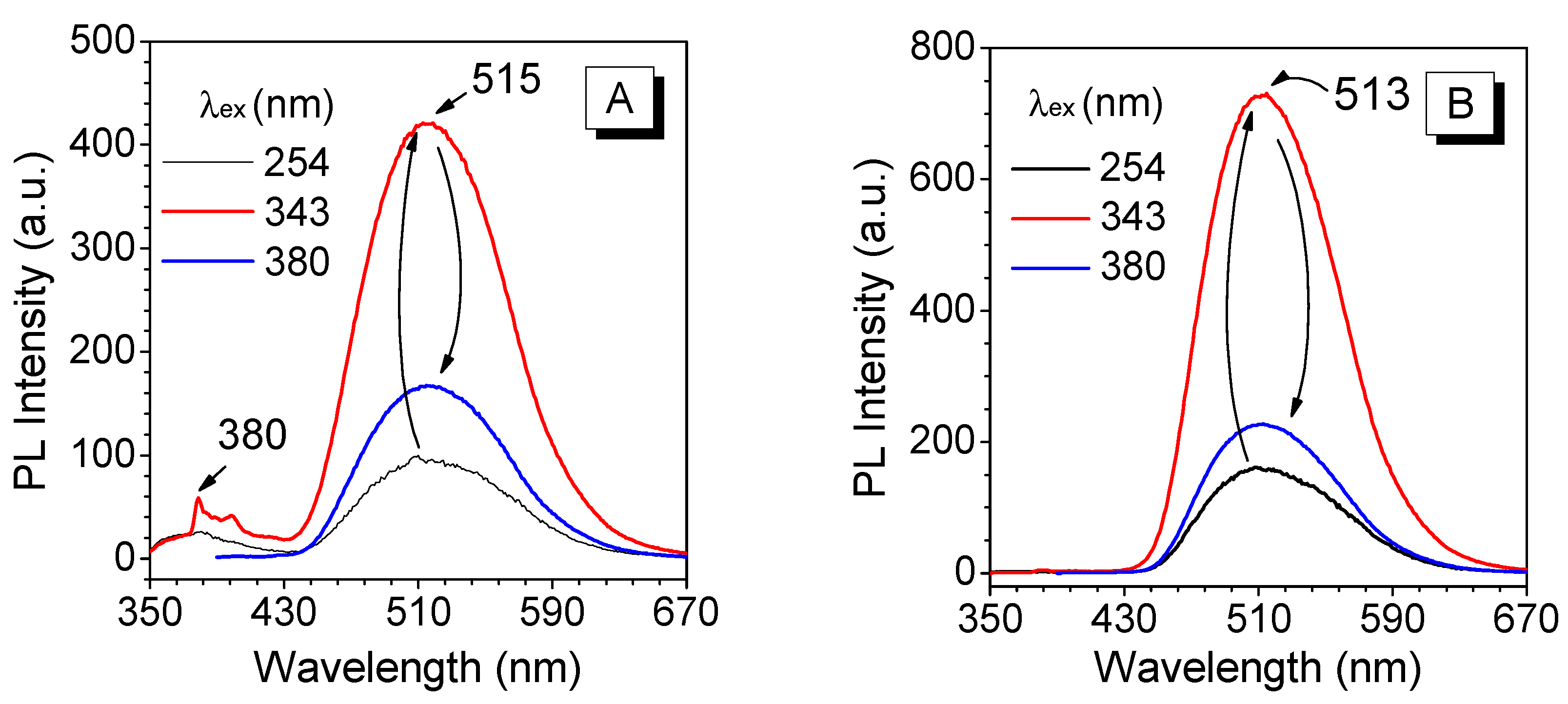
3.3. Absorption and Emission Behaviors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lam, J.W.Y.; Tang, B.Z. Functional polyacetylenes. Acc. Chem. Res. 2005, 38, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T. Substituted polyacetylenes. J. Polym. Sci. Part A 2007, 45, 165–180. [Google Scholar] [CrossRef]
- Liu, J.Z.; Lam, J.W.Y.; Tang, B.Z. Acetylenic polymers: Syntheses, structures, and functions. Chem. Rev. 2009, 109, 5799–5867. [Google Scholar] [CrossRef] [PubMed]
- Shiotsuki, M.; Sanda, F.; Masuda, T. Polymerization of substituted acetylenes and features of the formed polymers. Polym. Chem. 2011, 2, 1044–1058. [Google Scholar] [CrossRef]
- Rosen, B.M.; Wilson, C.J.; Wilson, D.A.; Peterca, M.; Imam, M.R.; Percec, V. Dendron-mediated self-assembly, disassembly, and self-organization of complex systems. Chem. Rev. 2009, 109, 6275–6540. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Z.; Qin, A.J.; Tang, B.Z. Functional polyacetylenes: Hybrids with carbon nanotubes. Polym. Chem. 2013, 4, 211–223. [Google Scholar] [CrossRef]
- Pauly, A.C.; Theato, P. Control of reactivity of constitutional isomers of pentafluorophenyl ethynylbenzoates for the synthesis of functional poly(phenylacetylenes). Polym. Chem. 2012, 3, 1769–1782. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Sun, J.Z.; Tang, B.Z. Synthesis of functional poly(disubstituted acetylene)s through the post-polymerization modification route. Chem. Rec. 2015, 15, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.A.; Chen, M.R.; Zhao, H.; Gao, Y.; Wei, Q.; Zhang, S.; Qin, A.; Sun, J.Z.; Tang, B.Z. A facile synthetic route to functional poly(phenylacetylene)s with tunable structures and properties. Macromolecules 2011, 44, 6724–6737. [Google Scholar] [CrossRef]
- Zhang, X.A.; Qin, A.; Tong, L.; Zhao, H.; Zhao, Q.L.; Sun, J.Z.; Tang, B.Z. Synthesis of functional disubstituted polyacetylenes bearing highly polar functionalities via activated ester strategy. ACS Macro Lett. 2012, 1, 75–79. [Google Scholar] [CrossRef]
- Tong, L.; Qin, A.; Zhang, X.A.; Mao, Y.; Sun, J.Z.; Tang, B.Z. Post-functionalization of disubstituted polyacetylenes via click chemistry. Sci. China Chem. 2011, 54, 1948–1954. [Google Scholar] [CrossRef]
- Wang, X.; Hu, H.; Wang, W.J.; Qin, A.J.; Sun, J.Z.; Tang, B.Z. A throughway to functional poly(disubstituted acetylenes): A combination of the activated ester strategy with click reaction. Polym. Chem. 2015, 6, 7958–7963. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Tong, L.; Qin, A.J.; Sun, J.Z.; Tang, B.Z. A new strategy of post-polymerization modification to prepare functionalized poly(disubstituted acetylenes). Polym. Chem. 2014, 5, 2309–2319. [Google Scholar] [CrossRef]
- Wang, W.J.; Shi, Y.; Wang, X.; Qin, A.; Sun, J.Z.; Tang, B.Z. A novel post-polymerization modification route to functional poly(disubstituted acetylenes) through Phenol-Yne click reaction. Polym. Chem. 2017, 8, 2630–2639. [Google Scholar] [CrossRef]
- Wang, X.; Sun, J.Z.; Tang, B.Z. Poly(disubstituted acetylene)s: Advances in polymer preparation and materials application. Prog. Polym. Sci. 2018, 79, 98–120. [Google Scholar] [CrossRef]
- San Jose, B.A.; Akagi, K. Liquid crystalline polyacetylene derivatives with advanced electrical and optical properties. Polym. Chem. 2013, 4, 5144–5161. [Google Scholar] [CrossRef]
- Lam, J.W.Y.; Luo, J.D.; Dong, Y.P.; Cheuk, K.K.L.; Tang, B.Z. Functional polyacetylenes: Synthesis, thermal stability, liquid crystallinity, and light emission of polypropiolates. Macromolecules 2002, 35, 8288–8299. [Google Scholar] [CrossRef]
- Yashima, E.; Maeda, K.; Iida, H.; Furusho, Y.; Nagai, K. Helical polymers: Synthesis, structures, and functions. Chem. Rev. 2009, 109, 6102–6211. [Google Scholar] [CrossRef] [PubMed]
- San Jose, B.A.; Matsushita, S.; Akagi, K. Lyotropic chiral nematic liquid crystalline aliphatic conjugated polymers based on disubstituted polyacetylene derivatives that exhibit high dissymmetry factors in circularly polarized luminescence. J. Am. Chem. Soc. 2012, 134, 19795–19807. [Google Scholar] [CrossRef]
- Kwak, G.; Minakuchi, M.; Sakaguchi, T.; Masuda, T.; Fujiki, M. Poly(diphenylacetylene) bearing long alkyl side chain via silylene linkage: Its lyotropic liquid crystallinity and optical anisotropy. Chem. Mater. 2007, 19, 3654–3661. [Google Scholar] [CrossRef]
- Lee, W.E.; Kim, J.W.; Oh, C.J.; Sakaguchi, T.; Fujiki, M.; Kwak, G. Correlation of intramolecular excimer emission with lamellar layer distance in liquid-crystalline polymers: Verification by the film-swelling method. Angew. Chem. Int. Ed. 2010, 49, 1406–1409. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K. Helical Polyacetylene: Asymmetric Polymerization in a Chiral Liquid-Crystal Field. Chem. Rev. 2009, 109, 5354–5401. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.X.; Chen, Y.W.; Chen, L.; Li, F. Luminescent mesogen jacketed poly(1-alkyne) bearing lateral terphenyl with hexyloxy tail. J. Polym. Sci. Part A 2010, 48, 5679–5692. [Google Scholar] [CrossRef]
- Lee, W.E.; Lee, C.L.; Sakaguchi, T.; Fujiki, M.; Kwak, G. Piezochromic fluorescence in liquid crystalline conjugated polymers. Chem Commun. 2011, 47, 3526–3528. [Google Scholar] [CrossRef]
- Lam, J.W.Y.; Dong, Y.P.; Cheuk, K.K.L.; Tang, B.Z. Helical disubstituted polyacetylenes: Synthesis and chiroptical properties of poly(phenylpropiolate)s. Macromolecules 2003, 36, 7927–7938. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, Y.W.; Chen, L.; Li, F.; Nie, H.R.; Yao, K. Effects of substitution and terminal groups for liquid-crystallinity enhanced luminescence of disubstituted polyacetylenes carrying chromophoric terphenyl pendants. Sci. China Chem. 2010, 53, 1302–1315. [Google Scholar] [CrossRef]
- Lee, D.; Jin, Y.J.; Kim, H.; Suzuki, N.; Fujiki, M.; Sakaguchi, T.; Kim, S.K.; Lee, W.; Kwak, G. Solvent-to-polymer chirality transfer in intramolecular stack structure. Macromolecules 2012, 45, 5379–5386. [Google Scholar] [CrossRef]
- Kim, H.; Jin, Y.J.; Kim, B.S.; Aoki, T.; Kwak, G. Optically active conjugated polymer nanoparticles from chiral solvent annealing and nanoprecipitation. Macromolecules 2015, 48, 4754–4757. [Google Scholar] [CrossRef]
- Van Oosten, C.L.; Bastiaansen, C.W.M.; Broer, D.J. Printed artificial cilia from liquid-crystal network actuators modularly driven by light. Nat. Mater. 2009, 8, 677–682. [Google Scholar] [CrossRef]
- Zhao, B.; Pan, K.; Deng, J.P. Intense circularly polarized luminescence contributed by helical chirality of monosubstituted polyacetylenes. Macromolecules 2018, 51, 7104–7111. [Google Scholar] [CrossRef]
- Yu, H.L.; Deng, J.P. Alkynylated cellulose nanocrystals simultaneously serving as chiral source and stabilizing agent for constructing optically active helical polymer particles. Macromolecules 2016, 49, 7728–7736. [Google Scholar] [CrossRef]
- San Jose, B.A.; Yan, J.L.; Akagi, K. Dynamic switching of the circularly polarized luminescence of disubstituted polyacetylene by selective transmission through a thermotropic chiral nematic liquid crystal. Angew Chem. Int. Ed. 2014, 53, 10641–10644. [Google Scholar] [CrossRef]
- Kawakami, Y.; Tang, H.Z. Recent progress in the synthesis of conformationally optically active polymers. Des Monomers Polym. 2000, 3, 1–16. [Google Scholar] [CrossRef]
- Yin, S.C.; Xu, H.Y.; Su, X.Y.; Gao, Y.C.; Song, Y.L.; Jacky, W.Y.L.; Tang, B.Z.; Shi, W.F. Synthesis and optical properties of azobenzene-containing poly(1-alkyne)s with different spacer lengths and ring substituents. Polymer 2005, 46, 10592–10600. [Google Scholar] [CrossRef]
- Ye, C.; Lam, W.Y.; Liu, Z.-F.; Cheng, S.Z.D.; Chen, E.-Q.; Tang, B.Z. Frustrated molecular packing in highly ordered smectic phase of side-chain liquid crystalline polymer with rigid polyacetylene backbone. J. Am. Chem. Soc. 2005, 127, 7668–7669. [Google Scholar] [CrossRef]
- Mathew, A.; Siu, H.; Duhamel, J. A blob model to study chain folding by fluorescence. Macromolecules 1999, 32, 7100–7108. [Google Scholar] [CrossRef]
- Rivera, E.; Belletête, M.; Zhu, X.-X.; Durocher, G.; Giasson, R. Novel polyacetylenes containing pendant 1-pyrenyl groups: Synthesis, characterization, and thermal and optical properties. Polymer 2002, 43, 5059–5068. [Google Scholar] [CrossRef]
- Yuan, W.Z.; Sun, J.Z.; Dong, Y.; Häußler, M.; Yang, F.; Xu, H.P.; Qin, A.; Lam, J.W.Y.; Zheng, Q.; Tang, B.Z. Wrapping carbon nanotubes in pyrene-containing poly(phenylacetylene) chains: Solubility, stability, light emission, and surface photovoltaic properties. Macromolecules 2006, 39, 8011–8020. [Google Scholar] [CrossRef]
- Yuan, W.Z.; Mao, Y.; Zhao, H.; Sun, J.Z.; Xu, H.P.; Jin, J.K.; Zheng, Q.; Tang, B.Z. Electronic interactions and polymer effect in the functionalization and solvation of carbon nanotubes by pyrene- and ferrocene-containing poly(1-alkyne)s. Macromolecules 2008, 41, 701–707. [Google Scholar] [CrossRef]
- Zhang, X.A.; Zhao, H.; Gao, Y.; Tong, J.; Shan, L.; Chen, Y.; Zhang, S.; Qin, A.; Sun, J.Z.; Tang, B.Z. Functional poly(phenylacetylene)s carrying azobenzene pendants: Polymer synthesis, photoisomerization behaviors, and liquid-crystalline property. Polymer 2011, 52, 5290–5301. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Wang, W.J.; Qin, A.; Sun, J.Z.; Tang, B.Z. Different amine-functionalized poly(diphenylsubstituted acetylenes) from the same precursor. Polym. Chem. 2016, 7, 5312–5321. [Google Scholar] [CrossRef]
- Zaragoza-Galán, G.; Fowler, M.; Duhamel, J.; Rein, R.; Solladié, N.; Rivera, E. Synthesis and Characterization of Novel Pyrene-Dendronized Porphyrins Exhibiting Efficient Fluorescence Resonance Energy Transfer: Optical and Photophysical Properties. Langmuir 2012, 28, 11195–11205. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Polymer | Yield (%) | Mw2 | Mw/Mn2 |
|---|---|---|---|---|
| 1 | P0 | 61.2 | 13,400 | 2.13 |
| 2 | P1 | 94.1 | 12,100 | 2.10 |
| 3 | P2 | 95.6 | 29,400 | 1.99 |
| 4 | P3 | 89.2 | 65,200 | 3.14 |
| 5 | P4 | 94.8 | 91,400 | 2.13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, T.; Jiang, N.; Zhang, X.; He, L.; Lang, X.H.; Sun, J.Z.; Zhao, H. Pyrene-Functionalized Polyacetylenes: Synthesis and Photoluminescence Property. Polymers 2019, 11, 1366. https://doi.org/10.3390/polym11081366
Shen T, Jiang N, Zhang X, He L, Lang XH, Sun JZ, Zhao H. Pyrene-Functionalized Polyacetylenes: Synthesis and Photoluminescence Property. Polymers. 2019; 11(8):1366. https://doi.org/10.3390/polym11081366
Chicago/Turabian StyleShen, Tanxiao, Nan Jiang, Xiao’a Zhang, Lirong He, Xian Hua Lang, Jing Zhi Sun, and Hui Zhao. 2019. "Pyrene-Functionalized Polyacetylenes: Synthesis and Photoluminescence Property" Polymers 11, no. 8: 1366. https://doi.org/10.3390/polym11081366
APA StyleShen, T., Jiang, N., Zhang, X., He, L., Lang, X. H., Sun, J. Z., & Zhao, H. (2019). Pyrene-Functionalized Polyacetylenes: Synthesis and Photoluminescence Property. Polymers, 11(8), 1366. https://doi.org/10.3390/polym11081366