Effect of Graphene oxide or Functionalized Graphene Oxide on the Copolymerization Kinetics of Styrene/n-butyl Methacrylate




Abstract
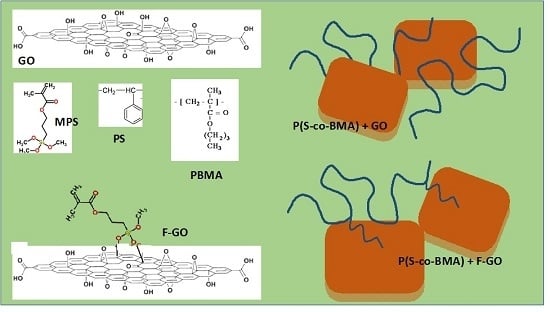
:
1. Introduction
2. Materials and Methods
2.1. Materials
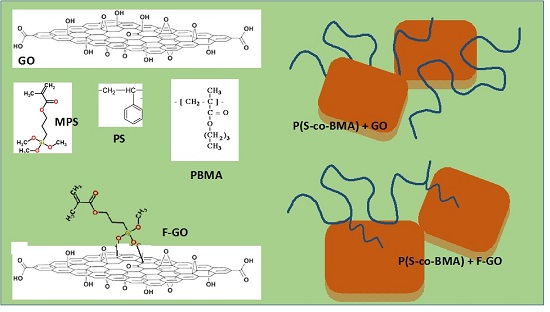
2.2. Synthesis of Graphite Oxide and Functionalized Graphite Oxide
2.3. Synthesis of P(S-co-BMA)/GO and P(S-co-BMA)/F-GO Nanocomposites by the In-Situ Bulk Radical Polymerization Technique
2.4. Measurements
3. Theoretical Section
- Chemical initiation:
- Thermal initiation of styrene:
- Chain Initiation:
- Propagation:
- Chain transfer to monomer:
- Chain transfer to polymer:
- Termination by combination or disproportionation:
4. Results and Discussion
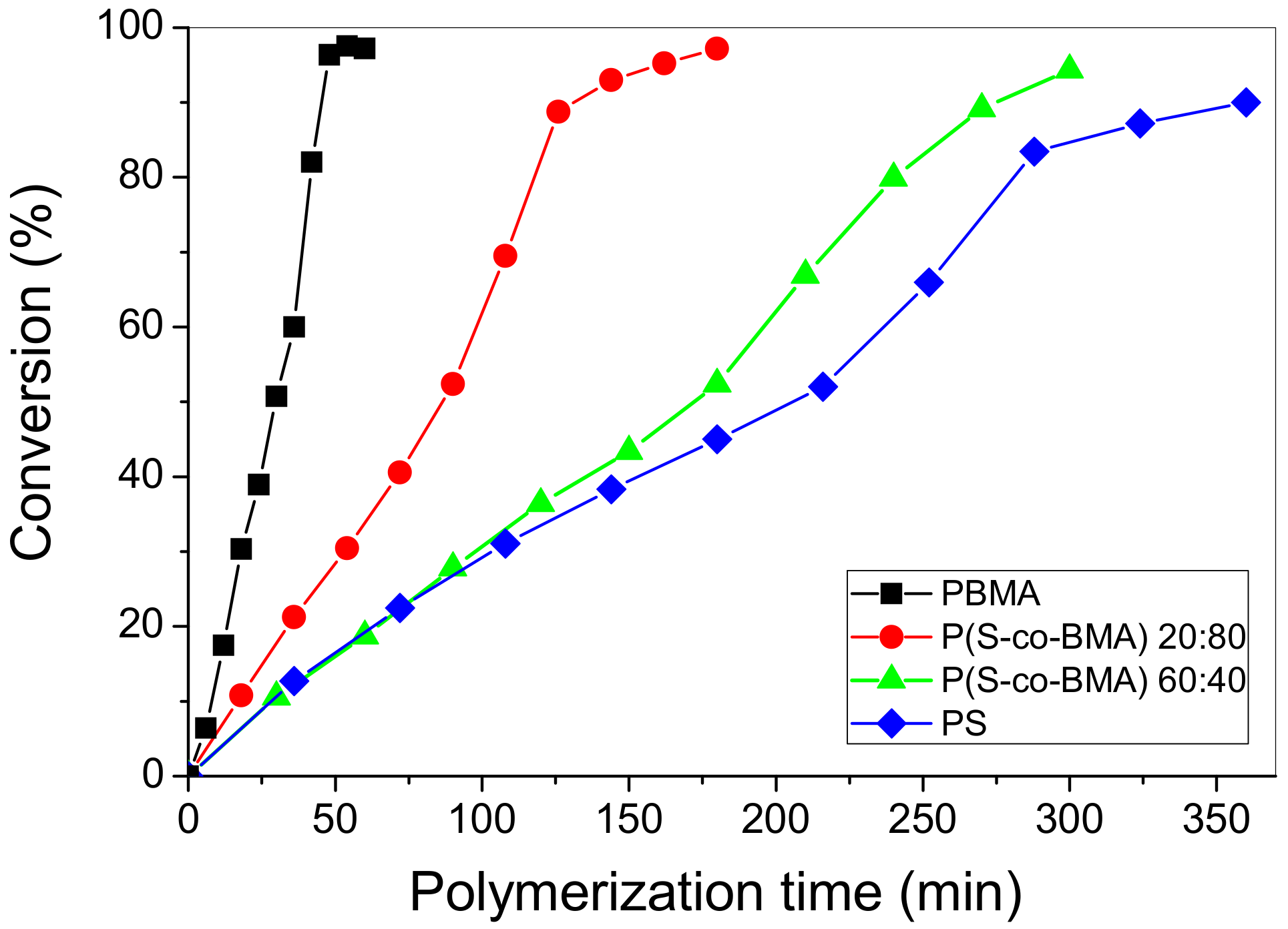
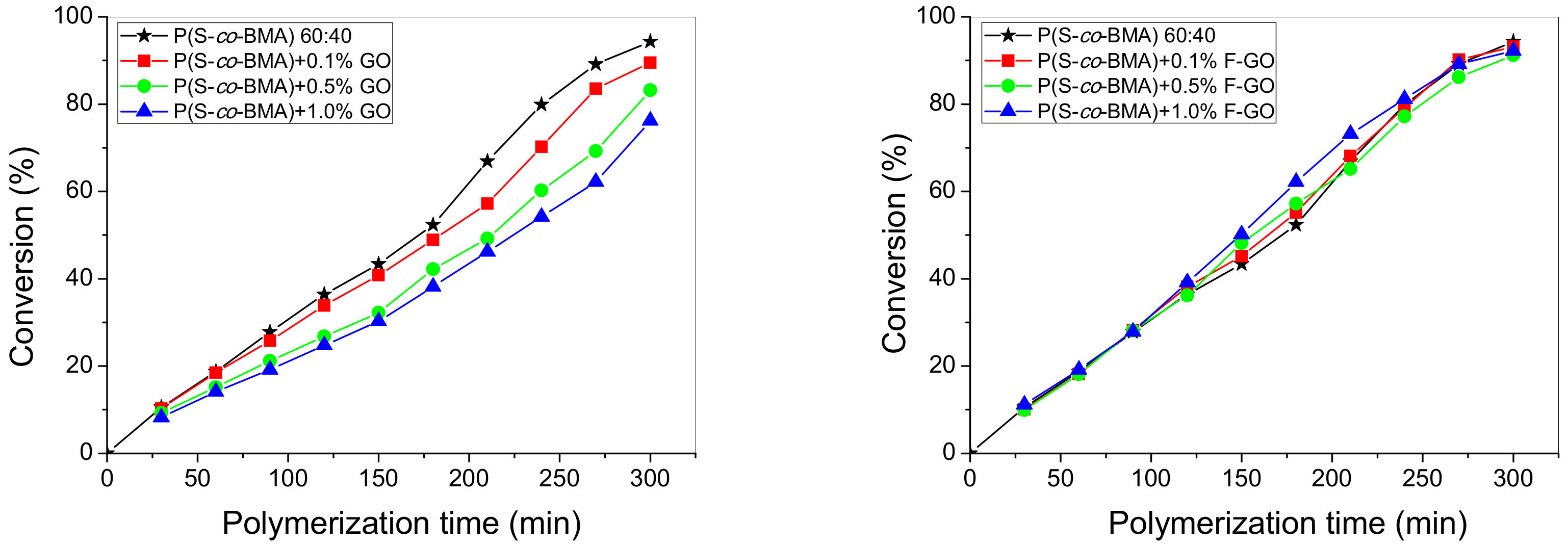
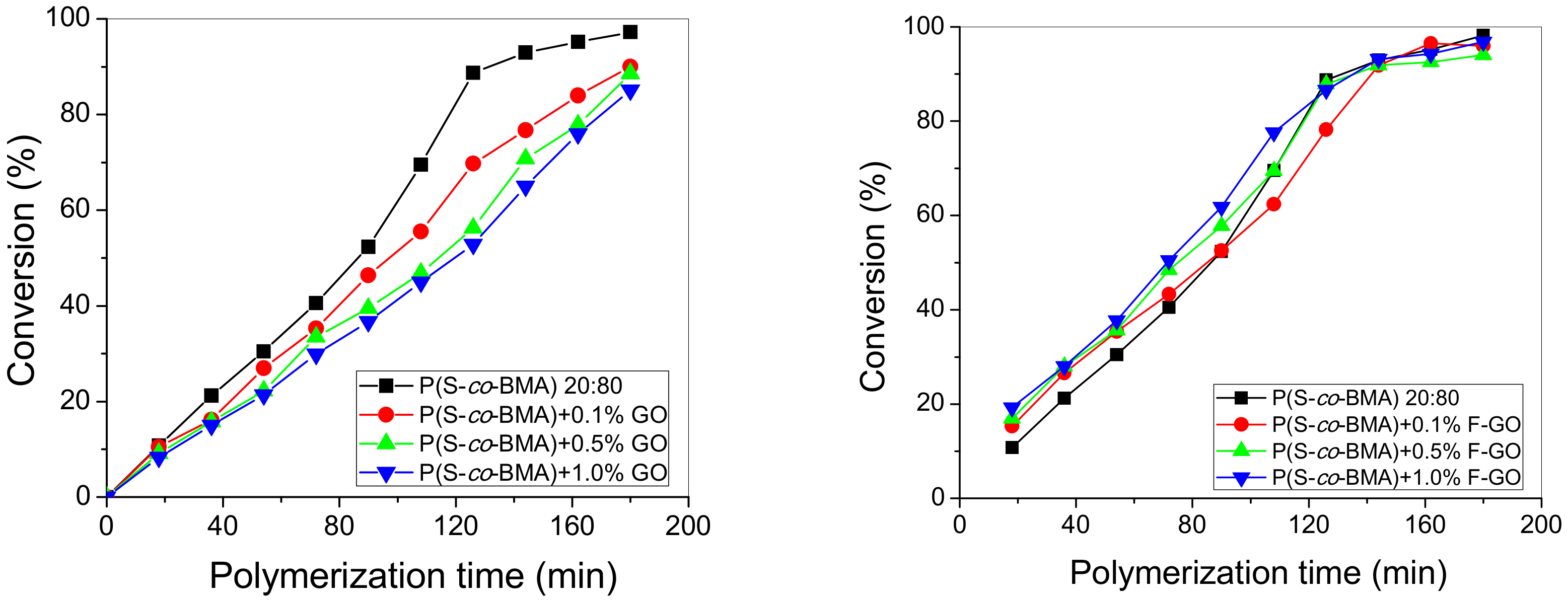
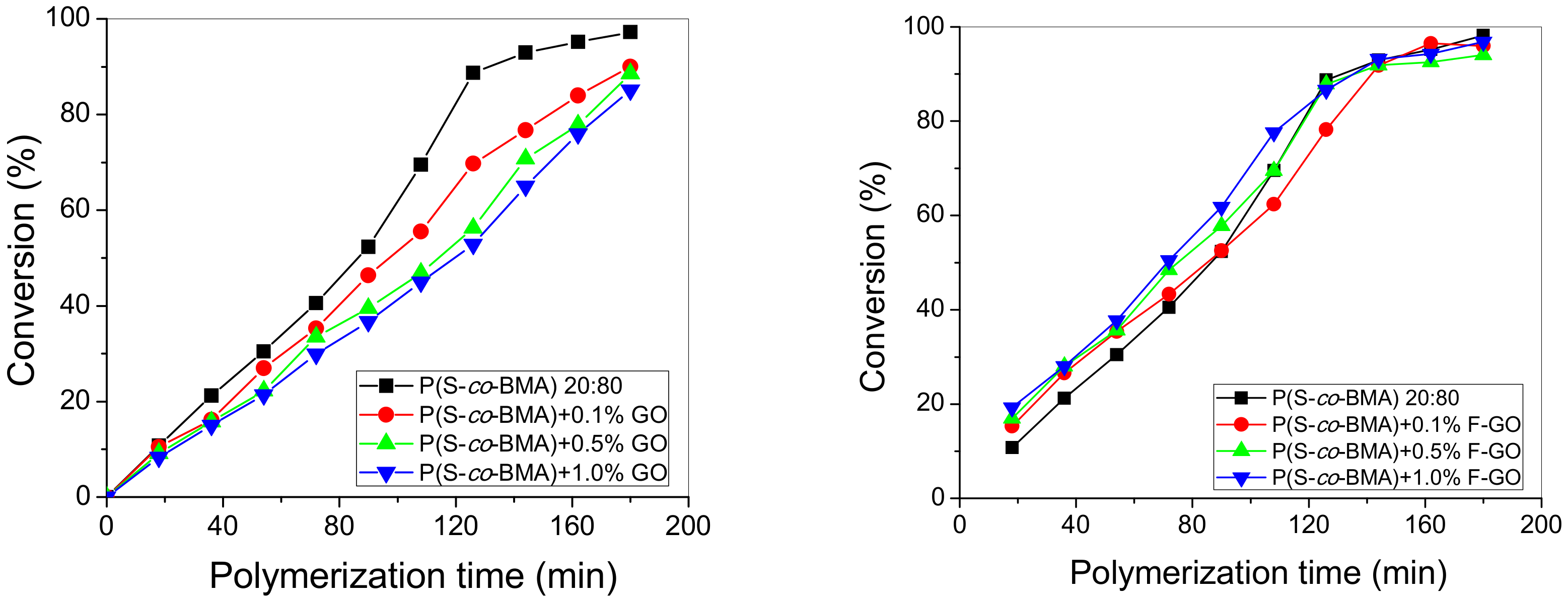
4.1. Experimental Polymerization Kinetics
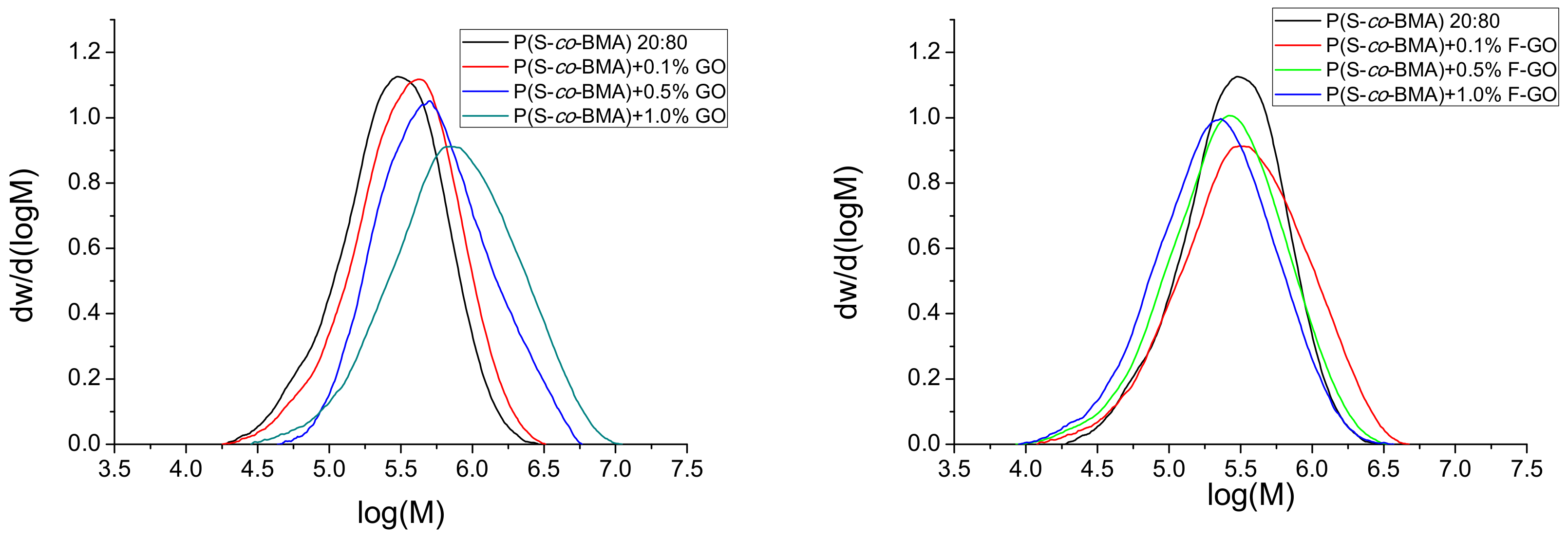
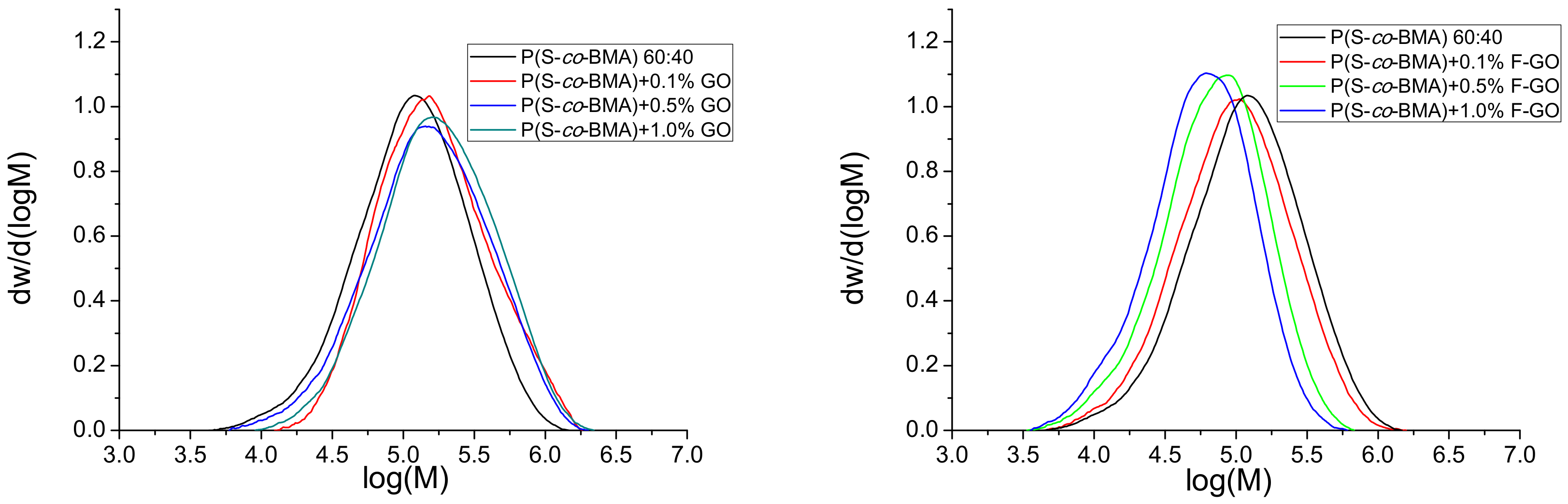
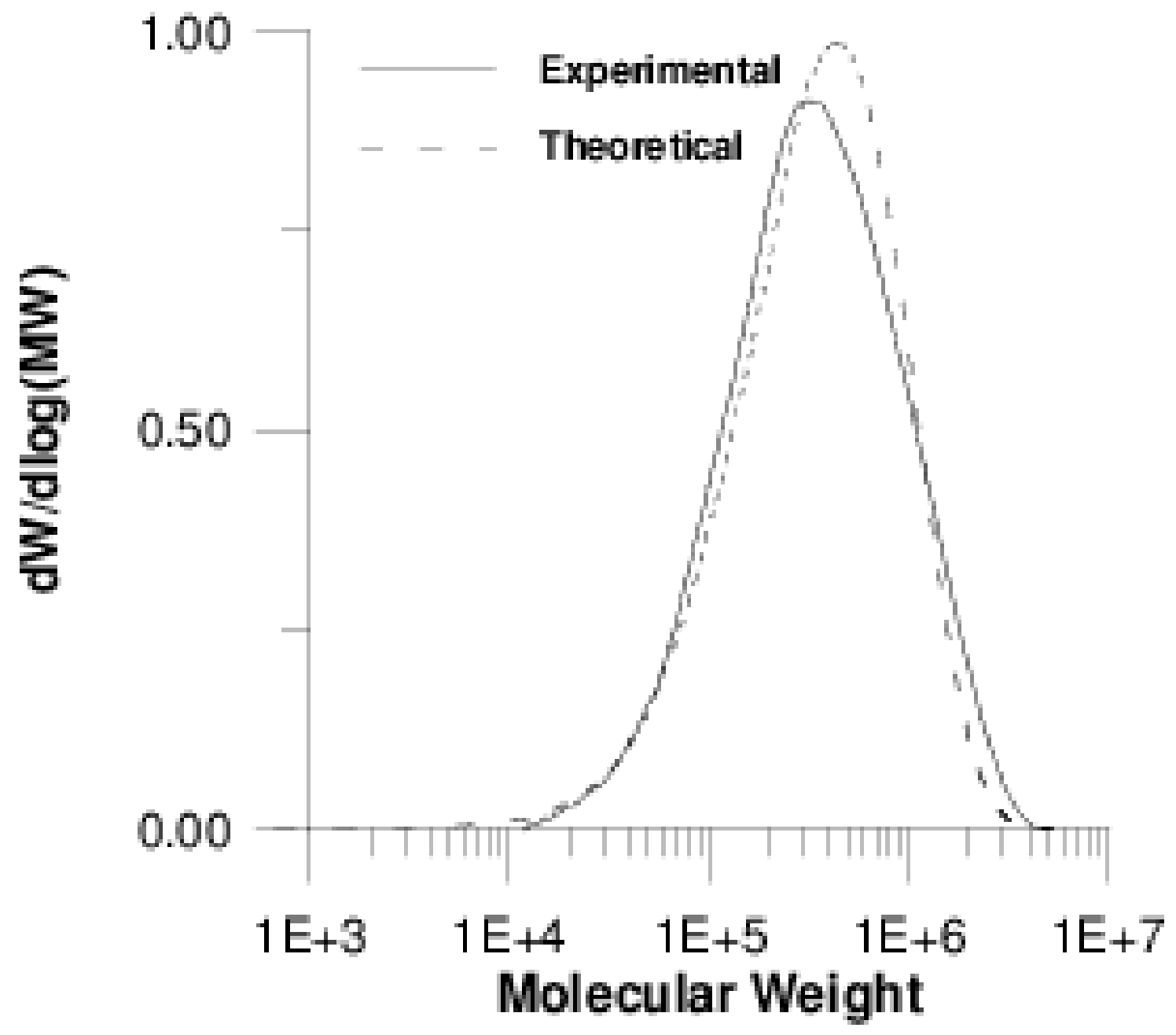
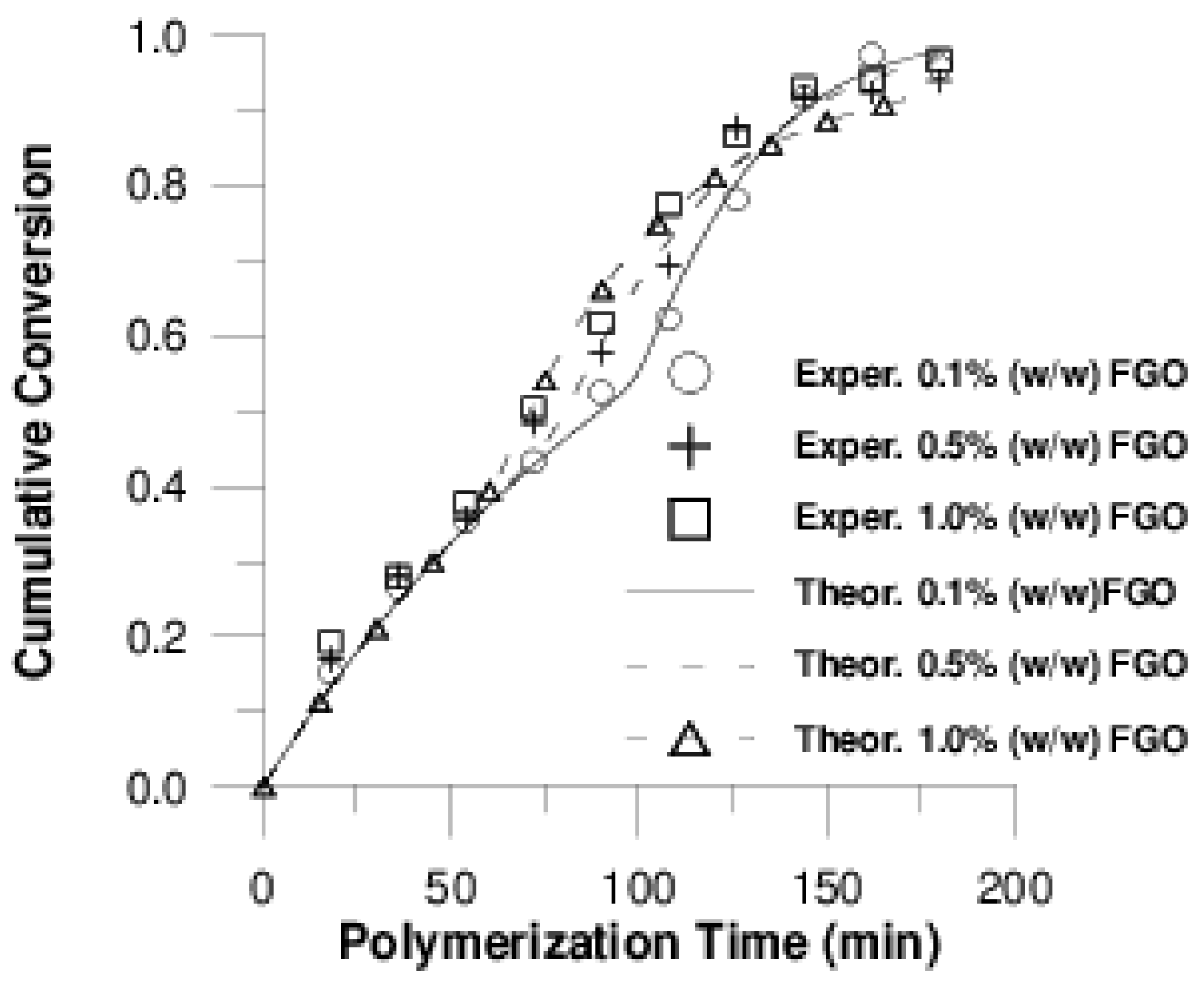
4.2 Simulation Results
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Nomenclature
| Aij | residual termination adjustable parameter between the i-th type and the j-th type radical |
| Dp,q,; Dp,q, | “dead” polymer having p BMA units and q styrene units; its concentration |
| b0 | free volume theory adjustable parameter set equal to unity |
| Bi | auxiliary parameters |
| CC | copolymer composition |
| D | polydispersity |
| DI0 | free volume theory pro-exponential parameter for primary radicals |
| free volume theory pro-exponential parameter for macroradicals self-diffusion for the termination reaction between the i-th and j-th type macroradicals | |
| C/DI0 | cage effect parameter |
| f | initiator efficiency |
| I; I | initiator; its concentration |
| kd | initiator decomposition kinetic rate constant |
| kfmij | chain transfer to monomer kinetic rate constant |
| kfpij | chain transfer to polymer kinetic rate constant |
| kpij | propagation rate kinetic rate constant |
| ktij | overall termination kinetic rate constant |
| kt0ij | intrinsic termination rate constant defined at zero conversion and involving two short chains |
| ktcij | termination by combination kinetic rate constant |
| ktdij | termination by disproportionation kinetic rate constant |
| kteij | diffusion-controlled termination kinetic rate constant |
| kt,reacij | reaction-diffusion controlled kinetic rate constant |
| Mi; Mi | i-th type monomer; its concentration |
| MW | molecular weight |
| radicals number average degree of polymerization | |
| number average molecular weight | |
| weight average molecular weight | |
| R•; R• | primary radical from the fragmentation of the initiator; its concentration |
| R | universal gas constant |
| , | macroradical having the i-th type monomer unit, consisting of p BMA units and of q styrene units; its concentration |
| t | time |
| T | temperature |
| Tg | glass transition temperature |
| V | reactor volume |
| wi | weight fraction of the i-th type monomer |
| free volume fraction | |
| Xcum | cumulative conversion |
| Yi | fractional i-th type monomer conversion |
| Greek symbols | |
| ε | volume contraction factor |
| λnm | n,m moments of “live” radicals chain length distribution-chain length-copolymer composition distribution |
| μnm | n,m moments of “dead” polymer chain length-copolymer composition distribution |
| ρ | density |
| Subscripts | |
| I | initiator |
| m | monomer |
| o | initial conditions |
| p | polymer |
References
- Du, J.; Cheng, H.M. The fabrication, properties, and uses of graphene/polymer composites. Macromol. Chem. Phys. 2012, 213, 1060–1077. [Google Scholar] [CrossRef]
- Kim, H.; Abdala, A.A.; Macosko, C.W. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar] [CrossRef]
- Park, S.; Ruoff, R.S. Chemical methods for the production of graphenes. Nat. Nanotechnol. 2009, 4, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; An, J.; Jung, I.; Piner, R.D.; An, S.J.; Li, X.; Velamakanni, A.; Ruoff, R.S. Colloidal suspensions of highly reduced graphene oxide in a wide variety of organic solvents. Nano Lett. 2009, 9, 1593–1597. [Google Scholar] [CrossRef] [PubMed]
- Ciszewski, M.; Mianowski, A. Badania nad procesem utleniania grafitu mieszaninami utleniającymi w kwasach nieorganicznych. Chemik 2013, 67, 267–274. [Google Scholar]
- Potts, J.R.; Lee, S.H.; Alam, T.M.; An, J.; Stoller, M.D.; Piner, R.D.; Ruoff, R.S. Thermomechanical properties of chemically modified graphene/poly (methyl methacrylate) composites made by in situ polymerization. Carbon 2011, 49, 2615–2623. [Google Scholar] [CrossRef]
- Heo, S.; Cho, S.Y.; Kim, D.H.; Choi, Y.; Park, H.H.; Jin, H.J. Improved thermal properties of graphene oxide-incorporated poly (methyl methacrylate) microspheres. J. Nanosci. Nanotechnol. 2012, 12, 5990–5994. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.N.; Saini, P.; Gupta, D.; Choudhary, V. Electrical and mechanical properties of PMMA/reduced graphene oxide nanocomposites prepared via in situ polymerization. J. Mater. Sci. 2013, 48, 6223. [Google Scholar] [CrossRef]
- Wang, J.; Hu, H.; Wang, X.; Xu, C.; Zhang, M.; Shang, X. Preparation and mechanical and electrical properties of graphene nanosheets–poly (methyl methacrylate) nanocomposites via in situ suspension polymerization. J. Appl. Polym. Sci. 2011, 122, 1866–1871. [Google Scholar] [CrossRef]
- Wang, Y.; Liao, X.; Li, S.; Luo, Y.; Yang, Q.; Li, G. Poly (methyl methacrylate) nanocomposites based on graphene oxide: A comparative investigation of the effects of surface chemistry on properties and foaming behavior. Polym. Int. 2016, 65, 1195–1203. [Google Scholar] [CrossRef]
- Feng, L.; Guan, G.; Li, C.; Zhang, D.; Xiao, Y.; Zheng, L.; Zhu, W. In situ synthesis of poly (methyl methacrylate)/graphene oxide nanocomposites using thermal-initiated and graphene oxide-initiated polymerization. J. Macromol. Sci. Part A 2013, 50, 720–727. [Google Scholar] [CrossRef]
- Morimune, S.; Nishino, T.; Goto, T. Ecological approach to graphene oxide reinforced poly (methyl methacrylate) nanocomposites. ACS Appl. Mater. Interfaces 2012, 4, 3596–3601. [Google Scholar] [CrossRef] [PubMed]
- Begum, F.; Simon, S.L. Modeling methyl methacrylate free radical polymerization in nanoporous confinement. Polymer 2011, 52, 1539–1545. [Google Scholar] [CrossRef]
- Zhao, H.; Simon, S.L. Methyl methacrylate polymerization in nanoporous confinement. Polymer 2011, 52, 4093–4098. [Google Scholar] [CrossRef]
- Begum, F.; Zhao, H.; Simon, S.L. Modeling methyl methacrylate free radical polymerization: Reaction in hydrophilic nanopores. Polymer 2012, 53, 3238–3244. [Google Scholar] [CrossRef]
- Begum, F.; Zhao, H.; Simon, S.L. Modeling methyl methacrylate free radical polymerization: Reaction in hydrophobic nanopores. Polymer 2012, 53, 4372–4378. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Yu, Z.N.; Begum, F.; Hedden, R.C.; Simon, S.L. The effect of nanoconfinement on methyl methacrylate polymerization: Tg, molecular weight, and tacticity. Polymer 2014, 55, 4959–4965. [Google Scholar] [CrossRef]
- Siddiqui, M.N.; Redhwi, H.H.; Verros, G.D.; Achilias, D.S. Evaluating the role of nanomontmorillonite in bulk in situ radical polymerization kinetics of butyl methacrylate through a simulation model. Ind. Eng. Chem. Res. 2014, 53, 11303–11311. [Google Scholar] [CrossRef]
- Verros, G.D.; Achilias, D.S. Toward the development of a mathematical model for the bulk in situ radical polymerization of methyl methacrylate in the presence of nano-additives. Can. J. Chem. Eng. 2016, 94, 1783–1791. [Google Scholar] [CrossRef]
- Michailidis, M.; Verros, G.D.; Deliyanni, E.; Andriotis, E.G.; Achilias, D.S. An experimental and theoretical study of butyl methacrylate in situ radical polymerization kinetics in the presence of graphene oxide nanoadditive. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1433. [Google Scholar] [CrossRef]
- Meng, X.; Wu, H.; Storti, G.; Morbidelli, M. Effect of dispersed polymeric nanoparticles on the bulk polymerization of methyl methacrylate. Macromolecules 2016, 49, 7758. [Google Scholar] [CrossRef]
- Herrera-Ordonez, J. On the diffusion-controlled rate coefficient for chemical reactions and collisions of nano-particles. Chem. Eng. Sci. 2017, 171, 481–484. [Google Scholar] [CrossRef]
- Sanz, B.; Ballard, N.; Asua, J.M.; Mijangos, C. Effect of confinement on the synthesis of PMMA in AAO templates and modeling of free radical polymerization. Macromolecules 2017, 50, 811. [Google Scholar] [CrossRef]
- Tsagkalias, I.S.; Papadopoulou, S.; Verros, G.D.; Achilias, D.S. Polymerization Kinetics of n-Butyl Methacrylate in the Presence of Graphene Oxide Prepared by Two Different Oxidation Methods with or without Functionalization. Ind. Eng. Chem. Res. 2018, 57, 2449–2460. [Google Scholar] [CrossRef]
- Li, D.; Li, N.; Hutchinson, R.A. High-temperature free radical copolymerization of styrene and butyl methacrylate with depropagation and penultimate kinetic effects. Macromolecules 2006, 39, 4366–4373. [Google Scholar] [CrossRef]
- Fukuda, T.; Ma, Y.; Inagaki, H. Free-radical copolymerization. 3. Determination of rate constants of propagation and termination for styrene/methyl methacrylate system. A critical test of terminal-model kinetics. Macromolecules 1985, 18, 17–26. [Google Scholar] [CrossRef]
- Wang, W.; Hutchinson, R.A.; Grady, M.C. Study of butyl methacrylate depropagation behavior using batch experiments in combination with modeling. Ind. Eng. Chem. Res. 2009, 48, 4810–4816. [Google Scholar] [CrossRef]
- Wang, W.; Hutchinson, R.A. A comprehensive kinetic model for high-temperature free radical production of styrene/methacrylate/acrylate resins. AIChE J. 2011, 57, 227–238. [Google Scholar] [CrossRef]
- Li, D.; Hutchinson, R.A. High temperature semibatch free radical copolymerization of butyl methacrylate and styrene. Macromol. Symp. 2006, 243, 24–34. [Google Scholar] [CrossRef]
- Siddiqui, M.N.; Redhwi, H.H.; Gkinis, K.; Achilias, D.S. Synthesis and characterization of novel nanocomposite materials based on poly (styrene-co-butyl methacrylate) copolymers and organomodified clay. Eur. Polym. J. 2013, 49, 353–365. [Google Scholar] [CrossRef]
- Tsagkalias, I.; Proskynitopoulou, V.; Verros, G.; Achilias, D.S. Effect of graphene oxide on the kinetics of the radical polymerization of styrene. Mater. Today. Proc. 2018, 5, 27517–27525. [Google Scholar] [CrossRef]
- Chanda, M. Introduction to Polymer Science and Chemistry; CRC Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Ray, W.H. On the mathematical modeling of polymerization reactors. J. Macromol. Sci. Rev. Macromol. Chem. 1972, 8, 1–56. [Google Scholar] [CrossRef]
- Arriola, D.J. Modeling of Addition Polymerization Systems. Ph.D. Thesis, University of Wisconsin, Madison, WI, USA, 1989. [Google Scholar]
- Achilias, D.S.; Kiparissides, C. Toward the development of a general framework for modeling molecular weight and compositional changes in free-radical copolymerization reactions. J. Macromol. Sci. Part C Polym. Rev. 1992, 32, 183–234. [Google Scholar] [CrossRef]
- Kiparissides, C.; Verros, G.; MacGregor, J.F. Mathematical modeling, optimization, and quality control of high-pressure ethylene polymerization reactors. J. Macromol. Sci. Part C Polym. Rev. 1993, 33, 437–527. [Google Scholar] [CrossRef]
- Achilias, D.S.; Sideridou, I.D. Kinetics of the benzoyl peroxide/amine initiated free-radical polymerization of dental dimethacrylate monomers: experimental studies and mathematical modeling for TEGDMA and Bis-EMA. Macromolecules 2004, 37, 4254–4265. [Google Scholar] [CrossRef]
- Fevotte, G.; McKenna, T.F.; Santos, A.M. Modelling of the glass transition temperature of free radical copolymers: An approach for control purposes. Chem. Eng. Sci. 1998, 53, 2241–2256. [Google Scholar] [CrossRef]
- Fevotte, G.; McKenna, T.F.; Othman, S.; Hammouri, H. Non-linear tracking of glass transition temperatures for free radical emulsion copolymers. Chem. Eng. Sci. 1998, 53, 773–786. [Google Scholar] [CrossRef]
- Cavin, L.; Rouge, A.; Meyer, T.; Renken, A. Kinetic modeling of free radical polymerization of styrene initiated by the bifunctional initiator 2, 5-dimethyl-2, 5-bis (2-ethyl hexanoyl peroxy) hexane. Polymer 2000, 41, 3925–3935. [Google Scholar] [CrossRef]
- Verros, G.D. Calculation of molecular weight distribution in non-linear free radical copolymerization. Polymer 2003, 44, 7021–7032. [Google Scholar] [CrossRef]
- Tsagkalias, I.S.; Manios, T.K.; Achilias, D.S. Effect of Graphene Oxide on the Reaction Kinetics of Methyl Methacrylate In Situ Radical Polymerization via the Bulk or Solution Technique. Polymers 2017, 9, 432. [Google Scholar] [CrossRef]
- Stewart, W.E.; Caracotsios, M.; Sorensen, J.P. Parameter estimation from multiresponse data. AIChE J. 1992, 38, 641–650. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Equations |
|---|---|
| Diffusion Controlled Limitations for Termination Reactions | |
| Gel Effect | |
| Residual Termination | |
| Diffusion Controlled Limitations for Initiation Reaction (Cage Effect) | |
| Free Volume Parameters |
| GO (w/w)%: | 0.1% | 0.5% | 1% |
| f0 | 0.52 ± 0.02 | 0.48 ± 0.03 | 0.41 ± 0.02 |
| a Dp00 × 10−17 | 0.075 ± 0.003 | 0.63 ± 0.03 | 0.63 ± 0.04 |
| a kfm11 | 0.97 ± 0.04 | 0.5 ± 0.03 | 0.41 ± 0.02 |
| (DI0/C) × 10−5 | 5.33 × 10−3 | 0.04 ± 0.003 | 0.02 ± 0.002 |
| F-GO (w/w)%: | 0.1% | 0.5 % | 1% |
| f0 | 1 | 1 | 1 |
| a Dp00 × 10−17 | 288.4 ± 10.5 | 1.1 ± 0.07 | 2.45 × 10−3 |
| a kfm11 | 0.95 ± 0.03 | 1.45 ± 0.11 | 1.81 ± 0.15 |
| a kfp | 0.45 ± 0.03 | 0.41 ± 0.02 | - |
| (DI0/C) × 10−5 | 31.62 ± 2.3 | 0.15 ± 0.01 | 0.014 ± 0.002 |
| Sample | Mn × 10−5 (g/mol) (exp.) | Mn × 10−5 (g/mol) (Model) | Mw × 10−5 (g/mol) (exp.) | Mw × 10−5 (g/mol) (Model) |
|---|---|---|---|---|
| neat | 1.93 | 1.934 | 5.71 | 4.04 |
| 0.1 wt.% GO | 2.335 | 2.331 | 4.6 | 4.78 |
| 0.5 wt.% GO | 3.74 | 3.740 | 7.73 | 8.026 |
| 1 wt.% GO | 4.40 | 4.397 | 11.53 | 9.523 |
| 0.1 wt.% F-GO | 1.96 | 1.998 | 5.13 | 5.193 |
| 0.5 wt.% F-GO | 1.57 | 1.530 | 3.72 | 3.765 |
| 1 wt.% F-GO | 1.325 | 1.334 | 3.11 | 2.729 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsagkalias, I.S.; Vlachou, A.; Verros, G.D.; Achilias, D.S. Effect of Graphene oxide or Functionalized Graphene Oxide on the Copolymerization Kinetics of Styrene/n-butyl Methacrylate. Polymers 2019, 11, 999. https://doi.org/10.3390/polym11060999
Tsagkalias IS, Vlachou A, Verros GD, Achilias DS. Effect of Graphene oxide or Functionalized Graphene Oxide on the Copolymerization Kinetics of Styrene/n-butyl Methacrylate. Polymers. 2019; 11(6):999. https://doi.org/10.3390/polym11060999
Chicago/Turabian StyleTsagkalias, Ioannis S., Afrodite Vlachou, George D. Verros, and Dimitris S. Achilias. 2019. "Effect of Graphene oxide or Functionalized Graphene Oxide on the Copolymerization Kinetics of Styrene/n-butyl Methacrylate" Polymers 11, no. 6: 999. https://doi.org/10.3390/polym11060999
APA StyleTsagkalias, I. S., Vlachou, A., Verros, G. D., & Achilias, D. S. (2019). Effect of Graphene oxide or Functionalized Graphene Oxide on the Copolymerization Kinetics of Styrene/n-butyl Methacrylate. Polymers, 11(6), 999. https://doi.org/10.3390/polym11060999