On the Dielectric Behavior of Amine and Anhydride Cured Epoxy Resins Modified Using Multi-Terminal Epoxy Functional Network Modifier


Abstract
:1. Introduction
2. Experimental
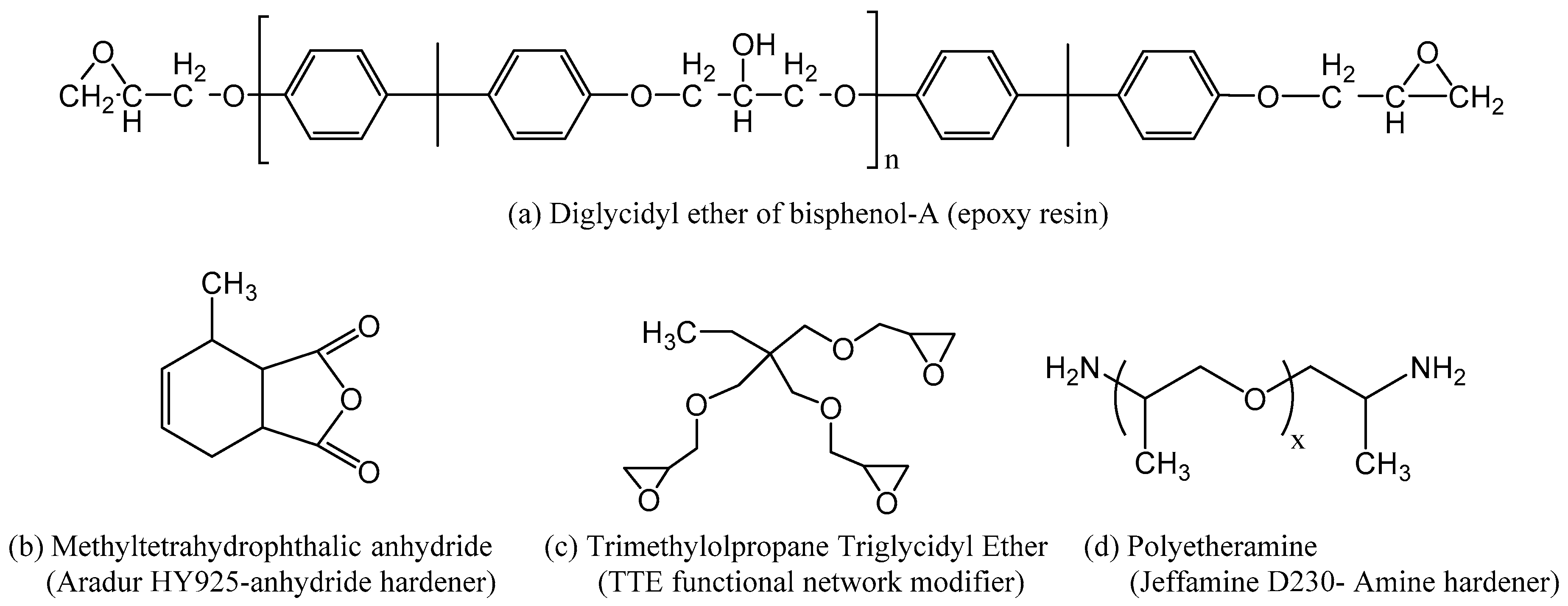
2.1. Materials
2.2. Sample Preparation
2.3. Characterization
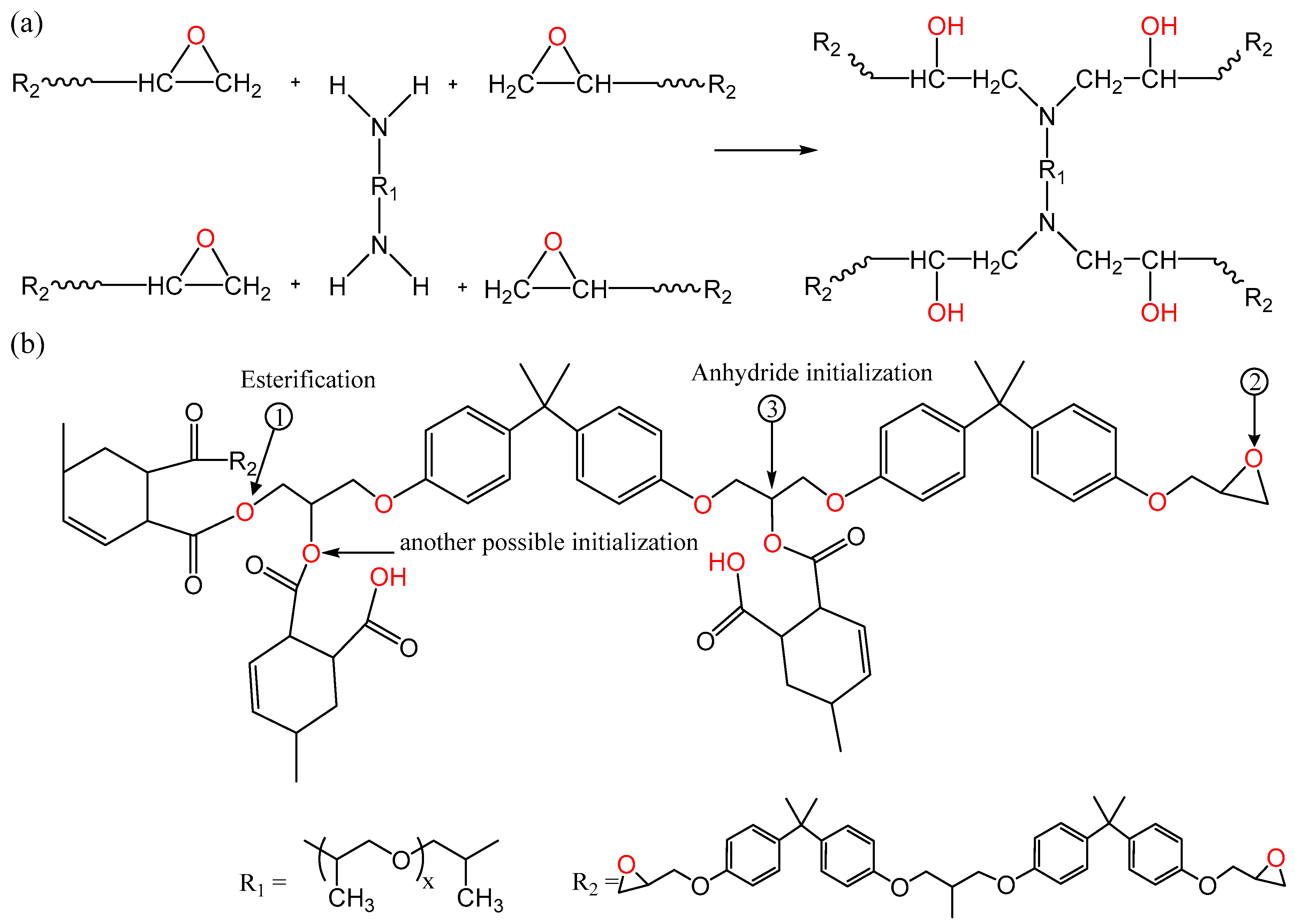
3. Results and Discussion
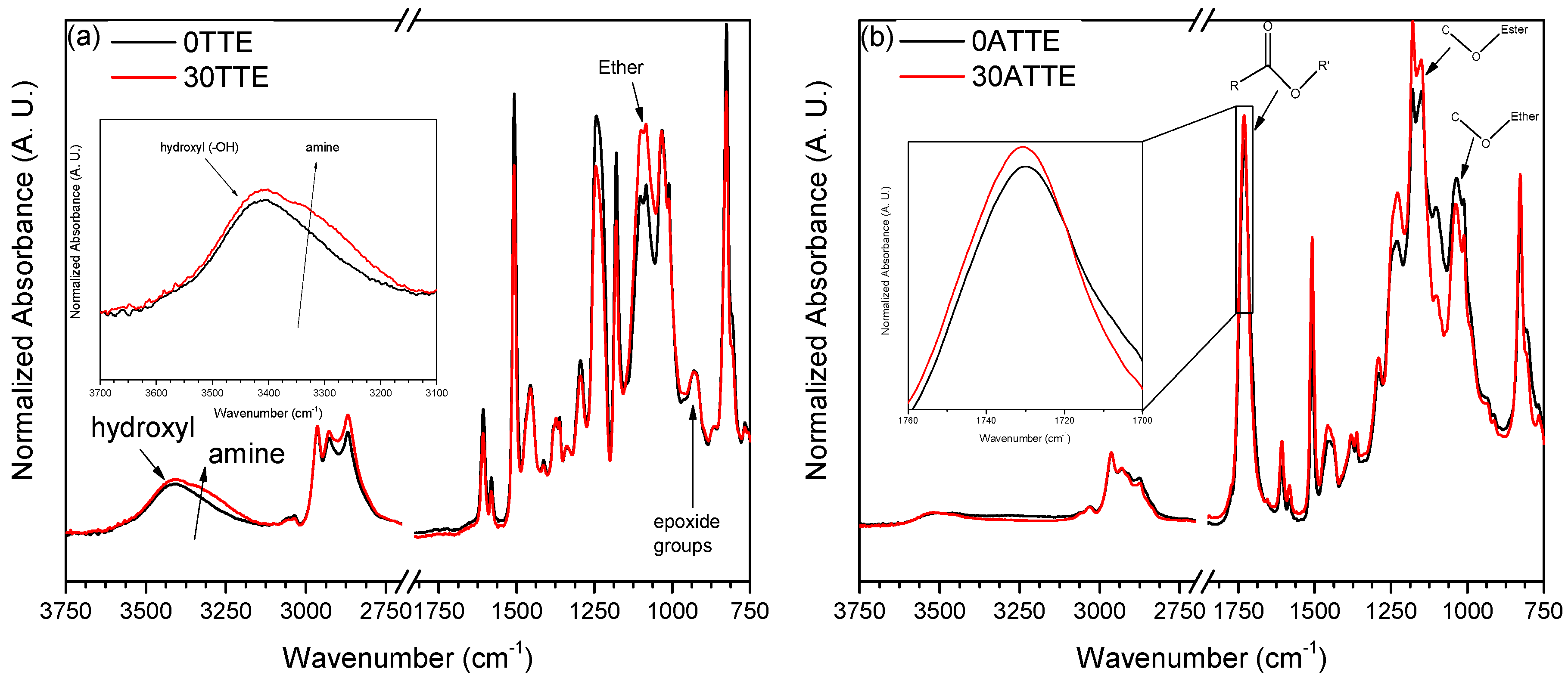
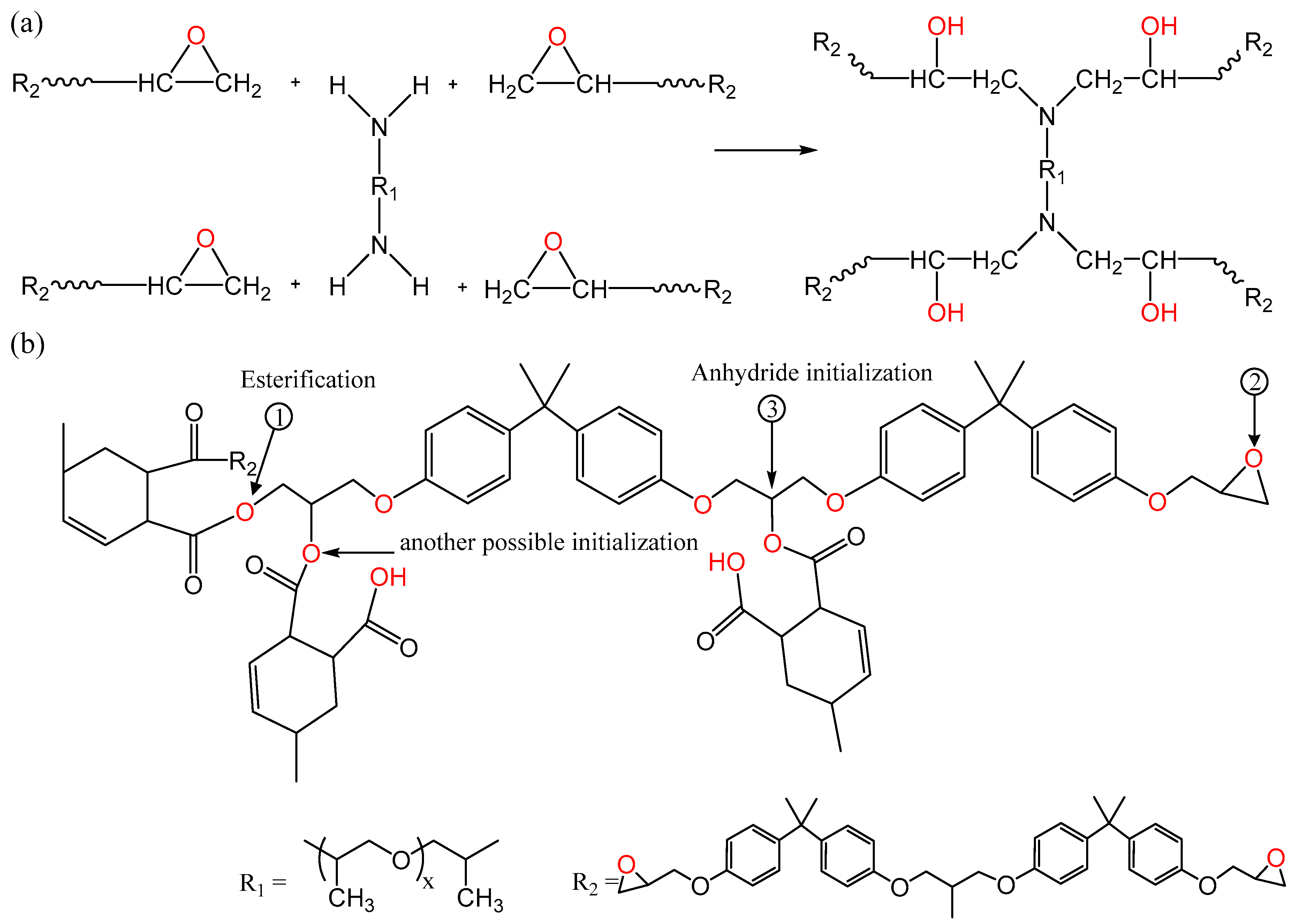
3.1. FTIR Spectroscopy
3.2. Differential Scanning Calorimetry
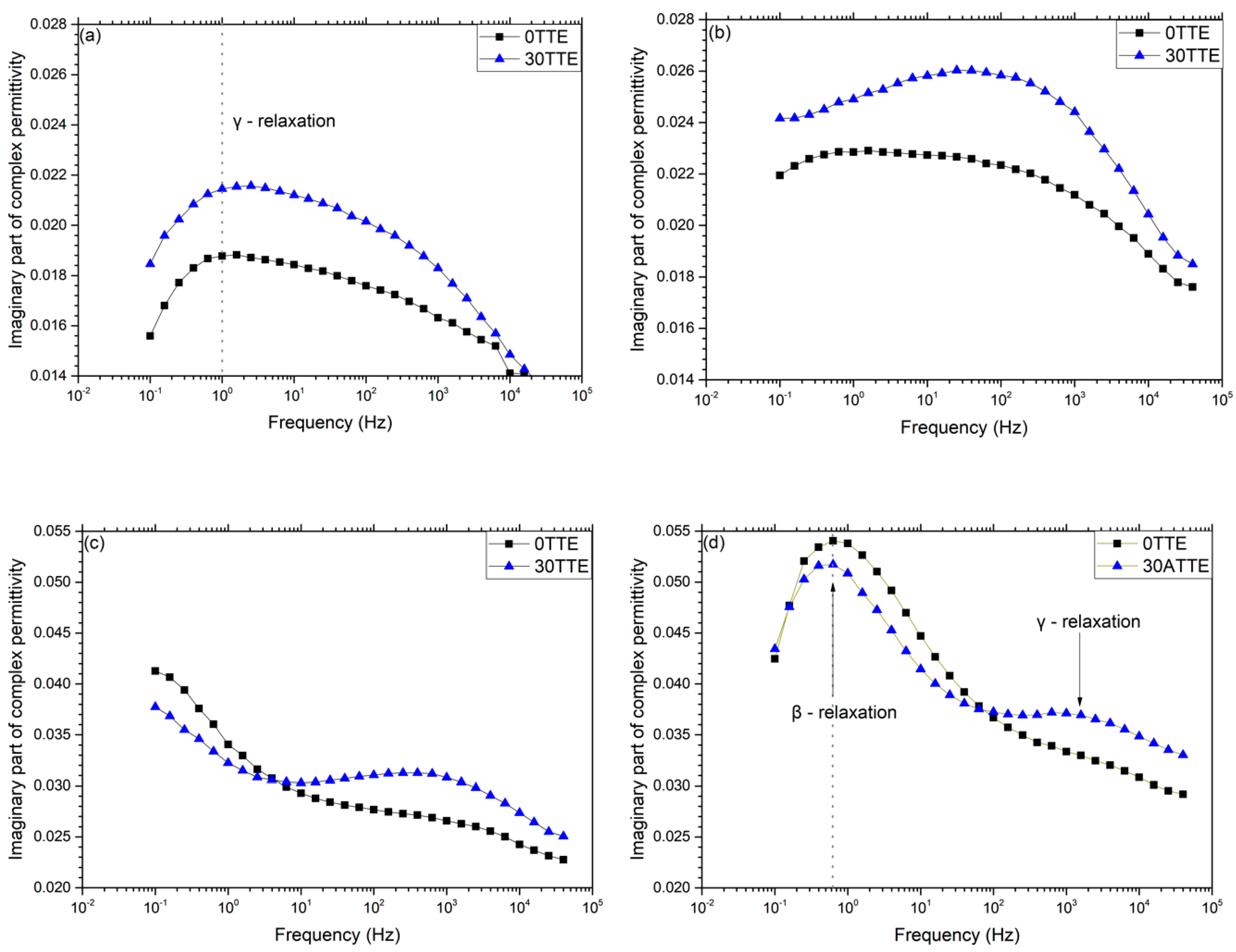
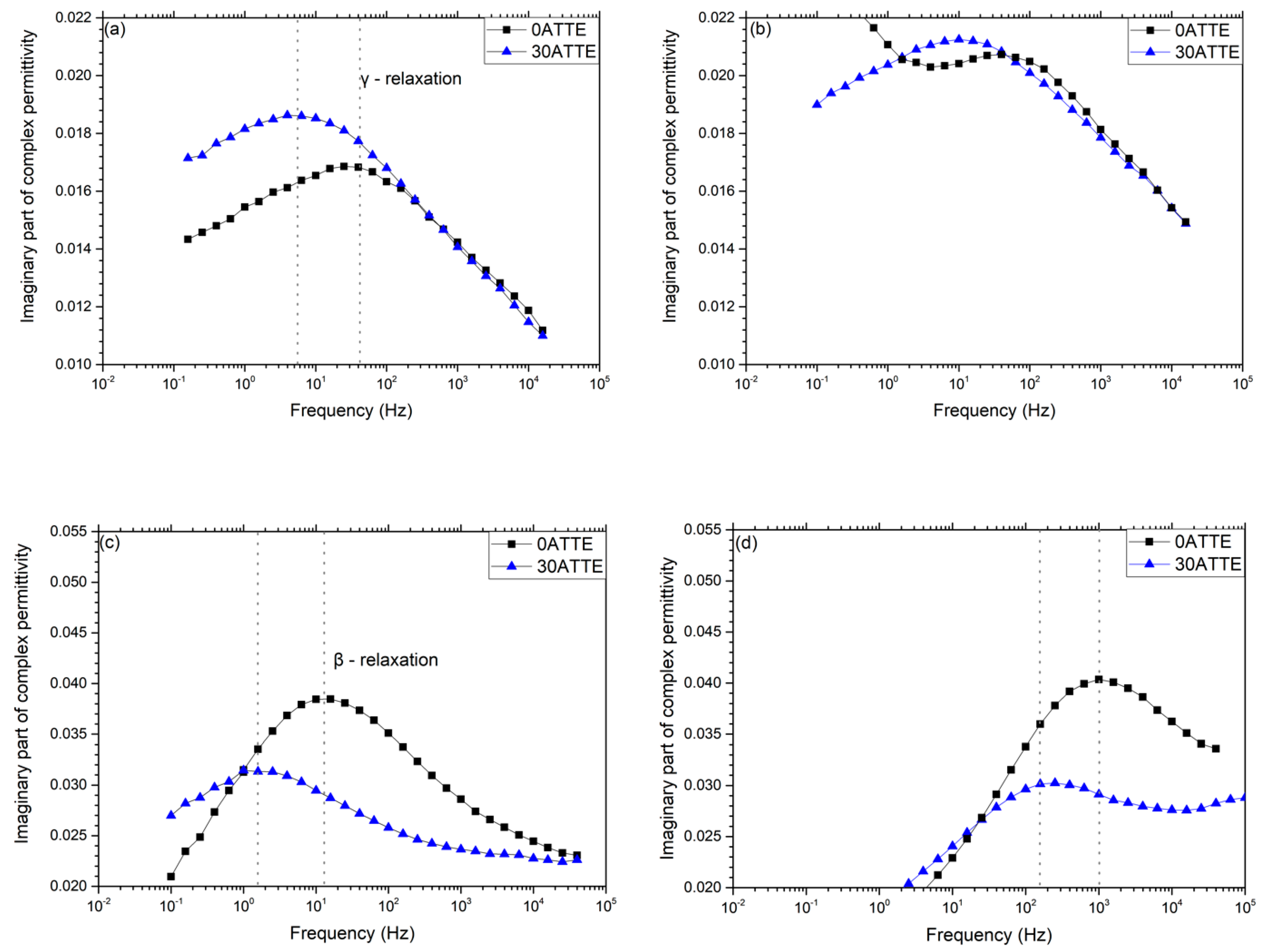
3.3. Dielectric Spectroscopy
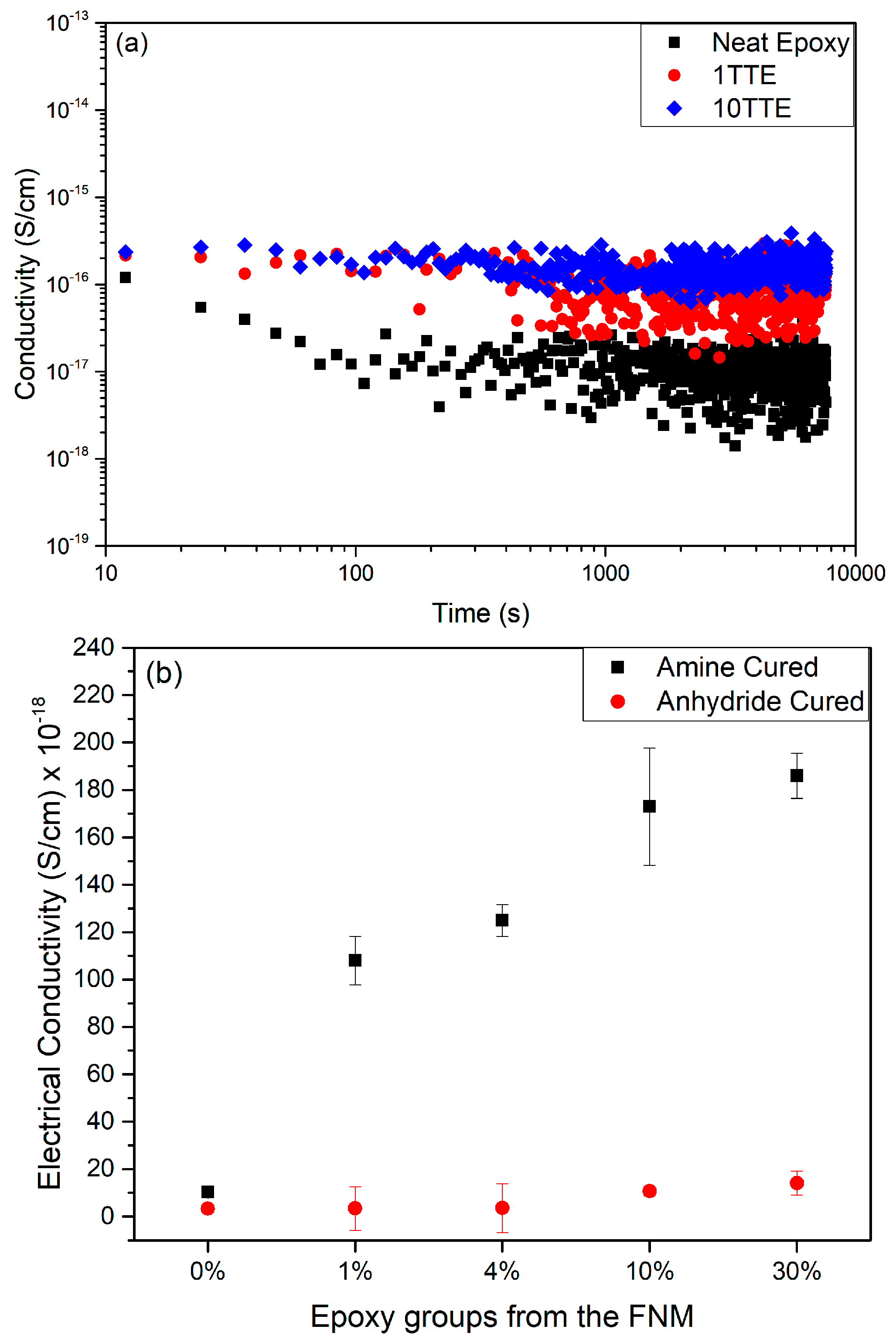
3.4. Electrical Conductivity
- Moieties explicitly introduced through the inclusion of the FNM;
- Variations in free volume in the various formulations;
- Changes in network dynamics that result from changes in network topology;
- The presence of specific unreacted functional groups from the resin and hardener.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Neville, K.; Lee, H. Handbook of Epoxy Resins; MacGraw-Hill: New York, NY, USA, 1967. [Google Scholar]
- May, C. Epoxy Resins: Chemistry and Technology; CRC Press: Boca Raton, FL, USA, 1987. [Google Scholar]
- Ellis, B. Chemistry and Technology of Epoxy Resins; Springer: Hall, UK, 1993. [Google Scholar]
- Nakka, J.; Jansen, K.; Ernst, L. Tailoring the viscoelasticity of epoxy thermosets. J. Appl. Polym. Sci. 2013, 128, 3794–3806. [Google Scholar] [CrossRef]
- Potter, W.G. Epoxide Resins; The Plastics Institute: London, UK, 1970. [Google Scholar]
- Stevens, G.C. Cure kinetics of a low epoxide/hydroxyl group-ratio bisphenol a epoxy resin–anhydride system by infrared absorption spectroscopy. J. Appl. Polym. Sci. 1981, 26, 4259–4278. [Google Scholar] [CrossRef]
- Steinmann, B. Investigations on the curing of epoxy resins with hexahydrophthalic anhydride. J. Appl. Polym. Sci. 1989, 37, 1753–1776. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kakiuchi, H. Study of epoxy compounds. Part VI. Curing reactions of epoxy resin and acid anhydride with amine, acid, alcohol, and phenol as catalysts. J. Polym. Sci. Part A Gen. Pap. 1964, 2, 3405–3430. [Google Scholar] [CrossRef]
- Carbas, R.; Marques, E.; Da Silva, L.; Lopes, A. Effect of cure temperature on the glass transition temperature and mechanical properties of epoxy adhesives. J. Adhes. 2014, 90, 104–119. [Google Scholar] [CrossRef]
- Carbas, R.; Da Silva, L.; Marques, E.; Lopes, A. Effect of post-cure on the glass transition temperature and mechanical properties of epoxy adhesives. J. Adhes. Sci. Technol. 2013, 27, 2542–2557. [Google Scholar] [CrossRef]
- Harani, H.; Fellahi, S.; Bakar, M. Toughening of epoxy resin using hydroxyl-terminated polyesters. J. Appl. Polym. Sci. 1999, 71, 29–38. [Google Scholar] [CrossRef]
- Tang, B.; Liu, X.; Zhao, X.; Zhang, J. Highly efficient in situ toughening of epoxy thermosets with reactive hyperbranched polyurethane. J. Appl. Polym. Sci. 2014, 131, 40614. [Google Scholar] [CrossRef]
- Gui, D.; Gao, X.; Hao, J.; Liu, J. Preparation and characterization of liquid crystalline polyurethane-imide modified epoxy resin composites. Polym. Eng. Sci. 2014, 54, 1704–1711. [Google Scholar] [CrossRef]
- Nguyen, V.; Vaughan, A.; Lewin, P.; Krivda, A. The effect of resin stoichiometry and nanoparticle addition on epoxy/silica nanodielectrics. IEEE Trans. Dielectr. Electr. Insul. 2015, 22, 895–905. [Google Scholar] [CrossRef]
- Alhabill, F.; Ayoob, R.; Andritsch, T.; Vaughan, A. Effect of resin/hardener stoichiometry on electrical behavior of epoxy networks. IEEE Trans. Dielectr. Electr. Insul. 2017, 24, 3739–3749. [Google Scholar] [CrossRef] [Green Version]
- Saeedi, I.A.; Vaughan, A.S.; Andritsch, T.; Virtanen, S. The effect of curing conditions on the electrical properties of an epoxy resin. IEEE Conf. Electr. Insul. Dielectr. Phenom. 2016, 461–464. [Google Scholar]
- Saeedi, I.A.; Vaughan, A.S.; Andritsch, T. Functional design of epoxy-based networks: Tailoring advanced dielectrics for next-generation energy systems. J. Phys. D Appl. Phys. 2019, 52, 205301. [Google Scholar] [CrossRef]
- Menczel, J.D.; Prime, R.B. Thermal Analysis of Polymers: Fundamentals and Applications; John Wiley and Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- Geyer, R.G. Dielectric Characterization and Reference Materials; Technical Note (NIST TN)-1338; NIST: Gaithersburg, MD, USA, 1990; pp. 90–92. [Google Scholar]
- Virtanen, S.; Vaughan, A.; Yang, S.; Saiz, F.; Quirke, N. Electrical Conductivity and Moisture Uptake Studies of Low Density Polyethylene Octylnanosilica Composite. In Proceedings of the Nordic Insulation Symposium, Trondheim, Norway, 3 October 2017. [Google Scholar]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.A. Introduction to Spectroscopy; Cengage Learning: Boston, MA, USA, 2008. [Google Scholar]
- González, M.G.; Cabanelas, J.C.; Baselga, J. Applications of FTIR on epoxy resins-identification, monitoring the curing process, phase separation and water uptake. Infrared Spectrosc. Mater. Sci. Eng. Technol. 2012, 2, 261–284. [Google Scholar]
- Lambert, J. Organic Structural Analysis; Macmillan Pub Co.: New York, NY, USA, 1976. [Google Scholar]
- Bellamy, L.J. The Infra-Red Spectra of Complex Molecules; Springer: London, UK, 1975. [Google Scholar]
- Foix, D.; Yu, Y.; Serra, A.; Ramis, X.; Salla, J.M. Study on the chemical modification of epoxy/anhydride thermosets using a hydroxyl terminated hyperbranched polymer. Eur. Polym. J. 2009, 45, 1454–1466. [Google Scholar] [CrossRef]
- Flores, M.; Fernández-Francos, X.; Ramis, X.; Serra, A. Novel epoxy-anhydride thermosets modified with a hyperbranched polyester as toughness enhancer. I. Kinetics study. Thermochim. Acta 2012, 544, 17–26. [Google Scholar] [CrossRef]
- Vryonis, O.; Virtanen, S.; Andritsch, T.; Vaughan, A.; Lewin, P. Understanding the cross-linking reactions in highly oxidized graphene/epoxy nanocomposite systems. J. Mater. Sci. 2018, 54, 3035–3051. [Google Scholar] [CrossRef] [Green Version]
- Ollier-Dureault, V.; Gosse, B. Photooxidation of anhydride-cured epoxies: FTIR study of the modifications of the chemical structure. J. Appl. Polym. Sci. 1998, 70, 1221–1237. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Elsevier Science: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Lin, S.-Y.; Cheng, W.-T.; Wei, Y.-S.; Lin, H.-L. DSC-FTIR microspectroscopy used to investigate the heat-induced intramolecular cyclic anhydride formation between Eudragit E and PVA copolymer. Polym. J. 2011, 43, 577–580. [Google Scholar] [CrossRef] [Green Version]
- Romão, B.; Diniz, M.F.; Azevedo, M.F.; Lourenço, V.L.; Pardini, L.C.; Dutra, R.C.; Burel, F. Characterization of the curing agents used in epoxy resins with TG/FT-IR technique. Polímeros 2006, 16, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Pissis, P.; Fragiadakis, D. Dielectric studies of segmental dynamics in epoxy nanocomposites. J. Macromol. Sci. Part B Phys. 2007, 46, 119–136. [Google Scholar] [CrossRef]
- Bakar, M.; Duk, R.; Przybyłek, M.; Kostrzewa, M. Mechanical and thermal properties of epoxy resin modified with polyurethane. J. Reinf. Plast. Compos. 2009, 28, 2107–2118. [Google Scholar] [CrossRef]
- Saeedi, I.; Hamad, A.; Vaughan, A.; Andritsch, T. Change in the electrical properties due to modification of the epoxy network structure using reactive diluents. In Proceedings of the 2016 IEEE Conference on Electrical Insulation and Dielectric Phenomena, Toronto, ON, Canada, 16–19 October 2016. [Google Scholar]
- Morgan, R.J.; Kong, F.-M.; Walkup, C.M. Structure-property relations of polyethertriamine-cured bisphenol-A-diglycidyl ether epoxies. Polymer 1984, 25, 375–386. [Google Scholar] [CrossRef]
- Alhabill, F.N.; Ayoob, R.; Andritsch, T.; Vaughan, A.S. Influence of filler/matrix interactions on resin/hardener stoichiometry, molecular dynamics, and particle dispersion of silicon nitride/epoxy nanocomposites. J. Mater. Sci. 2018, 53, 4144–4158. [Google Scholar] [CrossRef]
- Hassan, M.K.; Tucker, S.J.; Abukmail, A.; Wiggins, J.S.; Mauritz, K.A. Polymer chain dynamics in epoxy based composites as investigated by broadband dielectric spectroscopy. Arab. J. Chem. 2016, 9, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Jilani, W.; Mzabi, N.; Fourati, N.; Zerrouki, C.; Gallot-Lavallée, O.; Zerrouki, R.; Guermazi, H. A comparative study of structural and dielectric properties of diglycidyl ether of bisphenol A (DGEBA) cured with aromatic or aliphatic hardeners. J. Mater. Sci. 2016, 51, 7874–7886. [Google Scholar] [CrossRef]
- Shimbo, M.; Ochi, M.; Iesako, H. Mechanical relaxation mechanism of epoxide resins cured with acid anhydrides. J. Polym. Sci. Part B Polym. Phys. 1984, 22, 1461–1470. [Google Scholar] [CrossRef]
- Mikolajczak, G.; Cavaille, J.I. Dynamic mechanical behaviour and its dependence on operation method of structural epoxide resin. Polymer 1987, 28, 2023–2031. [Google Scholar] [CrossRef]
- Saeedi, I.; Vaughan, A.; Andritsch, T.; Dissado, L.; Chalashkanov, N. On molecular dynamics and charge transport in functionally modified epoxy networks. 2019, in press. [Google Scholar]
- Cuddihy, E.; Moacanin, J. Dynamic Mechanical Properties of Epoxies’ Beta-Transition Mechanism; American Chemical Society: Chicago, IL, USA, 1970. [Google Scholar]
- Ochi, M.; Iesako, H.; Nakajima, S.; Shimbo, M. Mechanical relaxation mechanism of epoxide resins cured with acid anhydrides. II. Effect of the chemical structure of the anhydrides on the β relaxation mechanism. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 251–261. [Google Scholar] [CrossRef]
- Mangion, M.B.; Johari, G. Relaxations in thermosets. 7. Dielectric effects during the curing and postcuring of an epoxide by mixed amines. Macromolecules 1990, 23, 3687–3695. [Google Scholar] [CrossRef]
- Varshney, V.; Patnaik, S.S.; Roy, A.K.; Farmer, B.L. A molecular dynamics study of epoxy-based networks: Cross-linking procedure and prediction of molecular and material properties. Macromolecules 2008, 41, 6837–6842. [Google Scholar] [CrossRef]
- Fan, H.B.; Yuen, M.M. Material properties of the cross-linked epoxy resin compound predicted by molecular dynamics simulation. Polymer 2007, 48, 2174–2178. [Google Scholar] [CrossRef]
- Wu, C.; Xu, W. Atomistic molecular modelling of crosslinked epoxy resin. Polymer 2006, 47, 6004–6009. [Google Scholar] [CrossRef]
- Yang, S.; Qu, J. Computing thermomechanical properties of crosslinked epoxy by molecular dynamic simulations. Polymer 2012, 53, 4806–4817. [Google Scholar] [CrossRef]
- McGonigle, E.-A.; Daly, J.H.; Jenkins, S.D.; Liggat, J.J.; Pethrick, R.A. Influence of physical aging on the molecular motion and structural relaxation in poly (ethylene terephthalate) and related polyesters. Macromolecules 2000, 33, 480–489. [Google Scholar] [CrossRef]
- Verot, S.; Battesti, P.; Perrier, G. Semi-empirical calculations and dielectric spectrometry of molecular units in PEEK. Polymer 1999, 40, 2605–2617. [Google Scholar] [CrossRef]
- Leonardi, A.; Dantras, E.; Dandurand, J.; Lacabanne, C. Dielectric relaxations in PEEK by combined dynamic dielectric spectroscopy and thermally stimulated current. J. Therm. Anal. Calorim. 2013, 111, 807–814. [Google Scholar] [CrossRef]
- Rault, J. Origin of the Vogel–Fulcher–Tammann law in glass-forming materials: The α–β bifurcation. J. Non Cryst. Solids 2000, 271, 177–217. [Google Scholar] [CrossRef]
- Saeedi, I.; Vaughan, A.; Andritsch, T. On the dielectric performance of modified epoxy networks. In Proceedings of the 2016 IEEE International Conference on Dielectrics (ICD), Montpellier, France, 3–7 July 2016; pp. 1044–1047. [Google Scholar]
- Wang, Y.; MacKernan, D.; Cubero, D.; Coker, D.F.; Quirke, N. Single electron states in polyethylene. J. Chem. Phys. 2014, 140, 154902. [Google Scholar] [CrossRef] [Green Version]
- Cubero, D.; Quirke, N.; Coker, D.F. Electronic transport in disordered n-alkanes: From fluid methane to amorphous polyethylene. J. Chem. Phys. 2003, 119, 2669–2679. [Google Scholar] [CrossRef] [Green Version]
- Cubero, D.; Quirke, N. Computer simulations of localized small polarons in amorphous polyethylene. J. Chem. Phys. 2004, 120, 7772–7778. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.M.P.; James, W. Surface Active Agents; Interscience Publishers, Inc.: New York, NY, USA, 1949. [Google Scholar]
- Meunier, M.; Quirke, N. Molecular modeling of electron trapping in polymer insulators. J. Chem. Phys. 2000, 113, 369–376. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tg (°C) Amine-Cured (±2 °C) | Tg (°C) Anhydride-Cured (±2 °C) |
|---|---|---|
| Reference | 85.3 | 102.9 |
| 1% | 83.4 | 100.5 |
| 4% | 80.1 | 99.0 |
| 10% | 78.5 | 94.15 |
| 30% | 68.6 | 87.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeedi, I.A.; Andritsch, T.; Vaughan, A.S. On the Dielectric Behavior of Amine and Anhydride Cured Epoxy Resins Modified Using Multi-Terminal Epoxy Functional Network Modifier. Polymers 2019, 11, 1271. https://doi.org/10.3390/polym11081271
Saeedi IA, Andritsch T, Vaughan AS. On the Dielectric Behavior of Amine and Anhydride Cured Epoxy Resins Modified Using Multi-Terminal Epoxy Functional Network Modifier. Polymers. 2019; 11(8):1271. https://doi.org/10.3390/polym11081271
Chicago/Turabian StyleSaeedi, Istebreq A., Thomas Andritsch, and Alun S. Vaughan. 2019. "On the Dielectric Behavior of Amine and Anhydride Cured Epoxy Resins Modified Using Multi-Terminal Epoxy Functional Network Modifier" Polymers 11, no. 8: 1271. https://doi.org/10.3390/polym11081271
APA StyleSaeedi, I. A., Andritsch, T., & Vaughan, A. S. (2019). On the Dielectric Behavior of Amine and Anhydride Cured Epoxy Resins Modified Using Multi-Terminal Epoxy Functional Network Modifier. Polymers, 11(8), 1271. https://doi.org/10.3390/polym11081271