Visualization of Polymer Crystallization by In Situ Combination of Atomic Force Microscopy and Fast Scanning Calorimetry



Abstract
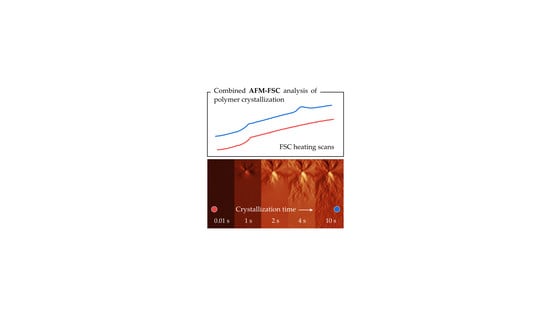
1. Introduction
2. Materials and Methods
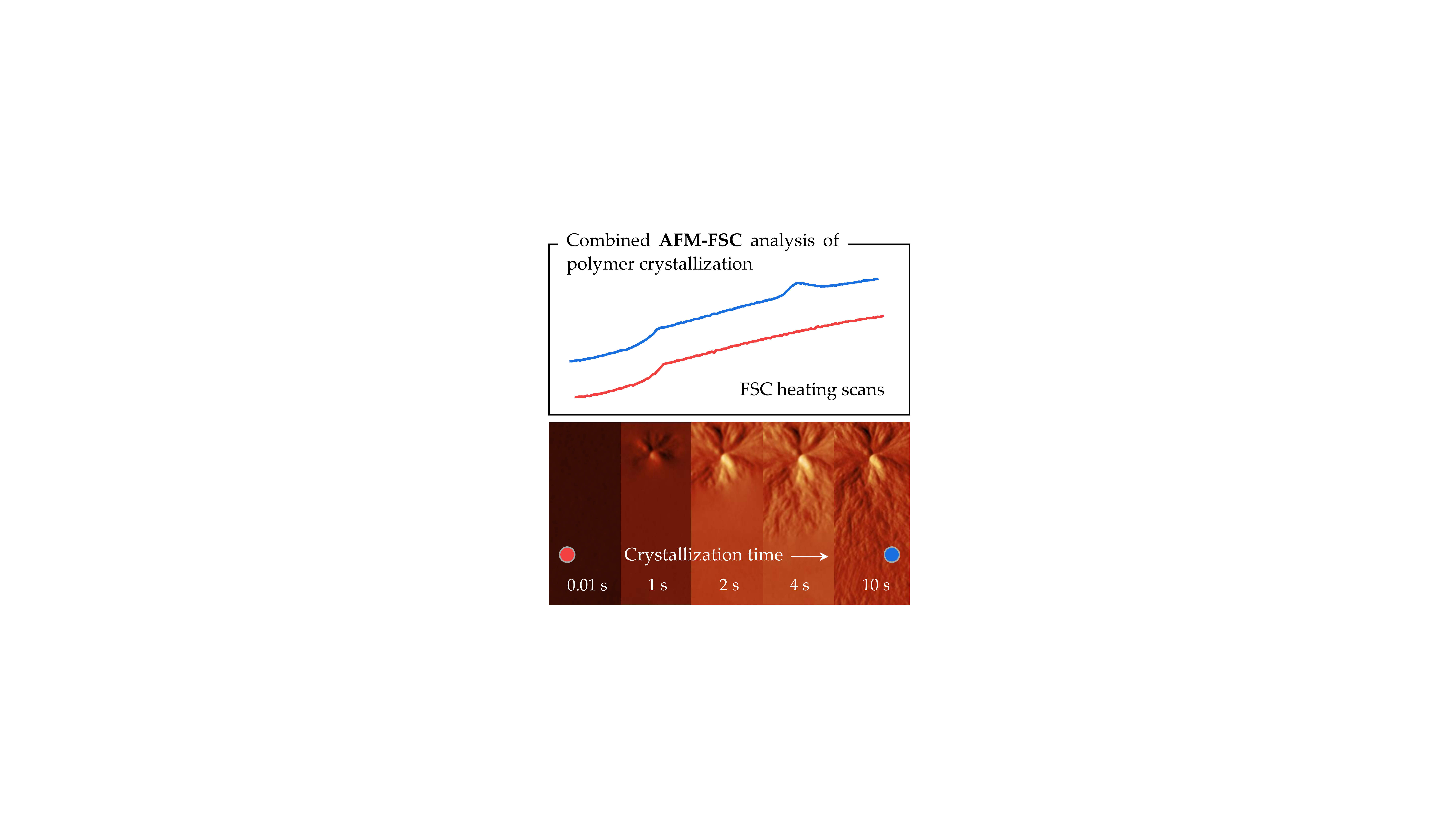
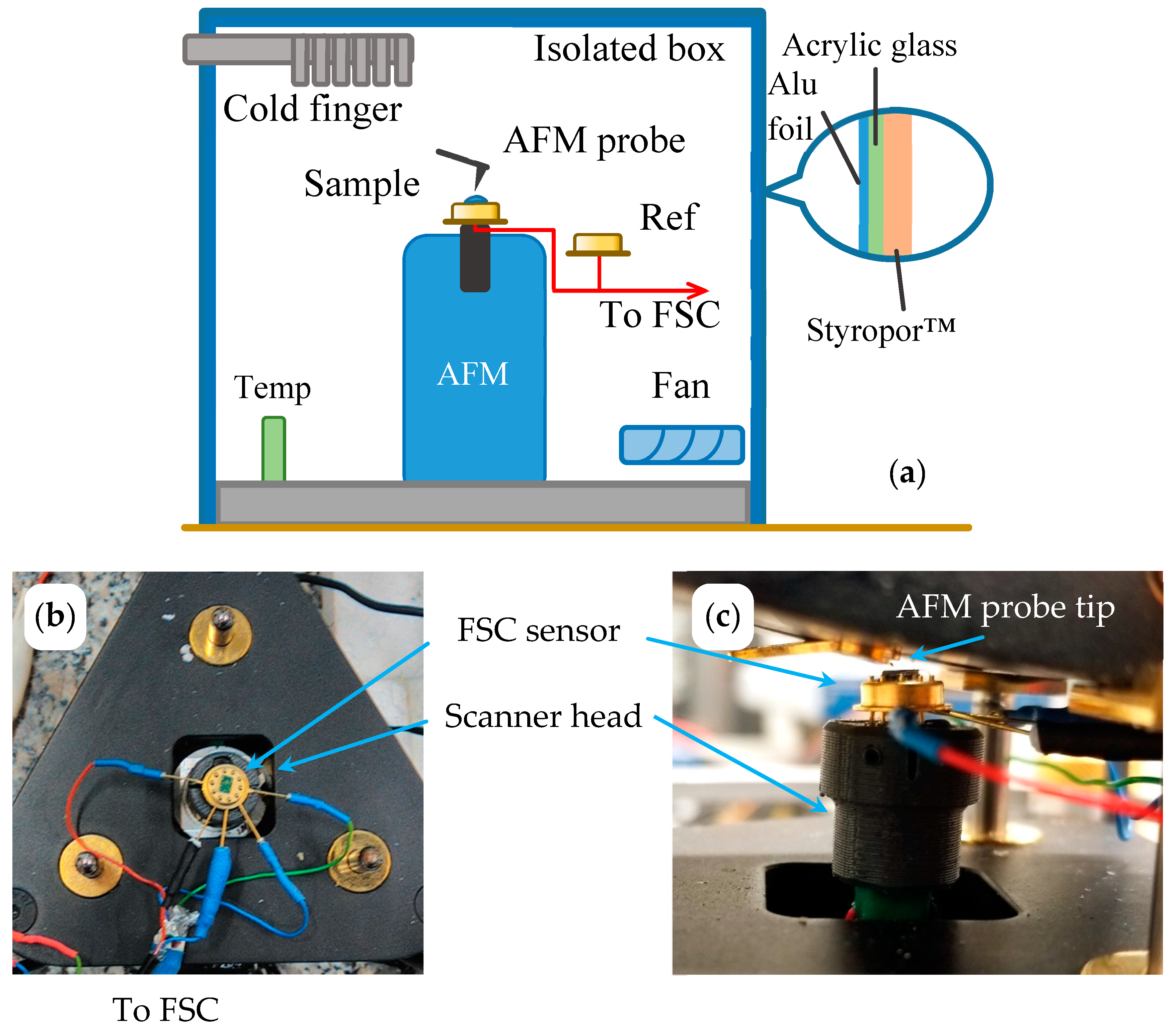
2.1. Combined AFM-FSC Device
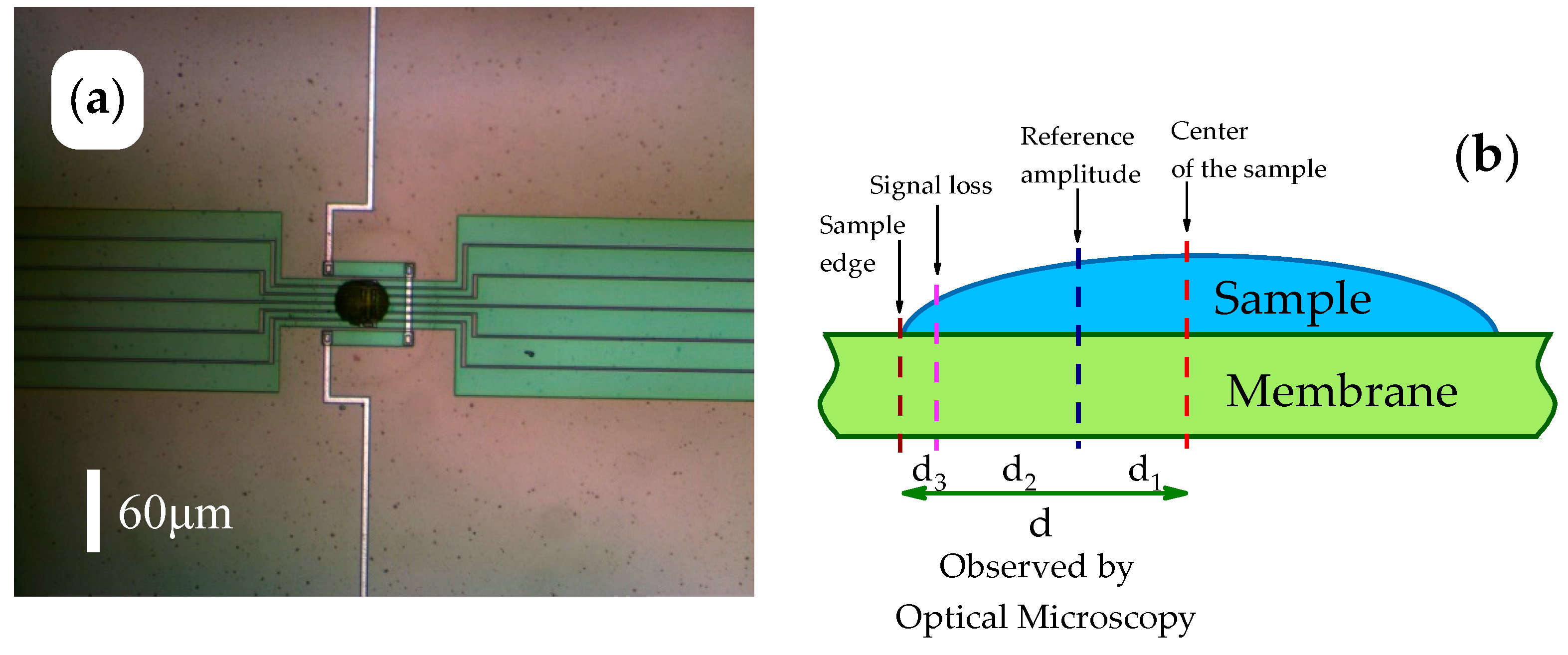
2.2. Sample Preparation and AFM Adjustment
2.3. Materials
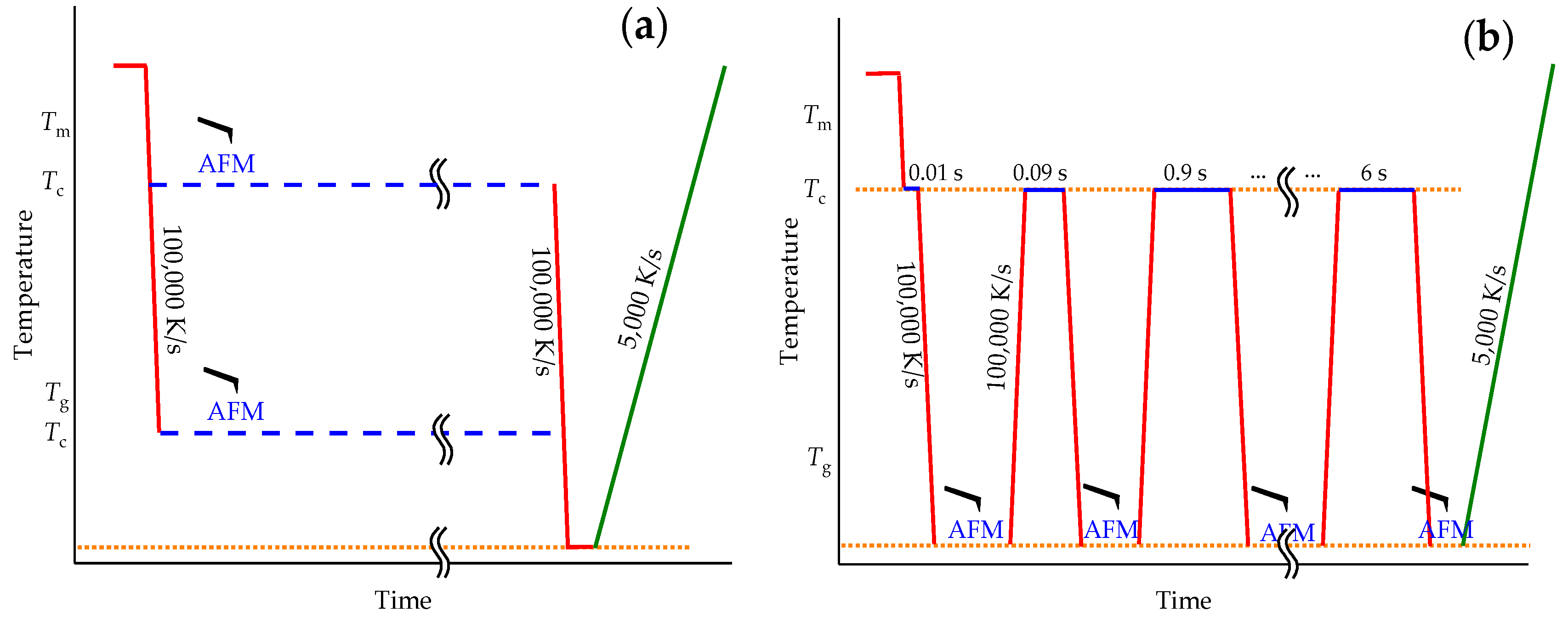
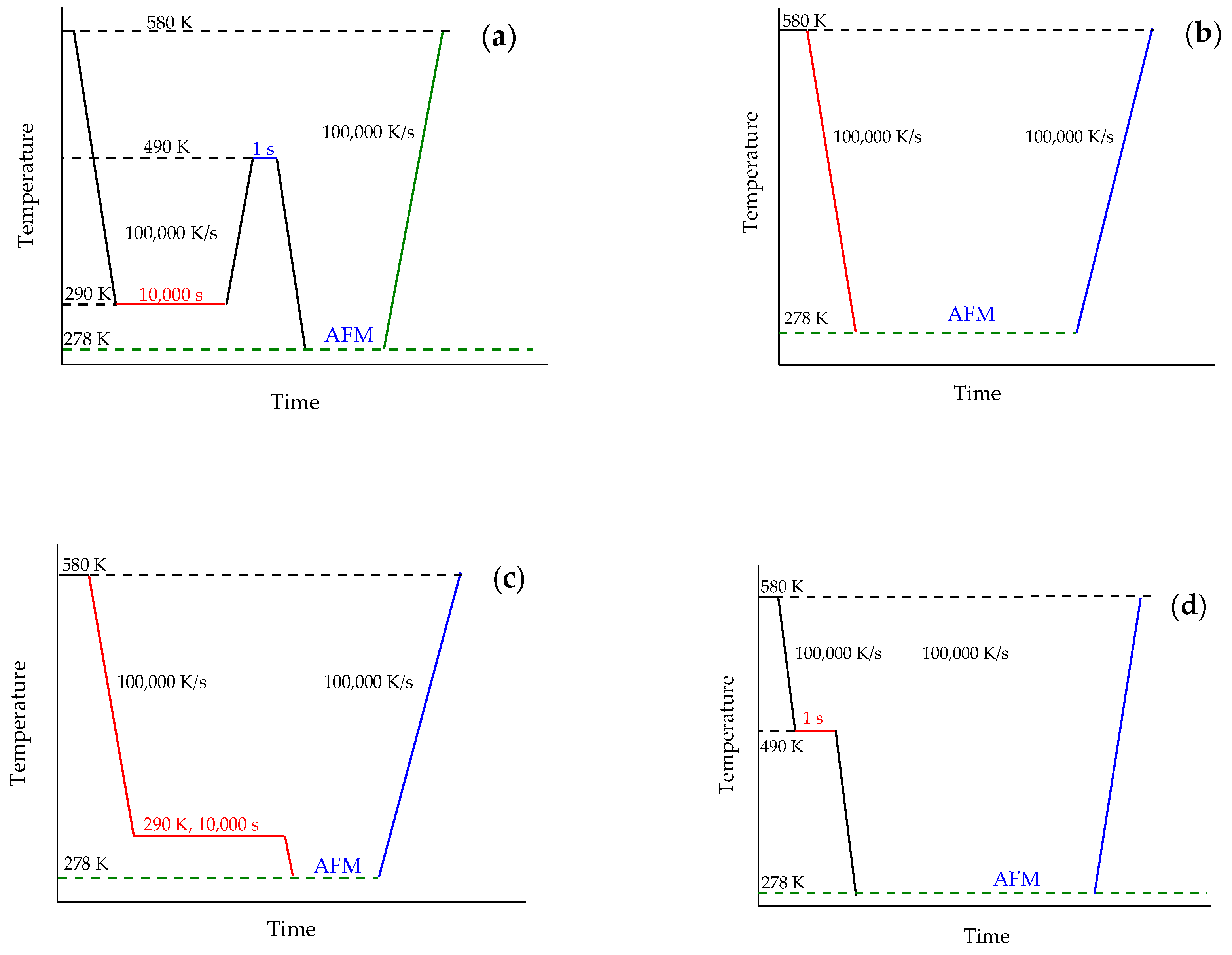
2.4. Measurement Strategy
3. Results
3.1. Crystallization at Low Undercooling
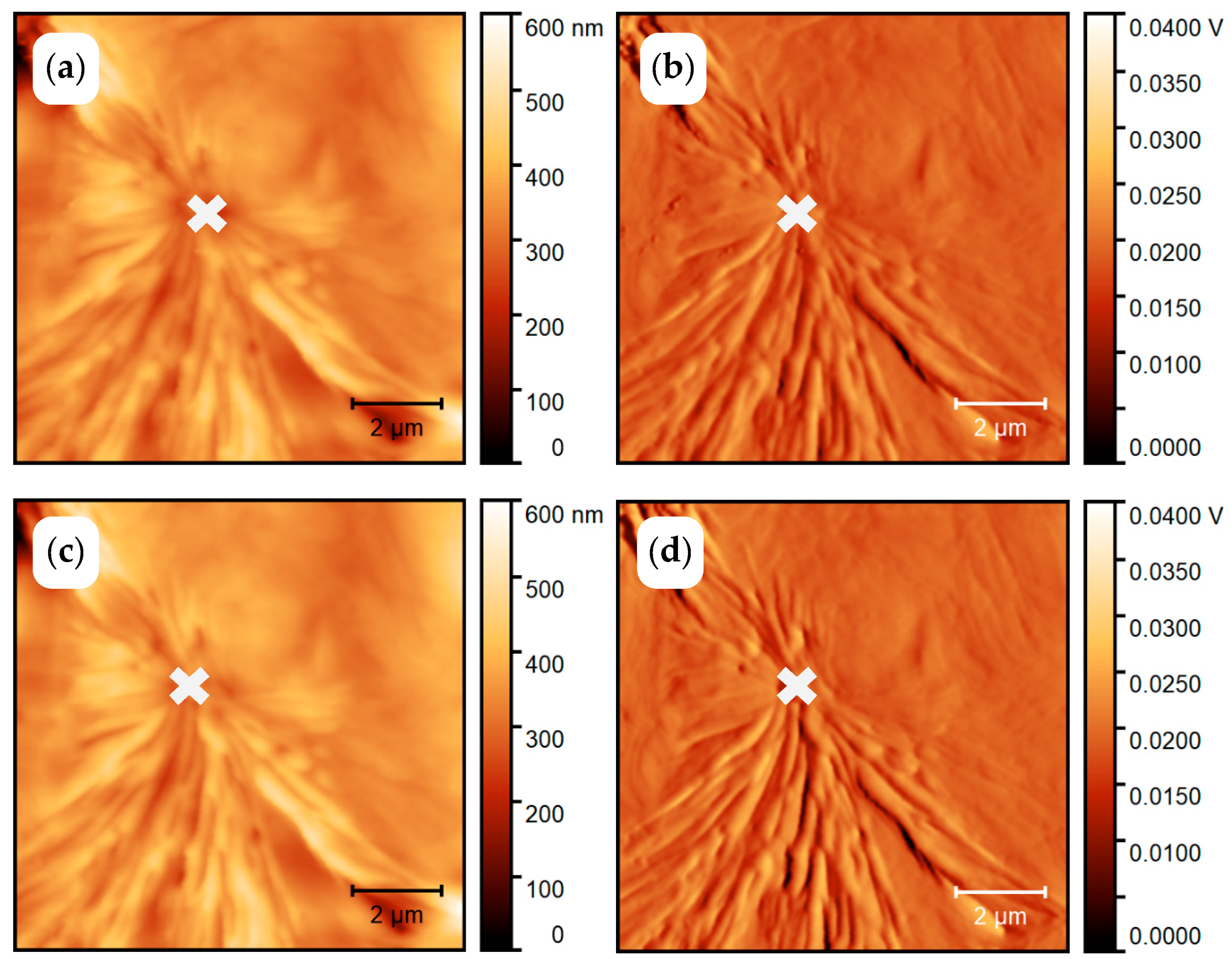
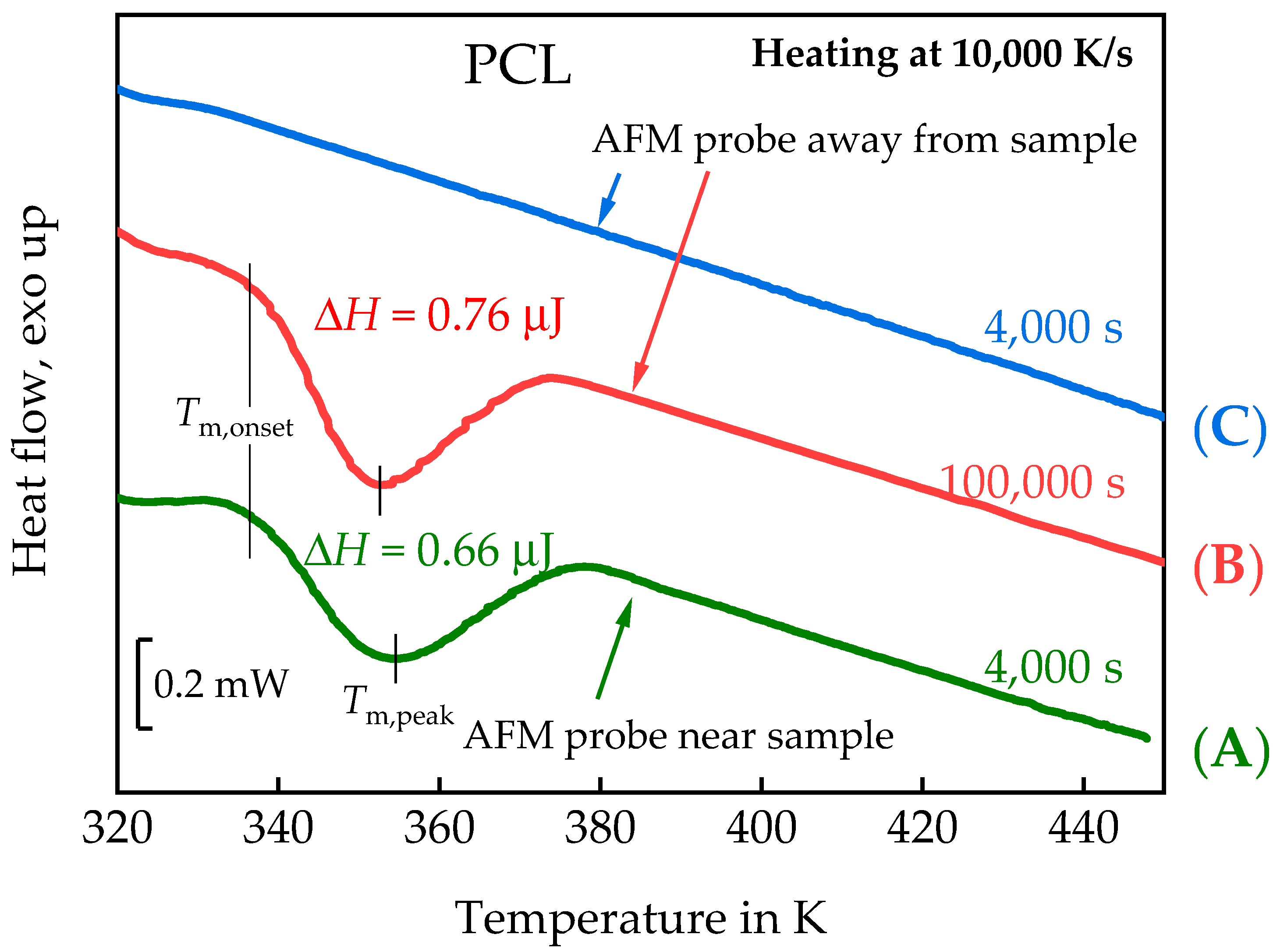
3.1.1. In Situ Study of PCL Crystallization
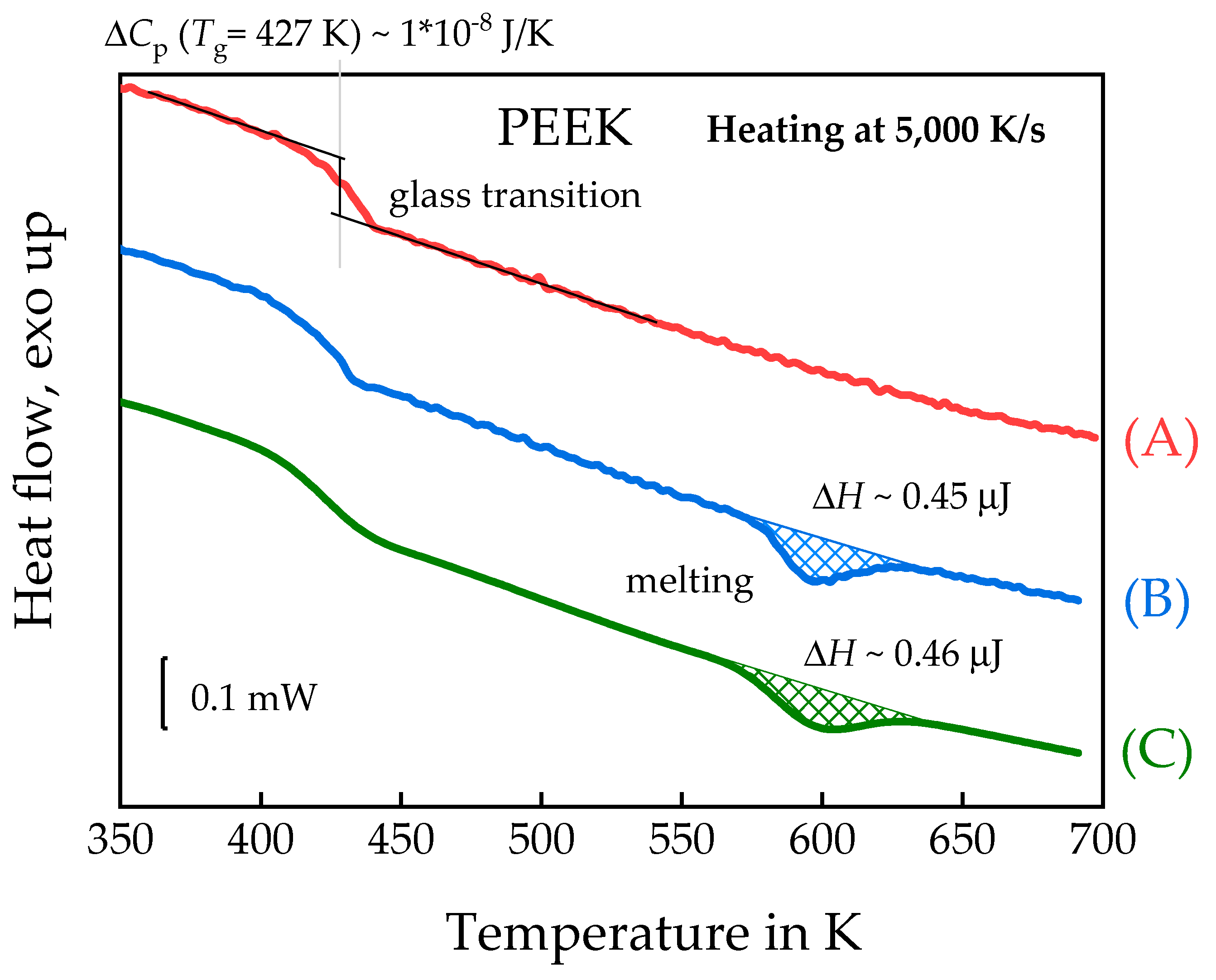
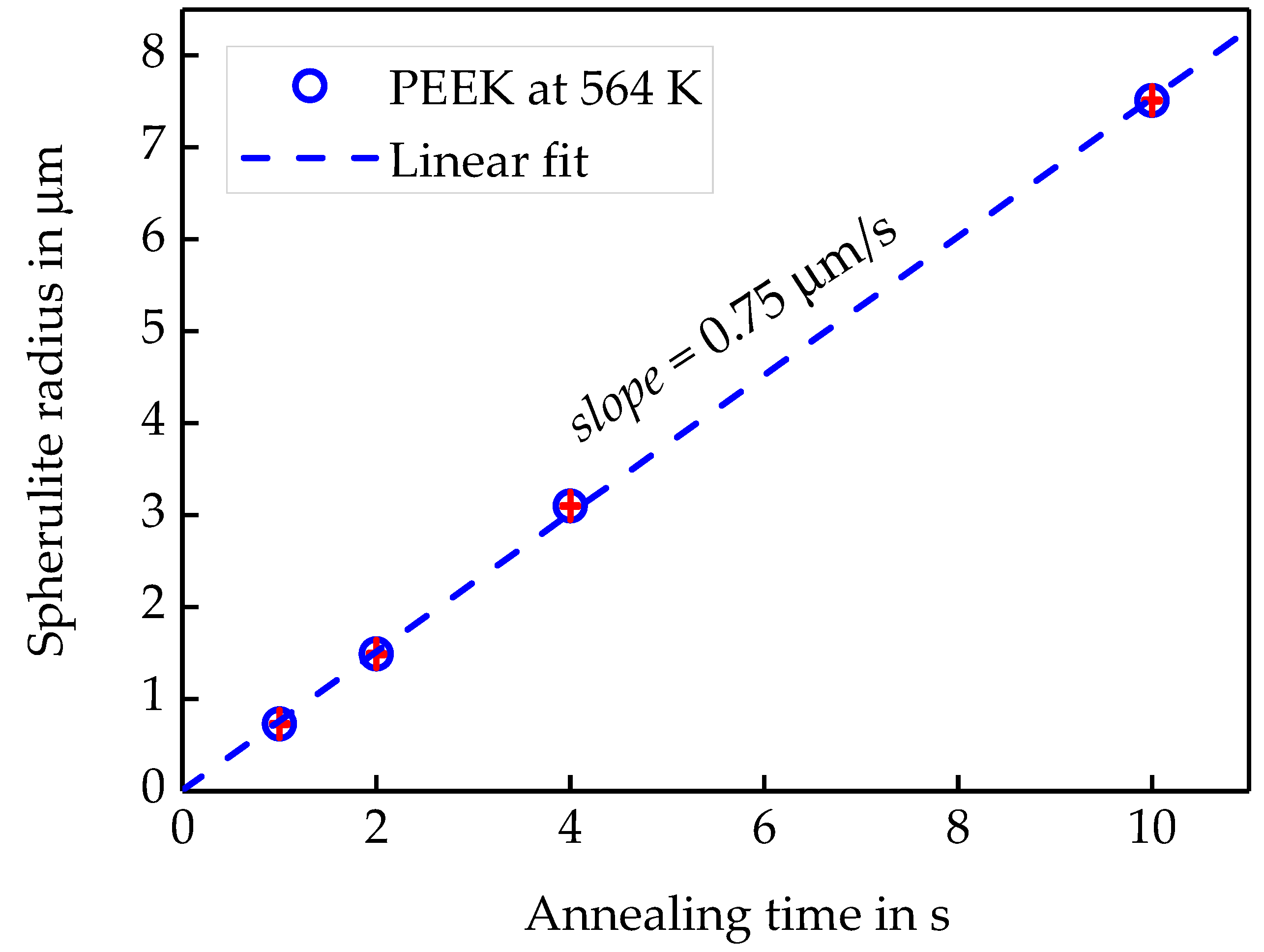
3.1.2. Linear Growth Rate of PEEK Spherulites
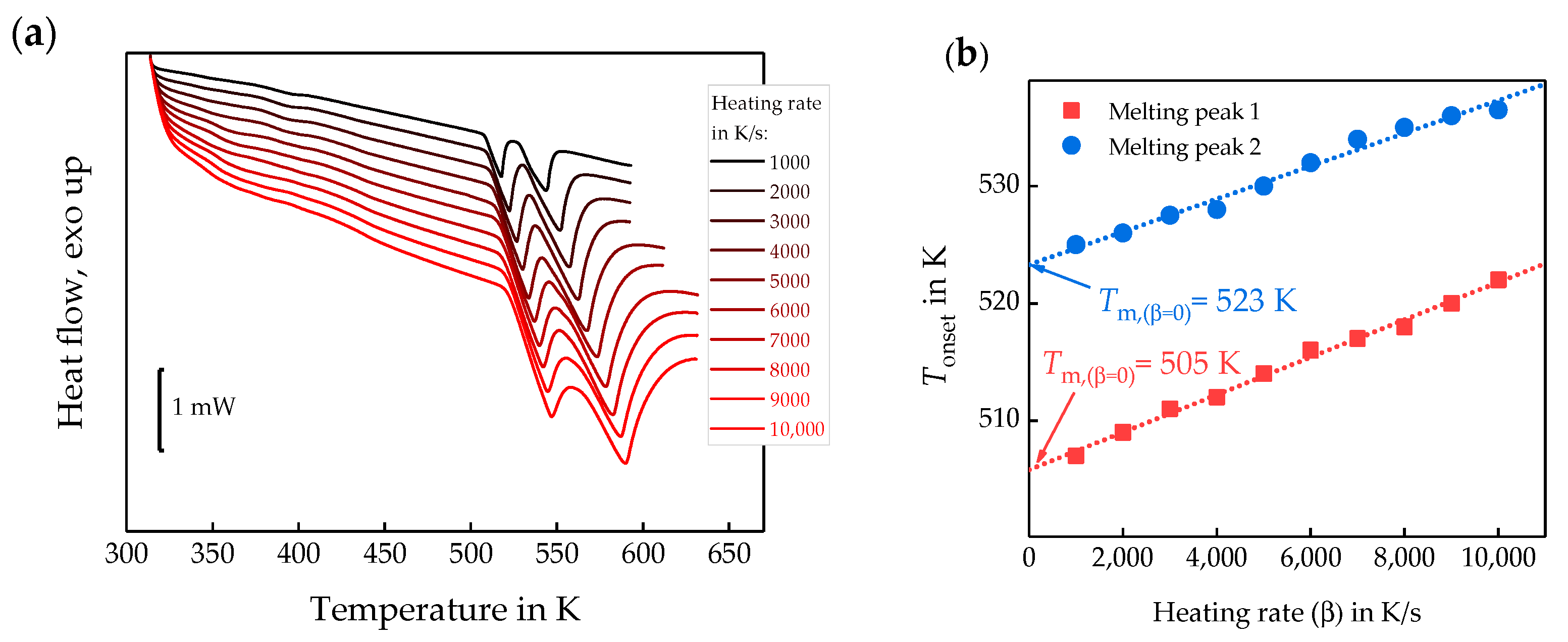
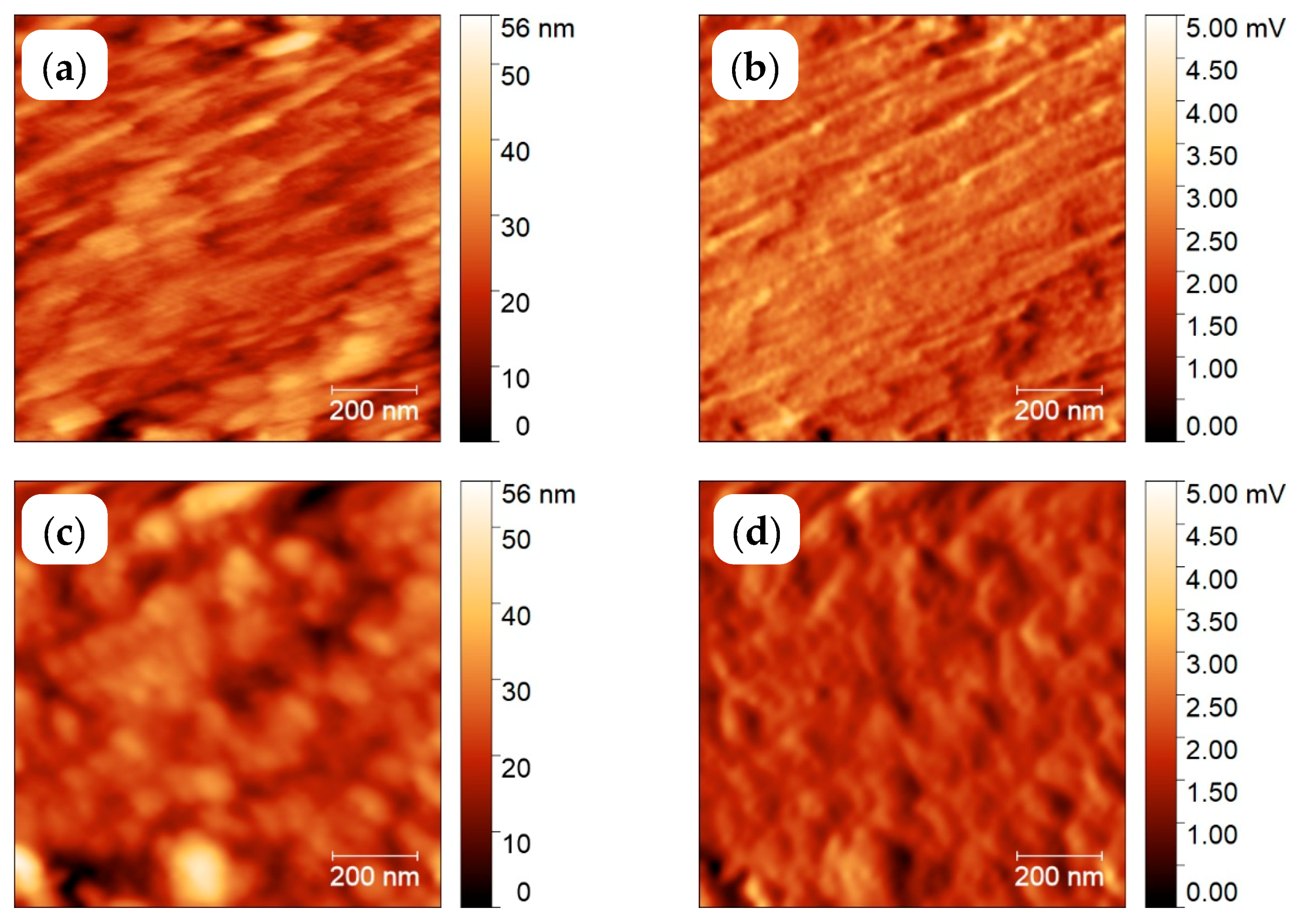
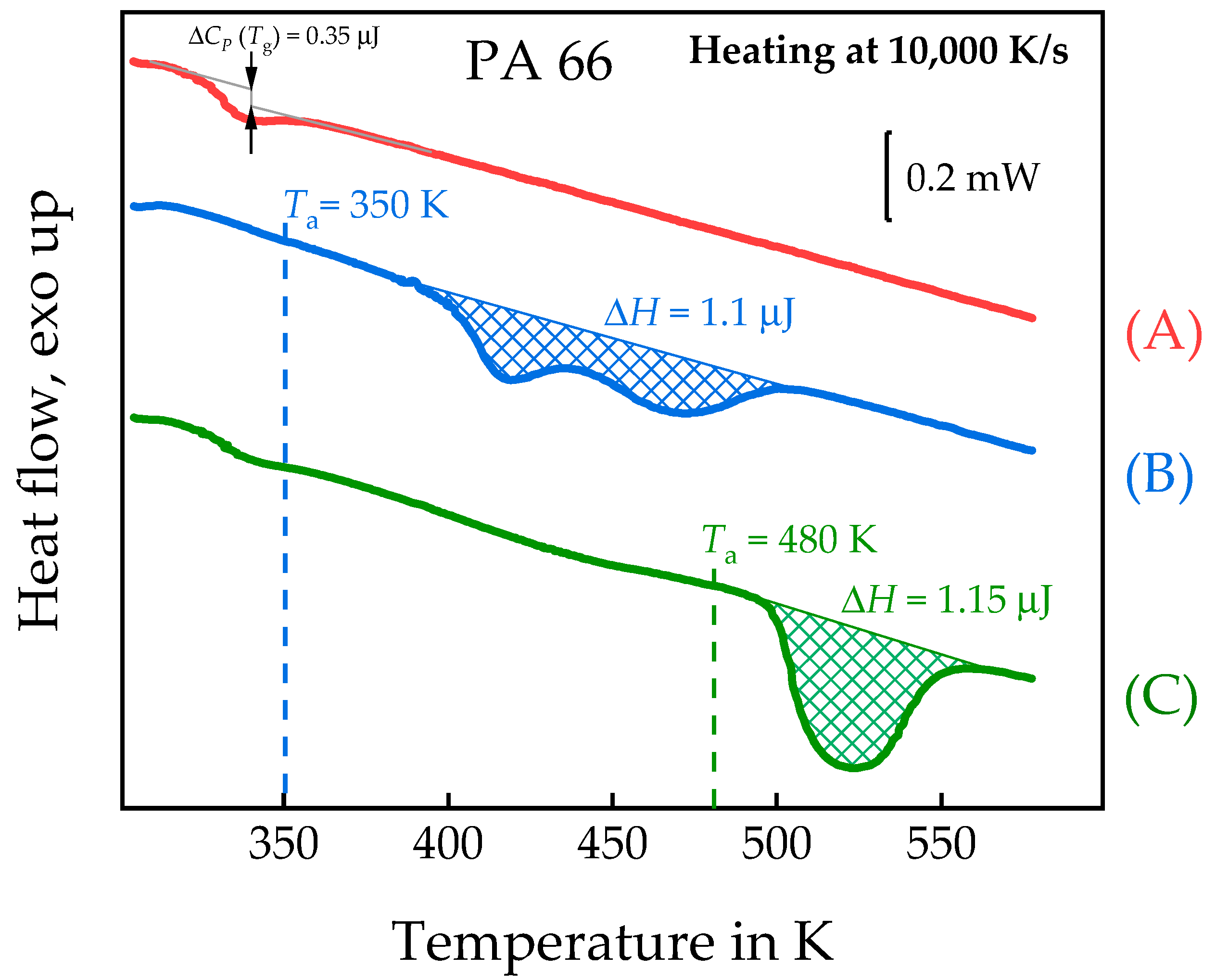
3.2. Semi-Crystalline Morphology of PA 66 at High and Low Supercooling
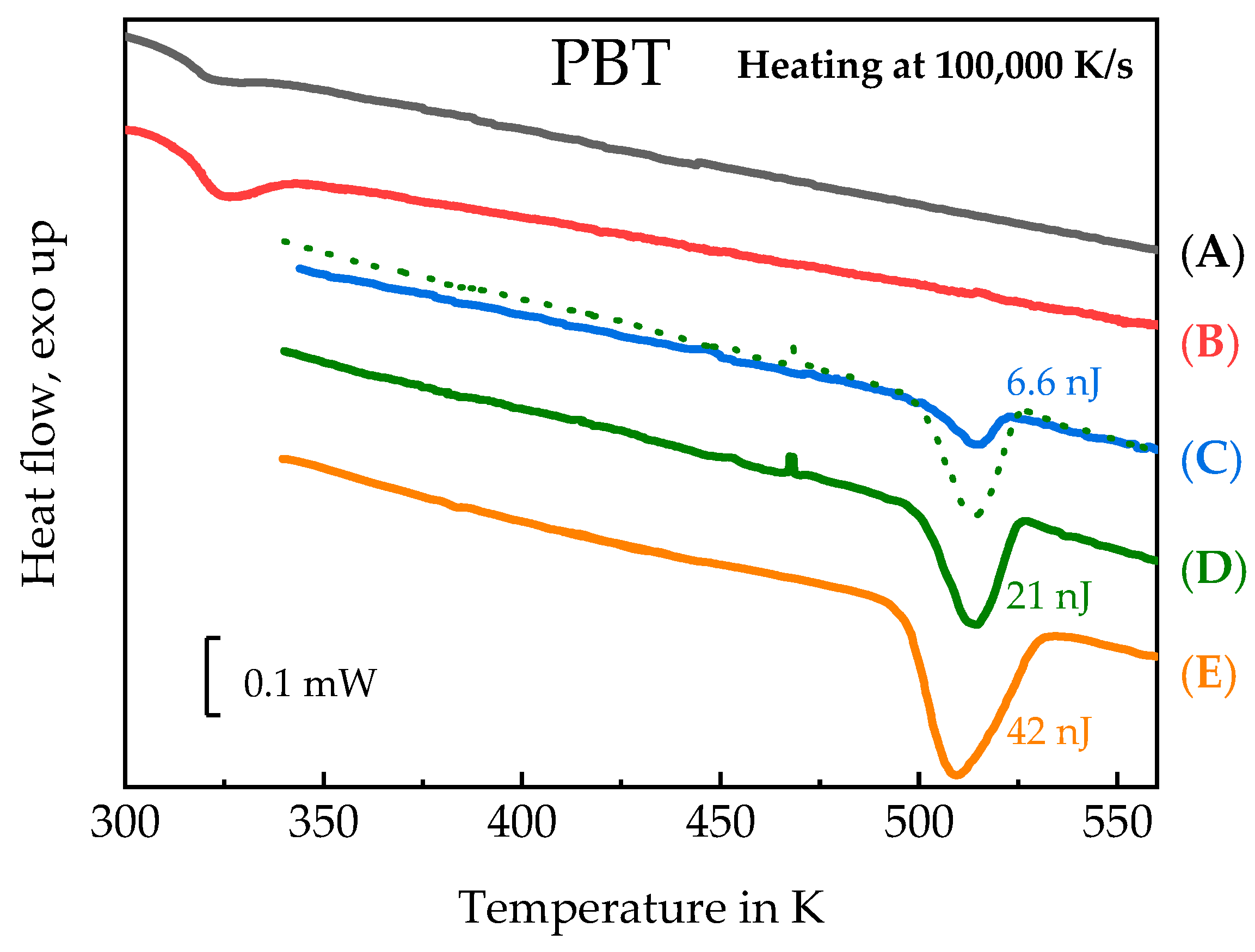
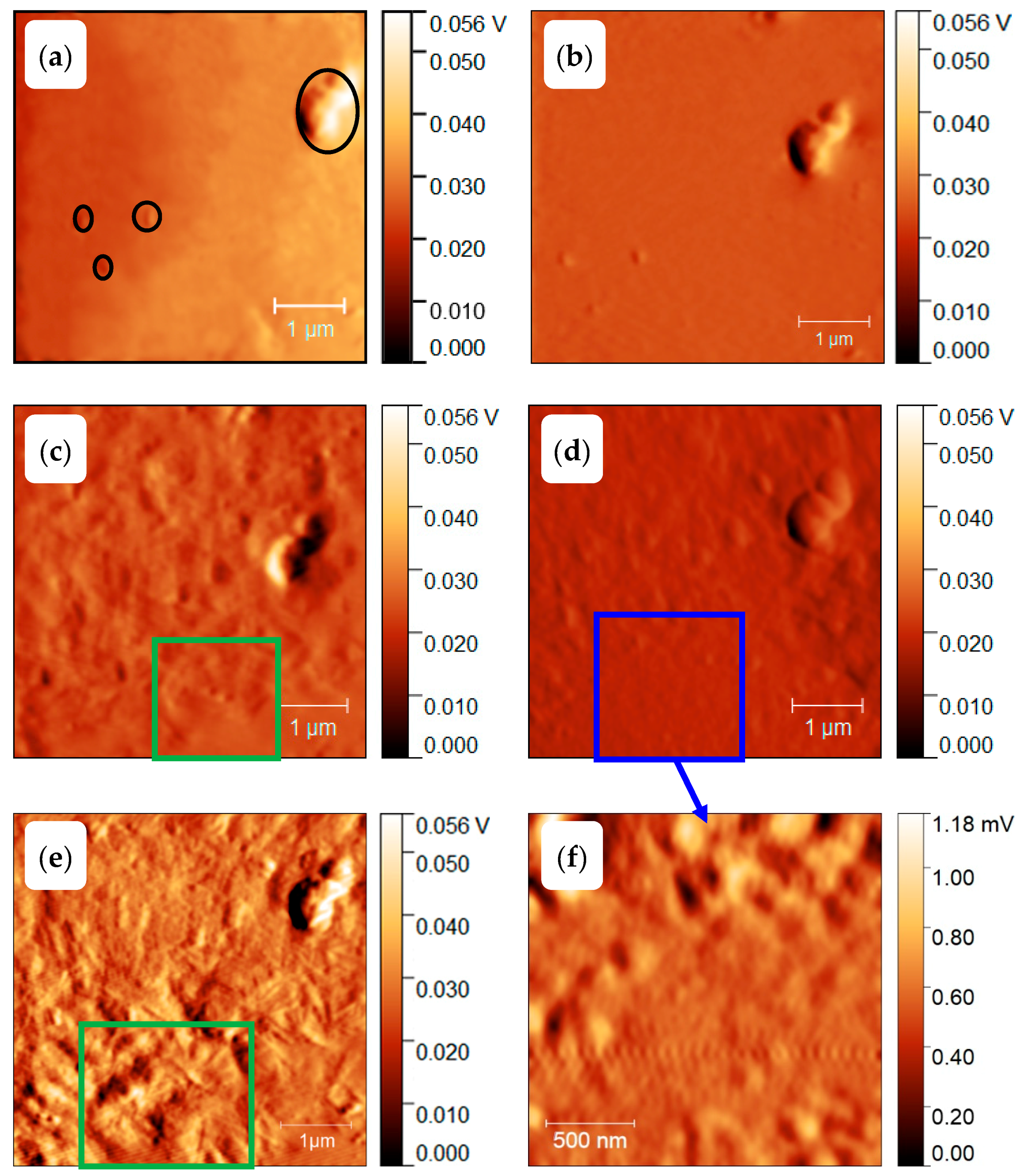
3.3. Homogenous Crystal Nucleation in PBT
4. Summary
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Androsch, R.; Di Lorenzo, M.L.; Schick, C. Optical Microscopy to Study Crystal Nucleation in Polymers Using a Fast Scanning Chip Calorimeter for Precise Control of the Nucleation Pathway. Macromol. Chem. Phys. 2017, 219, 1700479. [Google Scholar] [CrossRef]
- Schick, C.; Androsch, R.; Schmelzer, J.W.P. Homogeneous crystal nucleation in polymers (topical review). J. Phys. Condens. Matter 2017, 29, 453002. [Google Scholar] [CrossRef] [PubMed]
- Schick, C.; Mathot, V. Fast Scanning Calorimetry; Springer International Publishing AG: Basel, Switzerland, 2016; pp. 1–795. [Google Scholar]
- Toda, A.; Androsch, R.; Schick, C. Insights into polymer crystallization and melting from fast scanning chip calorimetry. Polymer 2016, 91, 239–263. [Google Scholar] [CrossRef]
- Androsch, R.; Schick, C. Crystal Nucleation of Polymers at High Supercooling of the Melt. Adv. Polym. Sci. 2015, 276, 257–288. [Google Scholar]
- Androsch, R.; Schick, C.; Di Lorenzo, M.L. Kinetics of Nucleation and Growth of Crystals of Poly(l-lactic acid). Adv. Polym. Sci. 2017, 279, 235–272. [Google Scholar]
- Schick, C.; Androsch, R. Nucleation-controlled semicrystalline morphology of bulk polymers. Polym. Cryst. 2018, 1, e10036. [Google Scholar] [CrossRef]
- Androsch, R.; Rhoades, A.M.; Stolte, I.; Schick, C. Density of heterogeneous and homogeneous crystal nuclei in poly (butylene terephthalate). Eur. Polym. J. 2015, 66, 180–189. [Google Scholar] [CrossRef]
- Zia, Q.; Androsch, R.; Radusch, H.-J.; Ingoliç, E. Crystal morphology of rapidly cooled isotactic polypropylene: A comparative study by TEM and AFM. Polym. Bull. 2008, 60, 791–798. [Google Scholar] [CrossRef]
- Zia, Q.; Androsch, R.; Radusch, H.-J. Effect of the Structure at the Micrometer and Nanometer Scales on the Light Transmission of Isotactic Polypropylene. J. Appl. Polym. Sci. 2010, 117, 1013–1020. [Google Scholar] [CrossRef]
- Sawyer, L.C.; Grubb, D.T.; Meyers, G.F. Polymer Microscopy; Springer: New York, NY, USA, 2008; pp. 1–540. [Google Scholar]
- Schick, C.; Androsch, R. New Insights into Polymer Crystallization by Fast Scanning Chip Calorimetry. In Fast Scanning Calorimetry; Mathot, V.B.F., Schick, C., Eds.; Springer: Berlin, Germany, 2016; pp. 463–535. [Google Scholar]
- Williams, D.B.; Carter, C.B. The Transmission Electron Microscope: A Textbook for Materials Science; Springer: Boston, MA, USA, 2009; pp. 1–760. [Google Scholar]
- Tencé-Girault, S.; Woehling, V.; Oikonomou, E.K.; Karpati, S.; Norvez, S. About the Art and Science of Visualizing Polymer Morphology using Transmission Electron Microscopy. Macromol. Chem. Phys. 2018, 219, 1700483. [Google Scholar] [CrossRef]
- Kim, S.; Jeong Park, M.; Balsara, N.P.; Liu, G.; Minor, A.M. Minimization of focused ion beam damage in nanostructured polymer thin films. Ultramicroscopy 2011, 111, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Zhai, Q.; Gao, Y. Formation of amorphous structure in Sn3.5Ag droplet by in situ fast scanning calorimetry controllable quenching. Appl. Phys. Lett. 2013, 103. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Yang, B.; Yan, M.; Zhai, Q.; Gao, Y. Structure observation of single solidified droplet by in situ controllable quenching based on nanocalorimetry. J. Alloy. Compd. 2013, 580, 386–391. [Google Scholar] [CrossRef]
- Egerton, R.F.; Li, P.; Malac, M. Radiation damage in the TEM and SEM. Micron 2004, 35, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Grapes, M.D.; LaGrange, T.; Friedman, L.H.; Reed, B.W.; Campbell, G.H.; Weihs, T.P.; LaVan, D.A. Combining nanocalorimetry and dynamic transmission electron microscopy for in situ characterization of materials processes under rapid heating and cooling. Rev. Sci. Instrum. 2014, 85. [Google Scholar] [CrossRef]
- Grapes, M.D.; Santala, M.K.; Campbell, G.H.; LaVan, D.A.; Weihs, T.P. A detailed study of the Al3Ni formation reaction using nanocalorimetry. Thermochim. Acta 2017, 658, 72–83. [Google Scholar] [CrossRef]
- Yi, F.; Stevanovic, A.; Osborn, W.A.; Kolmakov, A.; LaVan, D.A. A multi-environment nanocalorimeter with electrical contacts for use in a scanning electron microscope. Mater. Horiz. 2017, 4, 1128–1134. [Google Scholar] [CrossRef]
- Savage, R.C.; Mullin, N.; Hobbs, J.K. Molecular Conformation at the Crystal–Amorphous Interface in Polyethylene. Macromolecules 2015, 48, 6160–6165. [Google Scholar] [CrossRef]
- Morita, S.; Giessibl, F.J.; Meyer, E.; Wiesendanger, R. Noncontact Atomic Force Microscopy; Springer: Berlin, Germany, 2015; Volume 3. [Google Scholar]
- Hobbs, J.K. In-situ AFM of polymer crystallization. Chin. J. Polym. Sci. 2003, 21, 135–140. [Google Scholar]
- Hobbs, J.K.; Farrance, O.E.; Kailas, L. How atomic force microscopy has contributed to our understanding of polymer crystallization. Polymer 2009, 50, 4281–4292. [Google Scholar] [CrossRef]
- Zhou, J.-J.; Liu, J.-G.; Yan, S.-K.; Dong, J.-Y.; Li, L.; Chan, C.-M.; Schultz, J.M. Atomic force microscopy study of the lamellar growth of isotactic polypropylene. Polymer 2005, 46, 4077–4087. [Google Scholar] [CrossRef]
- Hobbs, J.K.; McMaster, T.J.; Miles, M.J.; Barham, P.J. Direct observations of the growth of spherulites of poly(hydroxybutyrate-co-valerate) using atomic force microscopy. Polymer 1998, 39, 2437–2446. [Google Scholar] [CrossRef]
- Li, L.; Chan, C.-M.; Yeung, K.L.; Li, J.-X.; Ng, K.-M.; Lei, Y. Direct Observation of Growth of Lamellae and Spherulites of a Semicrystalline Polymer by AFM. Macromolecules 2001, 34, 316–325. [Google Scholar] [CrossRef]
- Chen, E.-Q.; Jing, A.J.; Weng, X.; Huang, P.; Lee, S.-W.; Cheng, S.Z.D.; Hsiao, B.S.; Yeh, F. In situ observation of low molecular weight poly(ethylene oxide) crystal melting, recrystallization. Polymer 2003, 44, 6051–6058. [Google Scholar] [CrossRef]
- Mileva, D.; Androsch, R.; Zhuravlev, E.; Schick, C. Morphology of mesophase and crystals of polyamide 6 prepared in a fast scanning chip calorimeter. Polymer 2012, 53, 3994–4001. [Google Scholar] [CrossRef]
- Li, L.; Chan, C.-M.; Li, J.-X.; Ng, K.-M.; Yeung, K.L.; Weng, L.-T. A Direct Observation of the Formation of Nuclei and the Development of Lamellae in Polymer Spherulites. Macromolecules 1999, 32, 8240–8242. [Google Scholar] [CrossRef]
- Lei, Y.-G.; Chan, C.-M.; Wang, Y.; Ng, K.-M.; Jiang, Y.; Lin, L. Growth process of homogeneously and heterogeneously nucleated spherulites as observed by atomic force microscopy. Polymer 2003, 44, 4673–4679. [Google Scholar] [CrossRef]
- Humphris, A.D.L.; Miles, M.J.; Hobbs, J.K. A mechanical microscope: High-speed atomic force microscopy. Appl. Phys. lett. 2005, 86, 034106. [Google Scholar] [CrossRef]
- Kailas, L.; Vasilev, C.; Audinot, J.-N.; Migeon, H.-N.; Hobbs, J.K. A Real-Time Study of Homogeneous Nucleation, Growth, and Phase Transformations in Nanodroplets of Low Molecular Weight Isotactic Polypropylene Using AFM. Macromolecules 2007, 40, 7223–7230. [Google Scholar] [CrossRef]
- Zhao, B.; Howard-Knight, J.P.; Humphris, A.D.; Kailas, L.; Ratcliffe, E.C.; Foster, S.J.; Hobbs, J.K. Large scan area high-speed atomic force microscopy using a resonant scanner. Rev. Sci. Instrum. 2009, 80, 093707. [Google Scholar] [CrossRef]
- Jin, L.; Ball, J.; Bremner, T.; Sue, H.-J. Crystallization behavior and morphological characterization of poly(ether ether ketone). Polymer 2014, 55, 5255–5265. [Google Scholar] [CrossRef]
- Gohn, A.M.; Rhoades, A.M.; Wonderling, N.; Tighe, T.; Androsch, R. The effect of supercooling of the melt on the semicrystalline morphology of PA 66. Thermochim. Acta 2017, 655, 313–318. [Google Scholar] [CrossRef]
- Mollova, A.; Androsch, R.; Mileva, D.; Schick, C.; Benhamida, A. Effect of Supercooling on Crystallization of Polyamide 11. Macromolecules 2013, 46, 828–835. [Google Scholar] [CrossRef]
- Van den Brande, N.; Van Assche, G.; Van Mele, B. Isothermal crystallization of PC61BM in thin layers far below the glass transition temperature. Cryst. Growth Des. 2015, 15, 5614–5623. [Google Scholar] [CrossRef]
- Anfatec. Available online: http://www.anfatec.de/anfatec/mic.html (accessed on 1 May 2019).
- Micromasch. AFM Probe NSC 14. Available online: https://www.spmtips.com/afm-probes-14-series.afm (accessed on 1 May 2019).
- Nunano. Available online: https://www.nunano.com/store/scout-70r (accessed on 1 May 2019).
- La Spina, L.; Ovchinnikov, D.; Wien, W.H.A.; van Herwaarden, A.W.; Goudena, E.J.G.; Loos, J.; Nanver, L.K. Reduction of surface roughness of a silicon chip for advanced nanocalorimetry. Sens. Actuators A Phys. 2008, 144, 403–409. [Google Scholar] [CrossRef]
- van Herwaarden, A.W. Overview of calorimeter chips for various applications. Thermochim. Acta 2005, 432, 192–201. [Google Scholar] [CrossRef]
- Xensor-Integration. Xensor Product Overview. Available online: http://www.xensor.nl/index.php/products/product-overview (accessed on 1 May 2019).
- Zhuravlev, E.; Schick, C. Fast scanning power compensated differential scanning nano-calorimeter: 1. The device. Thermochim. Acta 2010, 505, 1–13. [Google Scholar] [CrossRef]
- Zhuravlev, E.; Schick, C. Fast scanning power compensated differential scanning nano-calorimeter: 2. Heat capacity analysis. Thermochim. Acta 2010, 505, 14–21. [Google Scholar] [CrossRef]
- Schick, C.; Mathot, V. Fast Scanning Calorimetry; Springer: Basel, Switzerland, 2016. [Google Scholar]
- Pyda, M. Poly(butylene terephthalate) (PBT) Heat Capacity, Enthalpy, Entropy, Gibbs Energy: Datasheet from “The Advanced THermal Analysis System (ATHAS) Databank—Polymer Thermodynamics”; Release 2014 in SpringerMaterials; Springer: Berlin, Germany, 2014; Available online: https://materials.springer.com/polymerthermodynamics/docs/athas_0106 (accessed on 1 May 2019).
- Pyda, M. Poly(oxy-1,4-phenylene-1,4-phenylene-carbonyl-1,4-phenylene) (PEEK) Heat Capacity, Enthalpy, Entropy, Gibbs Energy: Datasheet from “The Advanced THermal Analysis System (ATHAS) Databank—Polymer Thermodynamics”; Release 2014 in SpringerMaterials; Springer: Berlin, Germany, 2014; Available online: https://materials.springer.com/polymerthermodynamics/docs/athas_0108 (accessed on 1 May 2019).
- Pyda, M. Nylon 6,6 α (NYLON66) Heat Capacity, Enthalpy, Entropy, Gibbs Energy: Datasheet from “The Advanced THermal Analysis System (ATHAS) Databank—Polymer Thermodynamics”; Release 2014 in SpringerMaterials; Springer: Berlin, Germany, 2014; Available online: https://materials.springer.com/polymerthermodynamics/docs/athas_0027 (accessed on 1 May 2019).
- Pyda, M. Poly(epsilon-caprolactone) (PCL) Heat Capacity, Enthalpy, Entropy, Gibbs Energy: Datasheet from “The Advanced THermal Analysis System (ATHAS) Databank—Polymer Thermodynamics”; Release 2014 in SpringerMaterials; Springer: Berlin, Germany, 2014; Available online: https://materials.springer.com/polymerthermodynamics/docs/athas_0049 (accessed on 1 May 2019).
- Zhuravlev, E.; Madhavi, V.; Lustiger, A.; Androsch, R.; Schick, C. Crystallization of Polyethylene at Large Undercooling. ACS Macro Lett. 2016, 5, 365–370. [Google Scholar] [CrossRef]
- Cebe, P.; Hu, X.; Huang, W.; Kaplan, D.; Zhuravlev, E.; Wurm, A.; Arbeiter, D.; Schick, C. Using flash DSC for determining the liquid state heat capacity of silk fibroin. Abstr. Pap. 2015, 615, 8–14. [Google Scholar] [CrossRef]
- Wunderlich, B.; Pyda, M. Application of the Advanced Thermal Analysis System (ATHAS) to differential scanning calorimetry (DSC) and temperature—Modulated DSC of polymers. J. Reinf. Plast. Compos. 1999, 18, 487–498. [Google Scholar] [CrossRef]
- Zhuravlev, E.; Schmelzer, J.W.P.; Wunderlich, B.; Schick, C. Kinetics of nucleation and crystallization in poly(epsilon caprolactone) (PCL). Polymer 2011, 52, 1983–1997. [Google Scholar] [CrossRef]
- Pearce, R.; Vancso, G.J. Imaging of Melting and Crystallization of Poly(ethylene oxide) in Real-Time by Hot-Stage Atomic Force Microscopy. Macromolecules 1997, 30, 5843–5848. [Google Scholar] [CrossRef]
- Herd, M.; Prasad, A. On the Observation of a New Morphology in Poly(ary1ene ether ether ketone). A Further Examination of the Double Endothermic Behavior of Poly(ary1ene ether ether ketone). Macromolecules 1992, 25, 1731–1736. [Google Scholar]
- Ye Sun, H.X.; Shuang Chen, M.D.; Ediger, L.Y. Crystallization near Glass Transition: Transition from Diffusion-Controlled to Diffusionless Crystal Growth Studied with Seven Polymorphs. J. Phys. Chem. B 2008, 112, 5594–5601. [Google Scholar] [CrossRef] [PubMed]
- Toda, A.; Konishi, M. An Evaluation of Thermal Lags of Fast-Scan Microchip DSC with Polymer Film Samples. Thermochim. Acta 2014, 589, 262–269. [Google Scholar] [CrossRef]
- Höhne, G.W.H.; Cammenga, H.K.; Eysel, W.; Gmelin, E.; Hemminger, W. The temperature calibration of scanning calorimeters. Thermochim. Acta 1990, 160, 1–12. [Google Scholar]
- Burnett, B.B.; McDevit, W.F. Kinetics of Spherulite Growth in High Polymers. J. Appl. Phys. 1957, 28, 1101–1105. [Google Scholar] [CrossRef]
- Runt, J.; Miley, D.M.; Zhang, X.; Gallagher, K.P.; McFeaters, K.; Fishburn, J. Crystallization of Poly(butylene terephthalate) and Its Blends with Polyarylate. Macromolecules 1992, 25, 1929–1934. [Google Scholar] [CrossRef]
- Boyd, R.H. The dielectric constant of lamellar semicrystalline polymer. J. Polym. Sci. Polym. Phys. Ed. 1983, 21, 505–514. [Google Scholar] [CrossRef]
- Langhe, D.S.; Hiltner, A.; Baer, E. Transformation of isotactic polypropylene droplets from the mesophase into the α-phase. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1672–1682. [Google Scholar] [CrossRef]
- Zia, Q.; Androsch, R. Effect of atomic force microscope tip geometry on the evaluation of the crystal size of semicrystalline polymers. Meas. Sci. Technol. 2009, 20, 097003. [Google Scholar] [CrossRef]
- Furushima, Y.; Nakada, M.; Ishikiriyama, K.; Toda, A.; Androsch, R.; Zhuravlev, E.; Schick, C. Two crystal populations with different melting/reorganization kinetics of isothermally crystallized polyamide 6. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 2126–2138. [Google Scholar] [CrossRef]
- Tammann, G. Über die Abhängigkeit der Zahl der Kerne, welche sich in verschiedenen unterkühlten Flüssigkeiten bilden, von der Temperatur. Z. Phys. Chem. 1898, 25, 441–479. [Google Scholar]
- Furushima, Y.; Kumazawa, S.; Umetsu, H.; Toda, A.; Zhuravlev, E.; Schick, C. Melting and recrystallization kinetics of poly(butylene terephthalate). Polymer 2017, 109, 307–314. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Sample | Abbreviation | Molar Mass (g/mol) | Mw/Mn | (K) | Grade | Supplier |
|---|---|---|---|---|---|---|
| Poly(butylene terephthalate) | PBT | 558,600 | 2.25 | 518 [49] | Toray Industry, Inc, Japan | |
| Poly(ether ether ketone) | PEEK | 85,000 | 668 [50] | 150 G | Victrex plc. United Kingdom | |
| Polyamide 66 | PA 66 | 18,000 | 535 [51] | Zytel 101L | DuPont, USA | |
| Poly(ε-caprolactone) | PCL | 20,000 | 1.73 | 342 [52] | Sigma-Aldrich, USA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Zhuravlev, E.; Androsch, R.; Schick, C. Visualization of Polymer Crystallization by In Situ Combination of Atomic Force Microscopy and Fast Scanning Calorimetry. Polymers 2019, 11, 890. https://doi.org/10.3390/polym11050890
Zhang R, Zhuravlev E, Androsch R, Schick C. Visualization of Polymer Crystallization by In Situ Combination of Atomic Force Microscopy and Fast Scanning Calorimetry. Polymers. 2019; 11(5):890. https://doi.org/10.3390/polym11050890
Chicago/Turabian StyleZhang, Rui, Evgeny Zhuravlev, René Androsch, and Christoph Schick. 2019. "Visualization of Polymer Crystallization by In Situ Combination of Atomic Force Microscopy and Fast Scanning Calorimetry" Polymers 11, no. 5: 890. https://doi.org/10.3390/polym11050890
APA StyleZhang, R., Zhuravlev, E., Androsch, R., & Schick, C. (2019). Visualization of Polymer Crystallization by In Situ Combination of Atomic Force Microscopy and Fast Scanning Calorimetry. Polymers, 11(5), 890. https://doi.org/10.3390/polym11050890