On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate


Abstract
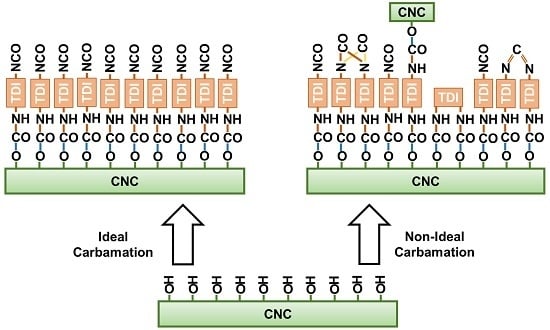
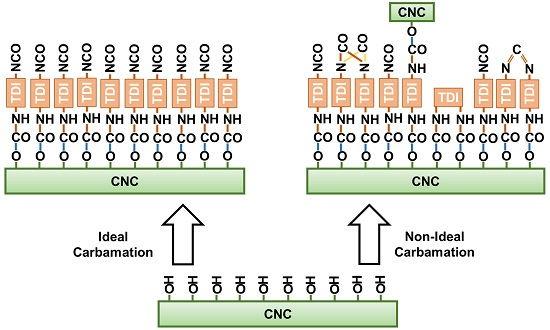
1. Introduction
2. Materials and Methods
2.1. Materials
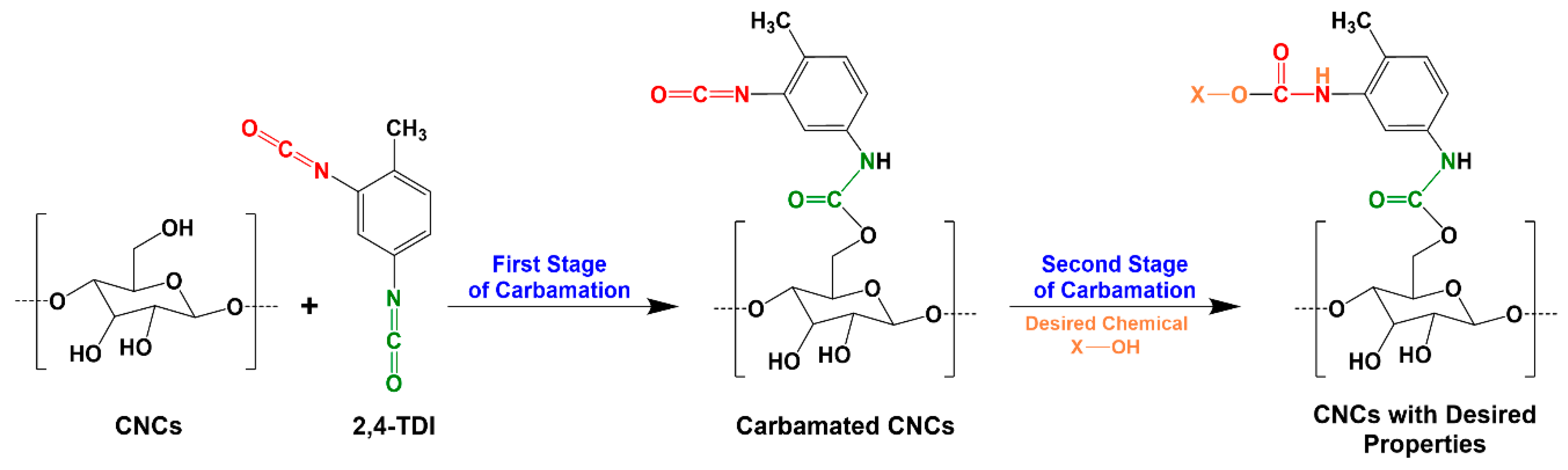
2.2. TDI-Carbamation of Cellulose Nanocrystals
2.3. Determination of the Degree of Substitution of TDI (DSTDI)
2.4. Determination of the Degree of Substitution of Free Isocyanates (DSNCO)
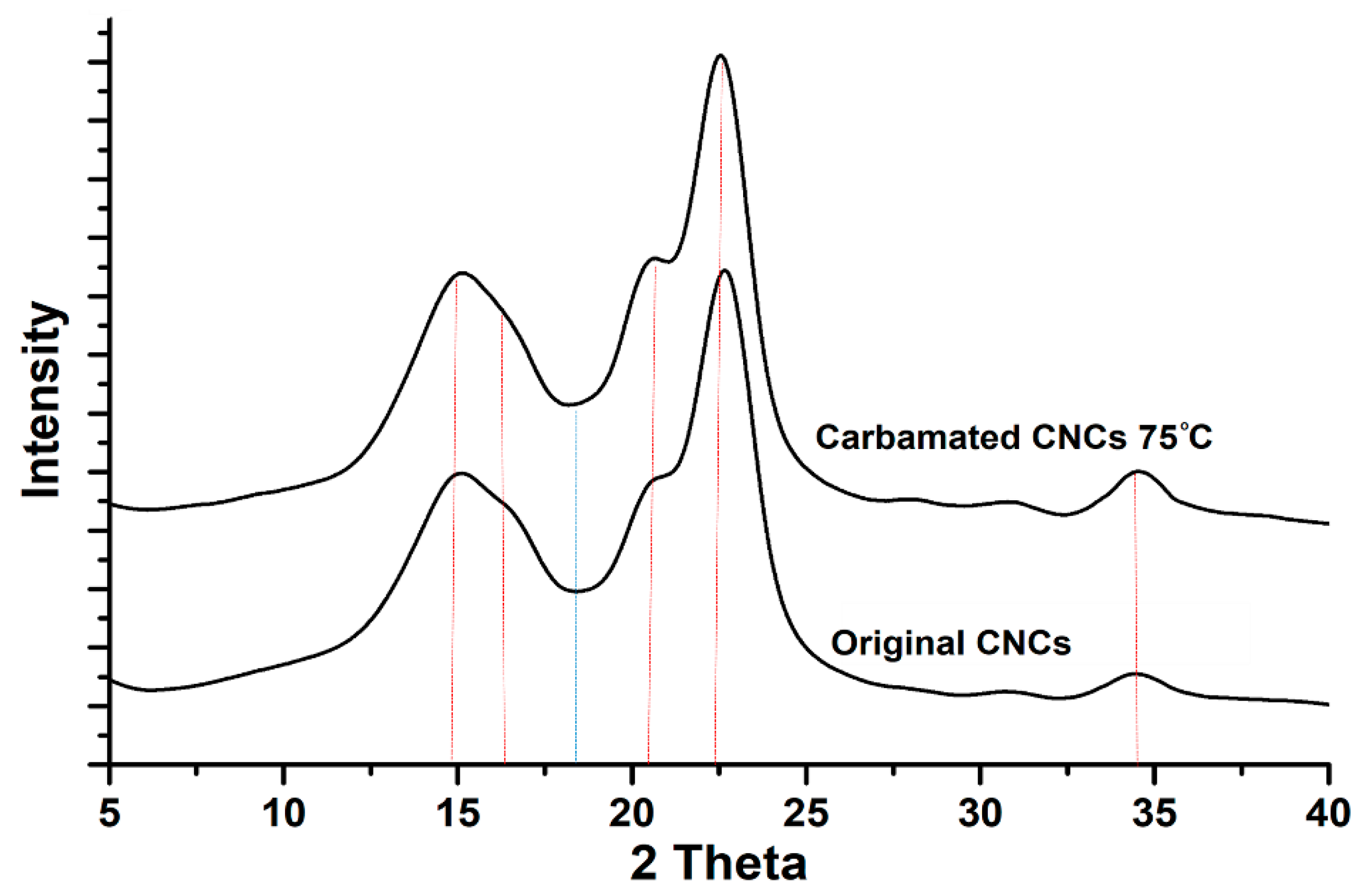
2.5. Morphological Characterization using X-Ray Diffraction (XRD)
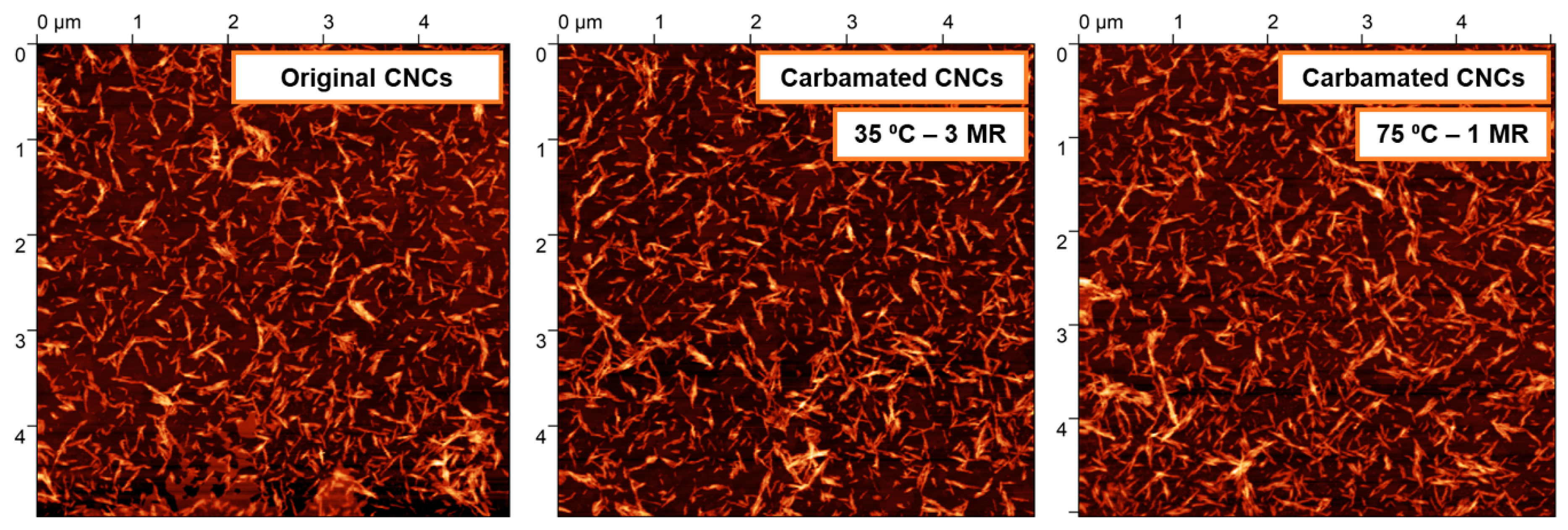
2.6. Morphological Characterization using Atomic Force Microscopy (AFM)
3. Results and Discussion
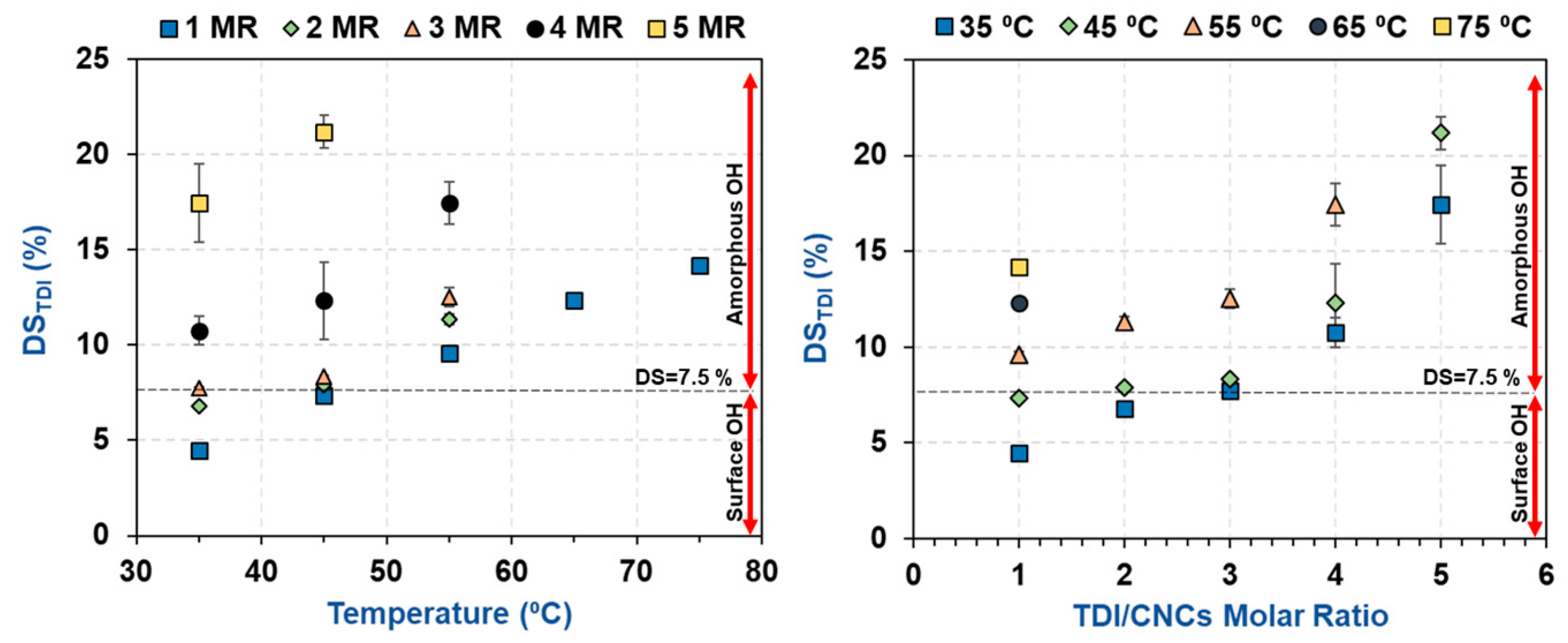
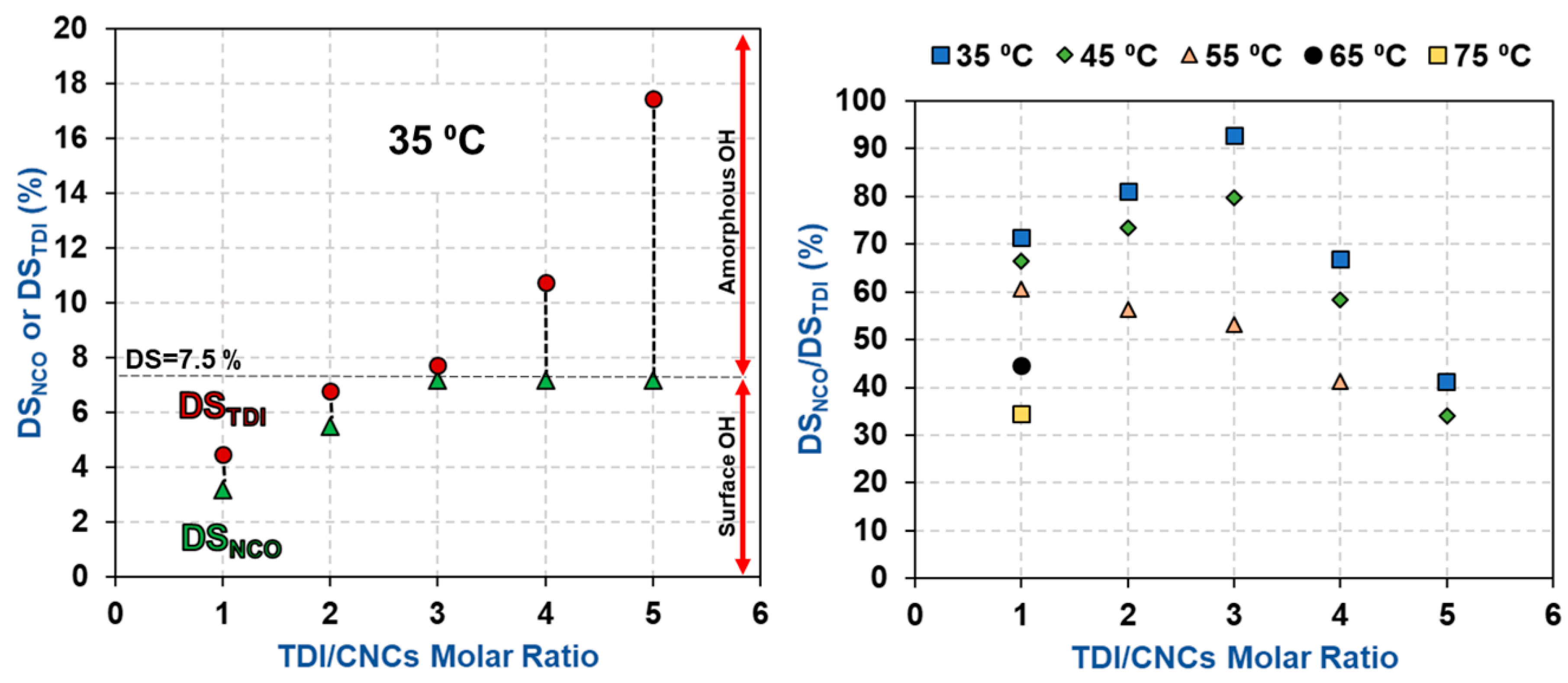
3.1. Estimation of the Percentage of Hydroxyl Groups on the CNC Surface
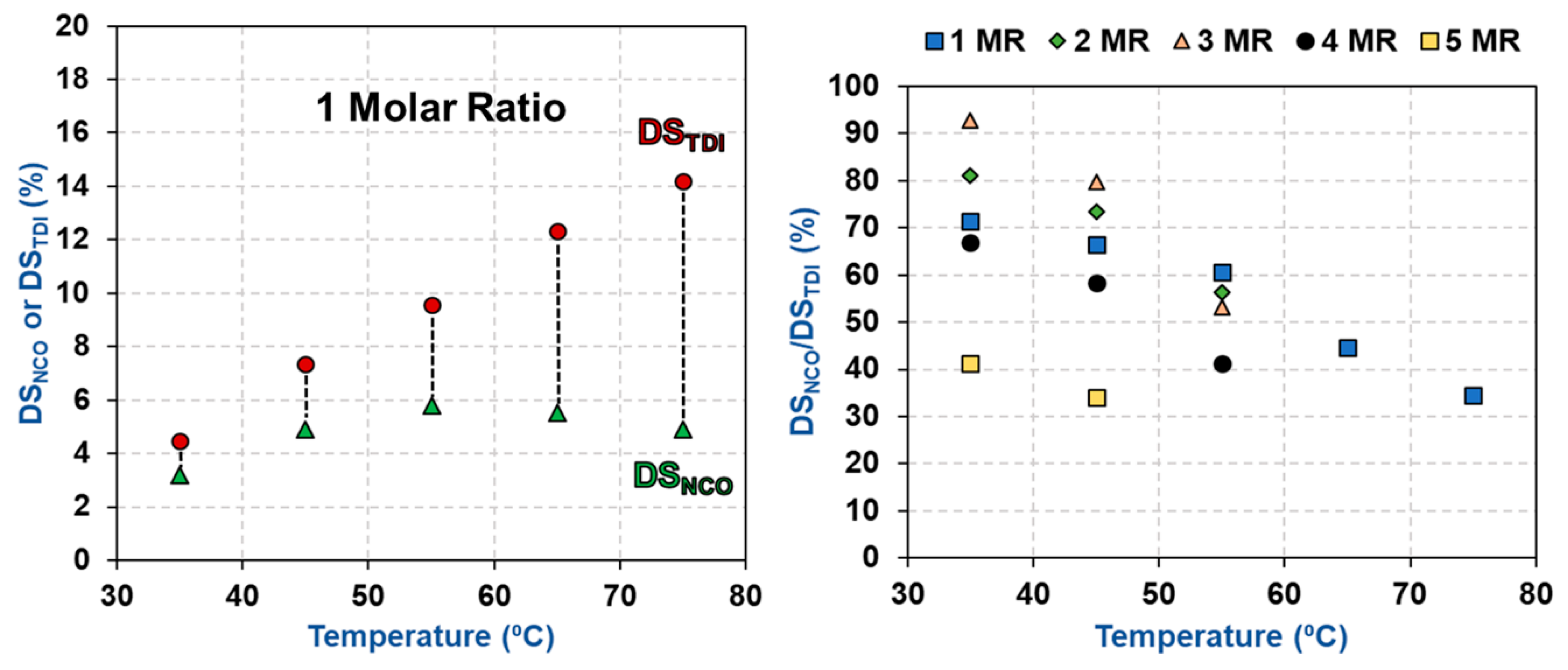
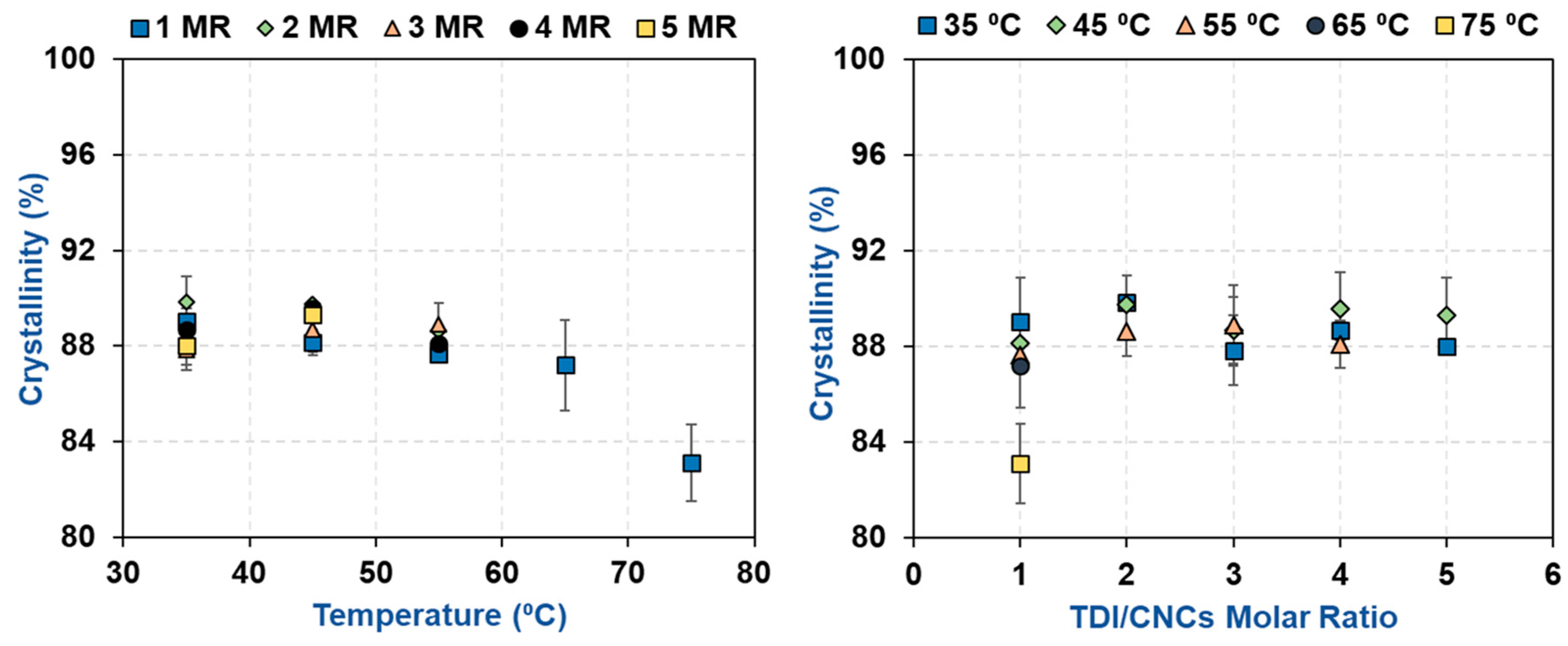
3.2. Impact of Temperature and TDI/CNCs Molar Ratio on Carbamation Efficiency
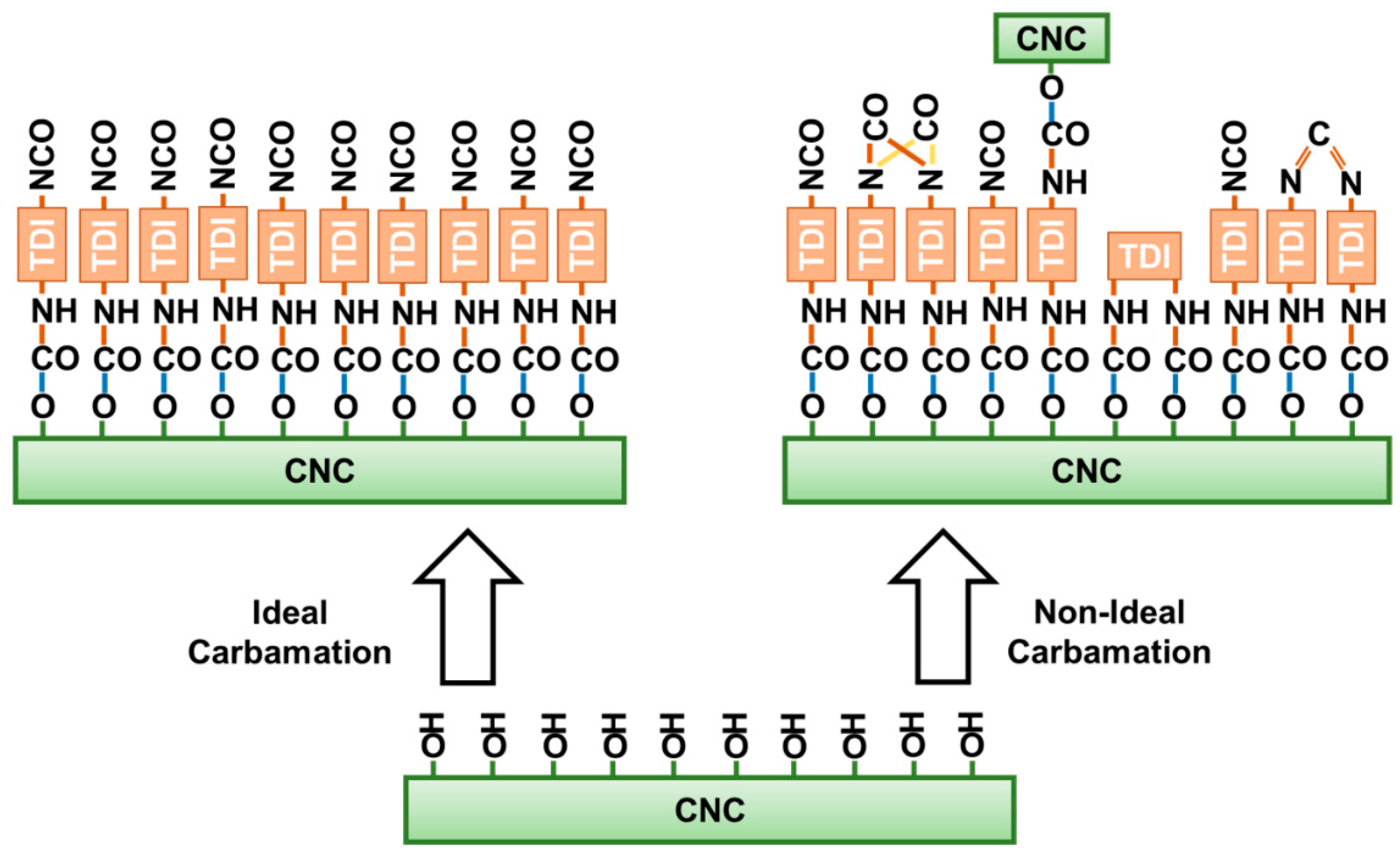
3.3. Possible Side Reactions during TDI-Based Carbamation
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Standard Terms and Their Definition for Cellulose Nanomaterial (ISO/TS 20477); International Organization for Standardization (ISO): Geneva, Switzerland, 2017.
- Bondeson, D.; Mathew, A.; Oksman, K. Optimization of the isolation of nanocrystals from microcrystalline cellulose by acid hydrolysis. Cellulose 2006, 13, 171. [Google Scholar] [CrossRef]
- Leung, C.W.; Luong, J.H.; Hrapovic, S.; Lam, E.; Liu, Y.; Male, K.B.; Mahmoud, K.; Rho, D. Cellulose nanocrystals from renewable biomass. U.S. Patent 8900706B2, 2 December 2014. [Google Scholar]
- Abushammala, H.; Krossing, I.; Laborie, M.P. Ionic liquid-mediated technology to produce cellulose nanocrystals directly from wood. Carbohydr. Polym. 2015, 134, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Abushammala, H.; Brown, N.; Laborie, M.-P. Comparative assessment of methods for producing cellulose I nanocrystals from cellulosic sources. In Nanocelluloses: Their Preparation, Properties, and Applications, ACS Symposium Series; ACS Publications: Washington, DC, USA, 2018. [Google Scholar]
- Salajková, M.; Berglund, L.A.; Zhou, Q. Hydrophobic cellulose nanocrystals modified with quaternary ammonium salts. J. Mater. Chem. 2012, 22, 19798–19805. [Google Scholar] [CrossRef]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-Y.; Qin, Z.Y. Surface grafting of cellulose nanocrystals with poly (3-hydroxybutyrate-co-3-hydroxyvalerate). Carbohydr. Polym. 2014, 101, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Morandi, G.; Thielemans, W. Synthesis of cellulose nanocrystals bearing photocleavable grafts by ATRP. Polym. Chem. 2012, 3, 1402–1407. [Google Scholar] [CrossRef]
- Hubmann, M.; Kong, X.; Curtis, J.M. Kinetic stabilization of cellulose nanocrystals in a photocurable prepolymer for application as an adhesion promoter in UV-curable coatings. Prog. Org. Coat. 2019, 129, 101–115. [Google Scholar] [CrossRef]
- Espino-Pérez, E.; Bras, J.; Ducruet, V.; Guinault, A.; Dufresne, A.; Domenek, S. Influence of chemical surface modification of cellulose nanowhiskers on thermal, mechanical, and barrier properties of poly (lactide) based bionanocomposites. Eur. Polym. J. 2013, 49, 3144–3154. [Google Scholar] [CrossRef]
- Pinheiro, I.; Ferreira, F.; Alves, G.; Rodolfo, A.; Morales, A.; Mei, L. Biodegradable PBAT-based nanocomposites reinforced with functionalized cellulose nanocrystals from Pseudobombax munguba: rheological, thermal, mechanical and biodegradability properties. J. Polym. Environ. 2019, 27, 757–766. [Google Scholar] [CrossRef]
- Habibi, Y.; Dufresne, A. Highly filled bionanocomposites from functionalized polysaccharide nanocrystals. Biomacromolecules 2008, 9, 1974–1980. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Peresin, M.S.; Habibi, Y.; Venditti, R.A.; Rojas, O.J. Reinforcing poly (ε-caprolactone) nanofibers with cellulose nanocrystals. ACS Appl. Mater. Interfaces 2009, 1, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Huang, J.; Luo, H.; Chang, P.R.; Feng, J.; Xie, G. Hydrophobic modification of cellulose nanocrystal via covalently grafting of castor oil. Cellulose 2013, 20, 179–190. [Google Scholar] [CrossRef]
- Dounis, D.V.; Wilkes, G.L. Structure-property relationships of flexible polyurethane foams. Polymer 1997, 38, 2819–2828. [Google Scholar] [CrossRef]
- Barikani, M.; Valipour Ebrahimi, M.; Seyed Mohaghegh, S. Preparation and characterization of aqueous polyurethane dispersions containing ionic centers. J. Appl. Polym. Sci. 2007, 104, 3931–3937. [Google Scholar] [CrossRef]
- Zhang, Y.; Qi, Y.H.; Zhang, Z.P. The influence of 2, 4 toluene diisocyanate content on the intrinsic self-healing performance of polyurethane at room-temperature. J. Polym. Res. 2015, 22, 94. [Google Scholar] [CrossRef]
- Belgacem, M.N.; Quillerou, J.; Gandini, A. Urethanes and polyurethanes bearing furan moieties—3. Synthesis, characterization and comparative kinetics of the formation of diurethanes. Eur. Polym. J. 1993, 29, 1217–1224. [Google Scholar] [CrossRef]
- Semsarzadeh, M.; Navarchian, A. Kinetic study of the bulk reaction between TDI and PPG in prescence of DBTDL and FEAA catalysts using quantitative FTIR spectroscopy. J. Polym. Eng. 2003, 23, 225–240. [Google Scholar] [CrossRef]
- Eyley, S.; Thielemans, W. Surface modification of cellulose nanocrystals. Nanoscale 2014, 6, 7764–7779. [Google Scholar] [CrossRef]
- Špírková, M.; Kubin, M.; Dušek, K. Side reactions in the formation of polyurethanes: Model reactions between phenylisocyanate and 1-butanol. J. Macromol. Sci. Chem. 1987, 24, 1151–1166. [Google Scholar] [CrossRef]
- Abushammala, H. A simple method for the quantification of free isocyanates on the surface of cellulose nanocrystals upon carbamation using toluene diisocyanate. Surfaces 2019, 2, 444–454. [Google Scholar] [CrossRef]
- Terinte, N.; Ibbett, R.; Schuster, K.C. Overview on native cellulose and microcrystalline cellulose I structure studied by X-ray diffraction (WAXD): Comparison between measurement techniques. Lenzing. Ber. 2011, 89, 118. [Google Scholar]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Verlhac, C.; Dedier, J.; Chanzy, H. Availability of surface hydroxyl groups in Valonia and bacterial cellulose. J. Polym. Sci. Part A Polym. Chem. 1990, 28, 1171–1177. [Google Scholar] [CrossRef]
- Xu, D.; Li, B.; Tate, C.; Edgar, K.J. Studies on regioselective acylation of cellulose with bulky acid chlorides. Cellulose 2011, 18, 405–419. [Google Scholar] [CrossRef]
- Marchessault, R.; Morehead, F.; Koch, M.J. Some hydrodynamic properties of neutral suspensions of cellulose crystallites as related to size and shape. J. Colloid Sci. 1961, 16, 327–344. [Google Scholar] [CrossRef]
- Beck, S.; Bouchard, J.; Berry, R. Dispersibility in water of dried nanocrystalline cellulose. Biomacromolecules 2012, 13, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Méthot, M.; Bouchard, J. General procedure for determining cellulose nanocrystal sulfate half-ester content by conductometric titration. Cellulose 2015, 22, 101–116. [Google Scholar] [CrossRef]
- Freire, C.; Silvestre, A.; Neto, C.P.; Belgacem, M.N.; Gandini, A. Controlled heterogeneous modification of cellulose fibers with fatty acids: Effect of reaction conditions on the extent of esterification and fiber properties. J. Appl. Polym. Sci. 2006, 100, 1093–1102. [Google Scholar] [CrossRef]
- Habibi, Y. Key advances in the chemical modification of nanocelluloses. Chem. Soc. Rev. 2014, 43, 1519–1542. [Google Scholar] [CrossRef]
- Van Maris, R.; Tamano, Y.; Yoshimura, H.; Gay, K.M. Polyurethane catalysis by tertiary amines. J. Cell. Plast. 2005, 41, 305–322. [Google Scholar] [CrossRef]
- Aranguren, M.I.; Williams, R.J. Kinetic and statistical aspects of the formation of polyurethanes from toluene diisocyanate. Polymer 1986, 27, 425–430. [Google Scholar] [CrossRef]
- Guo, Y.H.; Guo, J.J.; Li, S.C.; Li, X.; Wang, G.S.; Huang, Z. Properties and paper sizing application of waterborne polyurethane emulsions synthesized with TDI and IPDI. Colloids Surf. A Physicochem. Eng. Asp. 2013, 427, 53–61. [Google Scholar] [CrossRef]
- Zhan, S.; Chen, S.; Chen, L.; Hou, W. Preparation and characterization of polyurea microencapsulated phase change material by interfacial polycondensation method. Powder Technol. 2016, 292, 217–222. [Google Scholar] [CrossRef]
- Sarko, A.; Muggli, R. Packing analysis of carbohydrates and polysaccharides. III. Valonia cellulose and cellulose II. Macromolecules 1974, 7, 486–494. [Google Scholar] [CrossRef]
- Gardner, K.; Blackwell, J. The structure of native cellulose. Biopolym. Orig. Res. Biomol. 1974, 13, 1975–2001. [Google Scholar] [CrossRef]
- French, A.D. Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 2014, 21, 885–896. [Google Scholar] [CrossRef]
- Buckles, R.E.; McGrew, L. A kinetic study of the dimerization of phenyl isocyanate. J. Am. Chem. Soc. 1966, 88, 3582–3586. [Google Scholar] [CrossRef]
- Schwetlick, K.; Noack, R. Kinetics and catalysis of consecutive isocyanate reactions. Formation of carbamates, allophanates and isocyanurates. J. Chem. Soc. Perkin Trans. 2 1995, 2, 395–402. [Google Scholar] [CrossRef]
- Guo, J.; He, Y.; Xie, D.; Zhang, X. Process investigating and modelling for the self-polymerization of toluene diisocyanate (TDI)-based polyurethane prepolymer. J. Mater. Sci. 2015, 50, 5844–5855. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (°C) | TDI/CNCs Ratio | Mass TDI (g) | Volume TEA (mL) | Volume Toluene (mL) |
|---|---|---|---|---|
| 35 | 1 | 1.1 | 1.0 | 50.0 |
| 2 | 2.2 | 2.0 | 48.2 | |
| 3 | 3.3 | 3.0 | 46.3 | |
| 4 | 4.4 | 4.0 | 44.4 | |
| 5 | 5.5 | 5.0 | 42.5 | |
| 45 | 1 | 1.1 | 1.0 | 50.0 |
| 2 | 2.2 | 2.0 | 48.2 | |
| 3 | 3.3 | 3.0 | 46.3 | |
| 4 | 4.4 | 4.0 | 44.4 | |
| 5 | 5.5 | 5.0 | 42.5 | |
| 55 | 1 | 1.1 | 1.0 | 50.0 |
| 2 | 2.2 | 2.0 | 48.2 | |
| 3 | 3.3 | 3.0 | 46.3 | |
| 4 | 4.4 | 4.0 | 44.4 | |
| 65 | 1 | 1.1 | 1.0 | 50.0 |
| 75 | 1 | 1.1 | 1.0 | 50.0 |
| T (°C) | TDI/CNCs Ratio | DSNCO (%) | DSTDI (%) | DSNCO/DSTDI (%) |
|---|---|---|---|---|
| 35 | 1 | 3.2 ± 0.0 | 4.5 ± 0.1 | 72 |
| 2 | 5.5 ± 0.3 | 6.8 ± 0.2 | 81 | |
| 3 | 7.2 ± 0.3 | 7.7 ± 0.1 | 93 | |
| 4 | 7.2 ± 0.3 | 10.8 ± 0.8 | 67 | |
| 5 | 7.2 ± 0.3 | 17.4 ± 2.0 | 43 | |
| 45 | 1 | 4.9 ± 0.3 | 7.4 ± 0.2 | 66 |
| 2 | 5.8 ± 0.0 | 7.9 ± 0.2 | 73 | |
| 3 | 6.7 ± 0.3 | 8.4 ± 0.0 | 80 | |
| 4 | 7.2 ± 0.3 | 12.3 ± 2.0 | 58 | |
| 5 | 7.2 ± 0.3 | 21.2 ± 0.9 | 34 | |
| 55 | 1 | 5.8 ± 0.6 | 9.6 ± 0.2 | 61 |
| 2 | 6.4 ± 0.0 | 11.3 ± 0.3 | 56 | |
| 3 | 6.7 ± 0.3 | 12.5 ± 0.5 | 53 | |
| 4 | 7.2 ± 0.3 | 17.5 ± 1.1 | 41 | |
| 65 | 1 | 5.5 ± 0.3 | 12.3 ± 0.1 | 45 |
| 75 | 1 | 4.9 ± 0.3 | 14.2 ± 0.3 | 34 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abushammala, H. On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate. Polymers 2019, 11, 1164. https://doi.org/10.3390/polym11071164
Abushammala H. On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate. Polymers. 2019; 11(7):1164. https://doi.org/10.3390/polym11071164
Chicago/Turabian StyleAbushammala, Hatem. 2019. "On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate" Polymers 11, no. 7: 1164. https://doi.org/10.3390/polym11071164
APA StyleAbushammala, H. (2019). On the Para/Ortho Reactivity of Isocyanate Groups during the Carbamation of Cellulose Nanocrystals Using 2,4-Toluene Diisocyanate. Polymers, 11(7), 1164. https://doi.org/10.3390/polym11071164