Synthesis of Thermo-Responsive Block-Graft Copolymer Based on PCL and PEG Analogs, and Preparation of Hydrogel via Click Chemistry


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
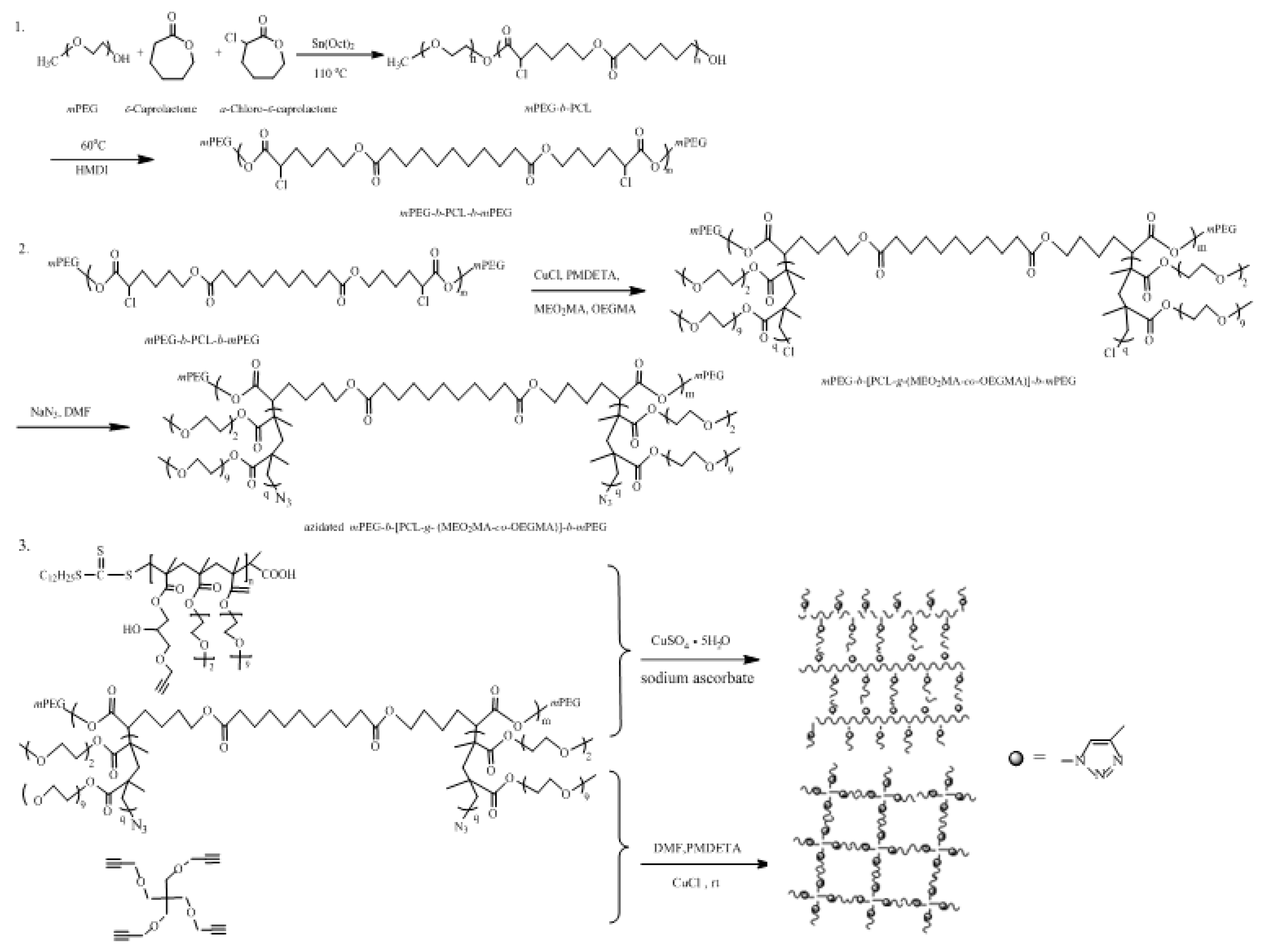
2.2. Synthesis
2.2.1. Synthesis of Block Copolymer mPEG-b-PCL-b-mPEG
2.2.2. Synthesis of mPEG-b-[PCL-g-(MEO2MA-co-OEGMA)]-b-mPEG and Azide End-Functionalized mPEG-b-[PCL-g-(MEO2MA-co-OEGMA)]-b-mPEG
2.2.3. Synthesis of Cross-Linker Alkyne-Terminated P(GMA-co-MEO2MA-co-OEGMA)
2.2.4. Synthesis of Cross-Linker TPOM
2.2.5. Preparation of Two Hydrogels via Click Chemistry
2.3. Characterization
2.3.1. Nuclear Magnetic Resonance Spectroscopy (1H NMR)
2.3.2. Fourier-transform infrared Spectroscopy (FTIR)
2.3.3. Gel Permeation Chromatography (GPC)
2.4. Water Solubility and Temperature Sensitivitys
2.5. Micelle Properties of Polymers
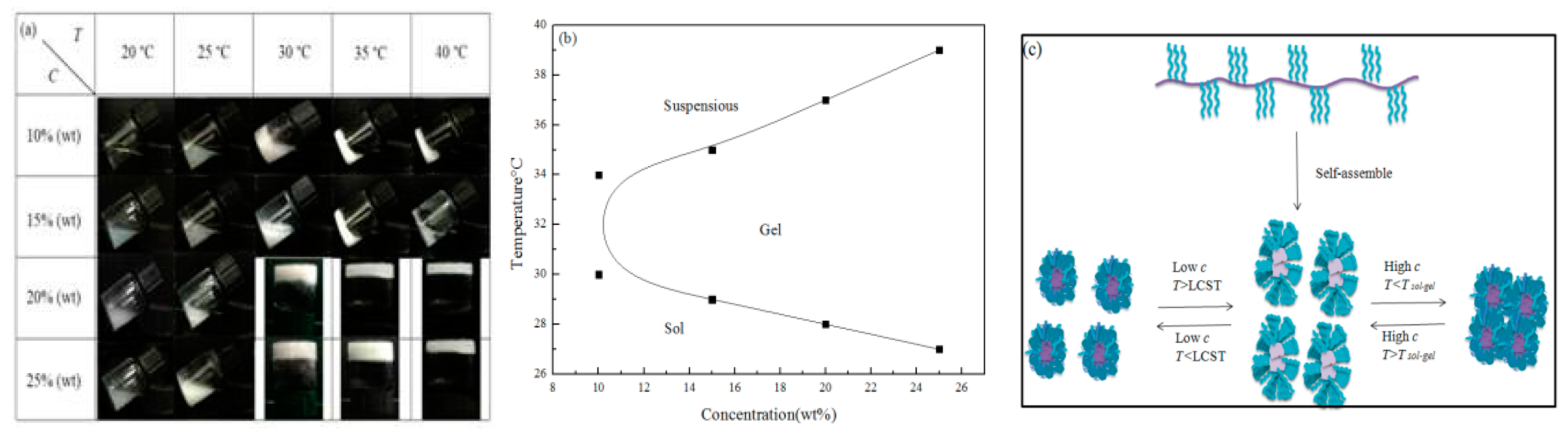
2.6. Sol–gel Transition
2.7. SEM Analysis of Gel
3. Results and Discussion
3.1. Synthesis of the Copolymers and Hydrogels
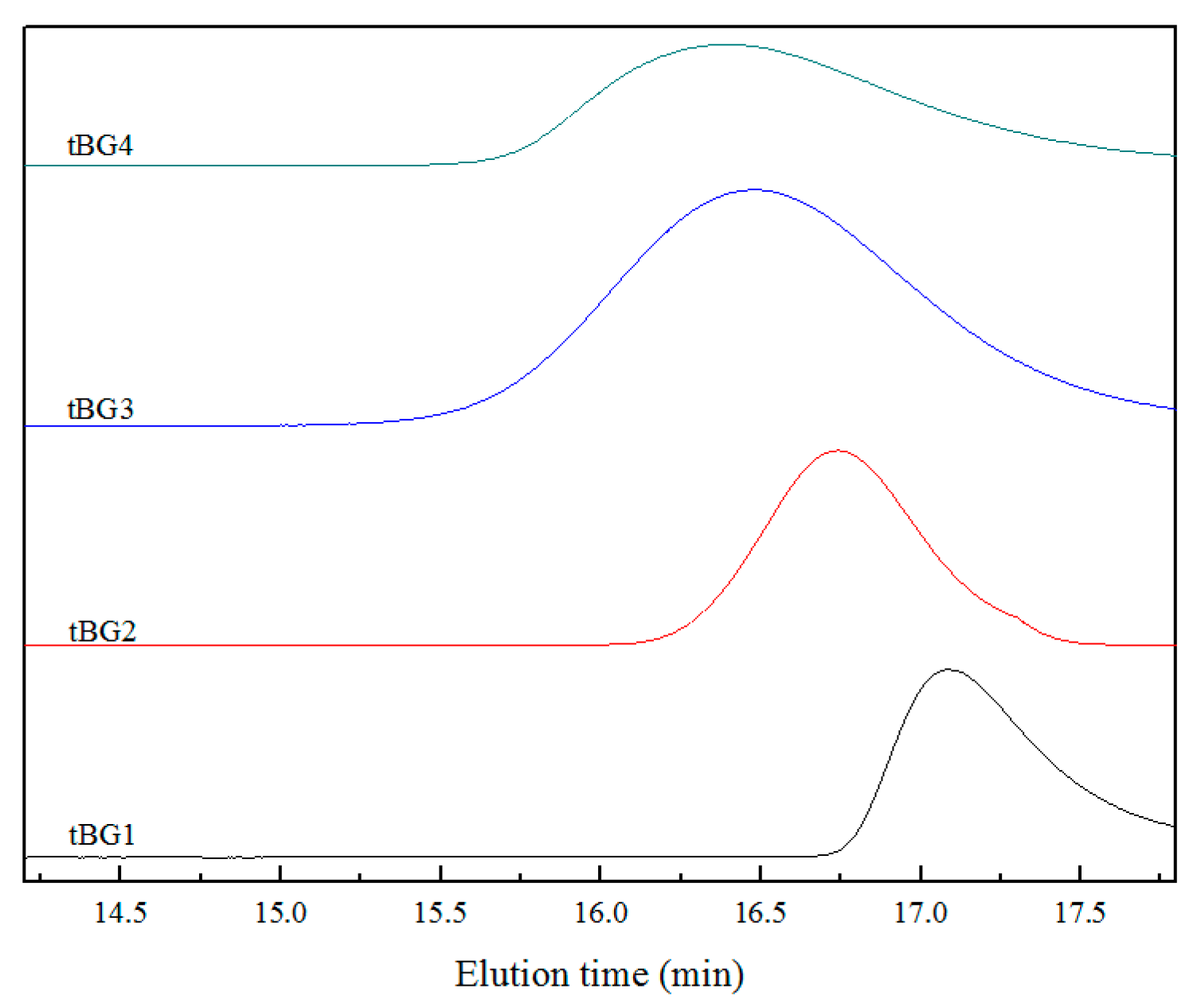
3.2. GPC Characterization of the Copolymers
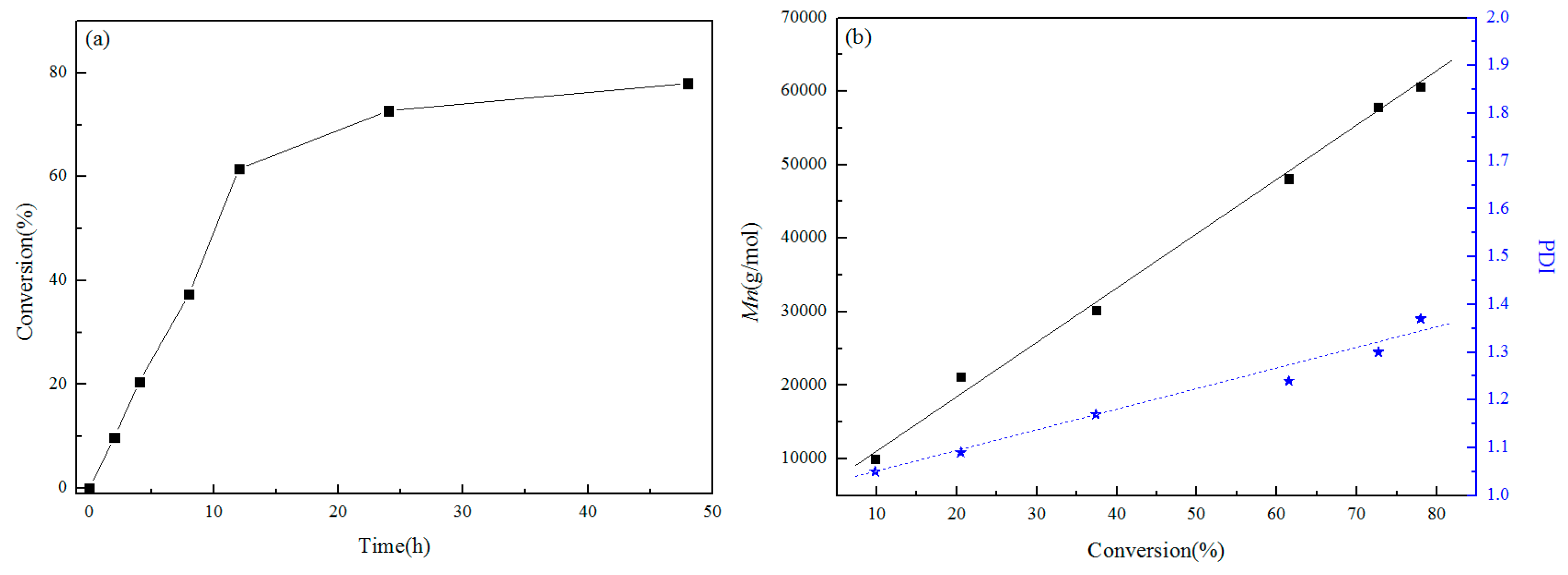
3.3. Relationship between Conversion Rate and Molecular Weight
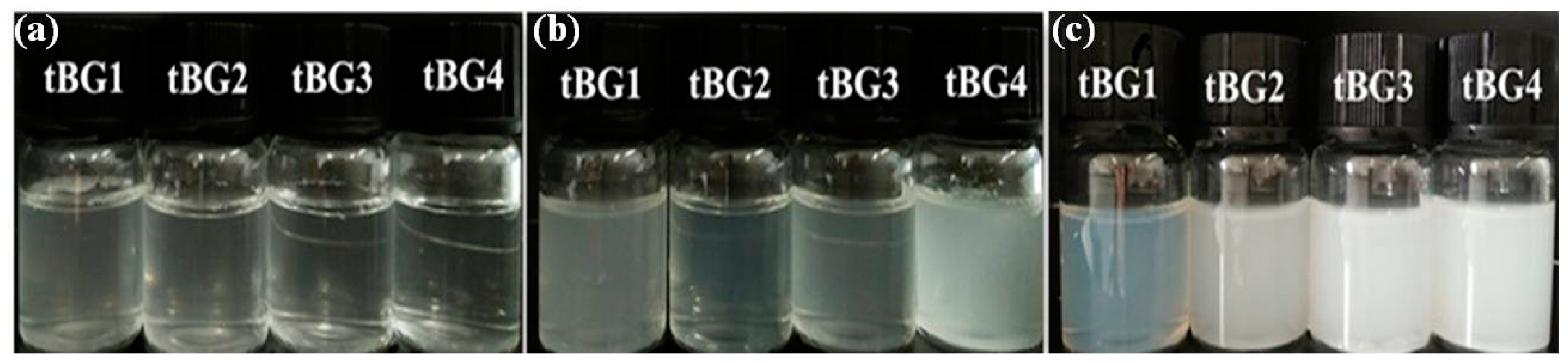
3.4. Water Solubility and Temperature Sensitivity
3.5. Critical Micellization Concentration Determination (CMC)
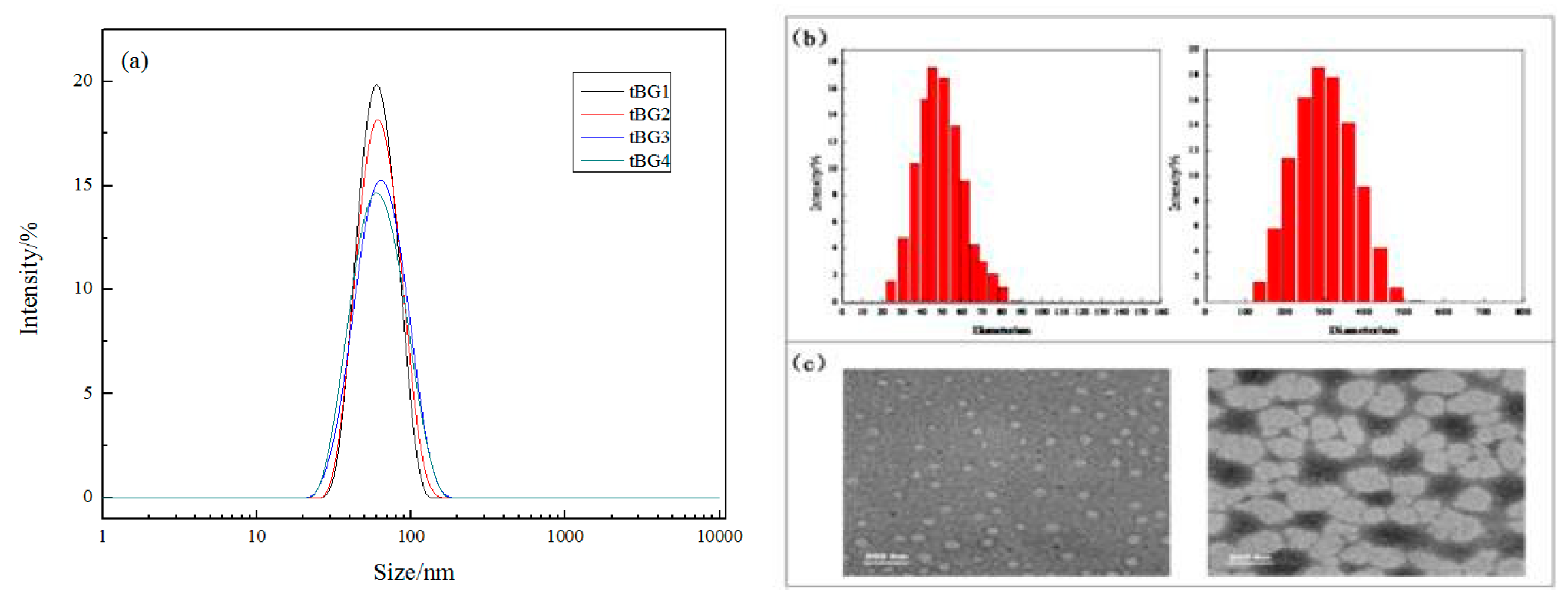
3.6. Particle Size of Copolymer Micelles
3.7. Sol–Gel Transition
3.8. SEM Analysis of Gel
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Yuan, Y.; Zhang, A.K.; Ling, J.; Yin, L.H.; Chen, Y.; Fu, G.D. Well-defined biodegradable amphiphilic conetworks. Soft Matter 2013, 9, 6309–6318. [Google Scholar] [CrossRef]
- Li, L.; Lu, B.B.; Wu, J.N.; Fan, Q.K.; Guo, X.H.; Liu, Z.Y. Synthesis and self-assembly behavior of thermo-responsive star-shaped POSS-(PCL- P(MEO2MA-co-PEGMA))16 inorganic/organic hybrid block copolymers with tunable lower critical solution temperature. New J. Chem. 2016, 40, 4761–4768. [Google Scholar] [CrossRef]
- Han, Y.; Li, Y.H.; Zeng, Q.Y.; Li, H.Y.; Peng, J.L.; Xu, Y.H.; Chang, J. Injectable bioactive akermanite/alginate composite hydrogels for in situ skin tissue engineering. J. Mater. Chem. B 2017, 5, 3315–3326. [Google Scholar] [CrossRef]
- Döring, A.; Birnbaum, W.G.; Kuckling, D. Responsive hydrogels-structurally and dimensionally optimized smart frameworks for applications in catalysis, micro-system technology and material science. Chem. Soc. Rev. 2013, 42, 7391–7420. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.Y.; Cosimbescu, L.; Karin, N.J.; Gutowska, A.; Tarasevich, B.J. Injectable and thermogelling hydrogels of PCL-g-PEG: Mechanisms, rheological and enzymatic degradation properties. J. Mater. Chem. B 2013, 1, 1249–1255. [Google Scholar] [CrossRef]
- Qian, S.S.; Zhou, C.; Xu, L.Q.; Yao, F.; Cen, L.; Fu, G.D. High strength biocompatible PEG single-network hydrogels. RSC Adv. 2014, 4, 25241–25250. [Google Scholar] [CrossRef]
- Kabir, M.H.; Hazama, T.; Watanabe, Y.; Gong, J.; Murase, K.; Suanda, T.; Furukawa, H. Smart hydrogel with shape memory for biomedical applications. J. Taiwan Inst. Chem. Eng. 2014, 45, 3134–3138. [Google Scholar] [CrossRef]
- Lim, H.L.; Hwang, Y.S.; Kar, M.; Varghese, S. Smart hydrogels as functional biomimetic systems. Biomater. Sci. 2014, 2, 603–618. [Google Scholar] [CrossRef]
- Yu, L.; Ding, J.D. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37, 1473–1481. [Google Scholar] [CrossRef]
- Ravichandran, R.; Astrand, C.; Patra, H.K.; Turner, A.P.F.; Chotteau, V.; Phopase, J. Intelligent ECM mimetic injectable scaffolds based on functional collagen building blocks for tissue engineering and biomedical applications. RSC Adv. 2017, 7, 21068–21078. [Google Scholar] [CrossRef]
- Che, Y.J.; Li, D.P.; Liu, Y.L.; Ma, Q.L.; Tan, Y.B.; Yue, Q.Y.; Meng, F.J. Physically cross-linked pH-responsive chitosan-based hydrogels with enhanced mechanical performance for controlled drug delivery. RSC Adv. 2016, 6, 106035–106045. [Google Scholar] [CrossRef]
- Ding, X.C.; Wang, Y.D. Weak bond-based injectable and stimuli responsive hydrogels for biomedical applications. J. Mater. Chem. B 2017, 5, 887–906. [Google Scholar] [CrossRef]
- Gong, Z.Y.; Zhang, G.P.; Zeng, X.L.; Li, J.H.; Li, G.; Huang, W.P.; Sun, R.; Wong, C.P. High-strength, tough, fatigue resistant, and self-healing hydrogel based on dual physically cross-linked network. ACS Appl. Mater. Interfaces 2016, 8, 24030–24037. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2004, 40, 2004–2021. [Google Scholar] [CrossRef]
- Es-haghi, S.S.; Leonov, A.I.; Weiss, R.A. Deconstructing the double-network hydrogels: The importance of grafted chains for achieving toughness. Macromolecules 2014, 47, 4769–4777. [Google Scholar] [CrossRef]
- Hemingway, M.G.; Gupta, R.B.; Elton, D.J. Hydrogel nanopowder production by inverse-miniemulsion polymerization and supercritical drying. Ind. Eng. Chem. Res. 2010, 49, 10094–10099. [Google Scholar] [CrossRef]
- Malkoch, M.; Vestberg, R.; Gupta, N.; Mespouille, L.; Dubois, P.; Mason, A.F.; Hedrick, J.L.; Liao, Q.; Frank, C.W.; Kingsbury, K.; Hawker, C.J. Synthesis of well-defined hydrogel networks using click chemistry. Chem. Commun. 2006, 2774–2776. [Google Scholar] [CrossRef]
- Crescenzi, V.; Cornelio, L.; Meo, C.D.; Nardecchia, S.; Lamanna, R. Novel hydrogels via click chemistry: Synthesis and potential biomedical applications. Biomacromolecules 2007, 8, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Ossipov, D.A.; Hilborn, J. Poly(vinyl alcohol)-based hydrogels formed by “click chemistry”. Macromolecules 2006, 39, 1709–1718. [Google Scholar] [CrossRef]
- Grover, G.N.; Lam, J.; Nguyen, T.H.; Segura, T.; Maynard, H.D. Bicompatible hydrogels by oxime click chemistry. Biomacromolecules 2012, 13, 3013–3017. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2592–3015. [Google Scholar] [CrossRef]
- Chang, L.L.; Deng, L.D.; Wang, W.W.; Lv, Z.S.; Hu, F.Q.; Dong, A.J.; Zhang, J.H. Poly(ethyleneglycol)-b-Poly(ε-caprolactone-co-γ-hydroxyl-ε-caprolactone) bearing pendant hydroxyl groups as nanocarriers for doxorubicin delivery. Biomacromolecules 2012, 13, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Thévenaz, D.C.; Monnier, C.A.; Balog, S.; Fiore, G.L. Luminescent nanoparticles with lanthanide-containing poly(ethylene glycol)-poly(ε-caprolactone) block copolymers. Biomacromolecules 2014, 15, 3994–4001. [Google Scholar] [CrossRef] [PubMed]
- Vidyasagar, A.; Ku, S.H.; Kim, M.; Kim, M.; Lee, H.S.; Pearce, T.R.; McCormick, A.V.; Bates, F.S.; Kokkoli, E. Design and characterization of a PVLA-PEG-PVLA thermosensitive and biodegradable hydrogel. ACS Macro Lett. 2017, 6, 1134–1139. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Kumar, C.U.; Jewrajka, S.K. Effect of polyethylene glycol on properties and drug encapsulation–release performance of biodegradable/cytocompatible agarose–polyethylene glycol–polycaprolactone amphiphilic co-network gels. ACS Appl. Mater. Interfacs. 2016, 8, 3182–3192. [Google Scholar] [CrossRef] [PubMed]
- He, J.P.; Chen, J.Z.; Lin, S.L.; Niu, D.C.; Hao, J.; Jia, X.B.; Li, N.; Gu, J.L.; Li, Y.S.; Shi, J.L. Synthesis of a pillar[5] arene-based polyrotaxane for enhancing the drug loading capacity of PCL-based supramolecular amphiphile as an excellent drug delivery platform. Biomacromolecules 2018, 19, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Jain, E.; Hill, L.; Canning, E.; Sell, S.A.; Zustiak, S.P. Control of gelation, degradation and physical properties of polyethylene glycol hydrogels through the chemical and physical identity of the crosslinker. J. Mater. Chem. B 2017, 5, 2679–2691. [Google Scholar] [CrossRef]
- Blackwell, C.J.; Haernvall, K.; Guebitz, G.M.; Groombridge, M.; Gonzales, D.; Khosravi, E. Enzymatic degradation of star poly(ε-caprolactone) with different central units. Polymers 2018, 10, 1266. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Li, X.; Guang, N.E.; Tian, L.; Mao, H.G.; Ning, W.Y. Novel amphiphilic temperature responsive graft copolymers PCL-g-P(MEO2MA-co- OEGME) via a combination of ROP and ATRP: Synthesis, characterization, and sol-gel transition. J. Polym. Res. 2016, 23, 141–153. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Liu, S.X.; Sheng, W.J.; Guang, N.E.; Li, X. Synthesis and sol-gel transition of novel temperature responsive ABA triblock-graft copolymers based on PCL and PEG analogues. Macromol. Res. 2015, 23, 607–617. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Weichenhan, K.; Akdemir, Ö.; Hoth, A. About the phase transitions in aqueous solutions of thermoresponsive copolymers and hydrogels based on 2-(2-methoxyethoxy)ethyl methacrylate and oligo(ethylene glycol) methacrylate. Macromolecules 2007, 40, 2503–2508. [Google Scholar] [CrossRef]
- Lin, W.C.; Liou, S.H.; Kotsuchibashi, Y. Development and characterisation of the imiquimod poly(2-(2-methoxyethoxy)ethyl methacrylate) hydrogel dressing for keloid therapy. Polymers 2017, 9, 579. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.T.; Wu, P.Y. On the thermally reversible dynamic hydration behavior of oligo(ethylene glycol) methacrylate-based polymers in water. Macromolecules 2013, 46, 236–246. [Google Scholar] [CrossRef]
- Xu, L.Q.; Yao, F.; Fu, G.D.; Kang, E.T. Interpenetrating network hydrogels via simultaneous “click chemistry” and atom transfer radical polymerization. Biomacromolecules 2010, 11, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, S.; Riva, R.; Lou, X.; Detrembleur, C.; Jérôme, R.; Lecomte, P. Ring-opening polymerization of α-chloro-ε-caprolactone and chemical modification of poly(α-chloro-ε-caprolactone) by atom transfer radical processes. Macromolecules 2004, 37, 4055–4061. [Google Scholar] [CrossRef]
- Chen, J.C.; Liu, M.Z.; Gao, C.M.; Lü, S.Y.; Zhang, X.Y.; Liu, Z. Self-assembly behavior of pH- and thermo-responsive hydrophilic ABCBA-type pentablock copolymers synthesized by consecutive RAFT polymerization. RSC Adv. 2013, 3, 15085–15093. [Google Scholar] [CrossRef]
- Lutz, J.F.; Hoth, A. Preparation of Ideal PEG Analogues with a Tunable Thermosensitivity by Controlled Radical Copolymerization of 2-(2-Methoxyethoxy) ethyl Methacrylate and Oligo(ethylene glycol) Methacrylate. Macromolecules 2006, 39, 893–896. [Google Scholar] [CrossRef]
- Mishra, A.K.; Vishwakarma, N.K.; Patel, V.K.; Biswas, C.S.; Paira, T.K.; Mandal, T.K.; Maiti, P.; Ray, B. Synthesis, characterization, and solution behavior of well-defined double hydrophilic linear amphiphilic poly (N-isopropylacrylamide)-b-poly (ε-caprolactone)-b-poly (N-isopropylacrylamide) triblock copolymers. Colloid Polym. Sci. 2014, 292, 1405–1418. [Google Scholar] [CrossRef]
- Ning, W.Y.; Shang, P.; Wu, J.; Shi, X.Y.; Liu, S.X. Novel amphiphilic, biodegradable, biocompatible, thermo-responsive ABA triblock copolymers based on PCL and PEG analogues via a combination of ROP and RAFT: Synthesis, characterization, and sustained drug release from self-assembled micelles. Polymers 2018, 10, 214. [Google Scholar] [CrossRef]
- Zhao, Y.P.; He, G.Q.; Guo, W.H.; Bao, L.L.; Yi, M.J.; Gong, Y.K.; Zhang, S.P. Self-assembled micelles prepared from amphiphilic copolymers bearing cell outer membrane phosphorylcholine zwitterions for a potential anti-phagocytic clearance carrier. Polym. Chem. 2016, 7, 5698–5708. [Google Scholar] [CrossRef]
- Li, S.S.; Chen, G.X.; Zhou, Z.; Li, Q.F. Stimuli-induced multiple dissociation and micellization transitions of random copolymers. RSC Adv. 2015, 5, 65847–65855. [Google Scholar] [CrossRef]
- Jin, N.X.; Zhang, H.; Jin, S.; Dadmun, M.D.; Zhao, B. Tuning of thermally induced sol-to-gel transitions of moderately concentrated aqueous solutions of doubly thermosensitive hydrophilic diblock copolymers poly(methoxytri(ethylene glycol) acrylate)-b-poly(ethoxydi(ethylene glycol) acrylate-co-acrylic acid). J. Phys. Chem. B 2012, 116, 3125–3137. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Song, W.J.; Liu, T.; Xin, J.N.; Zhang, J.W. Temperature and pH Responsive Hydrogels Using Methacrylated Lignosulfonate Cross-Linker: Synthesis, Characterization, and Properties. ACS Sustain. Chem. Eng. 2018, 6, 1763–1771. [Google Scholar] [CrossRef]
- Sheng, W.J.; Liu, T.; Liu, S.X.; Wang, Q.Q. Temperature and pH responsive hydrogels based on polyethylene glycol analogues and poly(methacrylic acid) via click chemistry. Polym. Int. 2015, 64, 1415–1424. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | [αClεCL]/[εCL]a | [αClεCL]/[MO]b | Time | Conversion | Mn, Theory | Mn, GPC | PDI |
|---|---|---|---|---|---|---|---|
| (n:n) | (n:n) | (h) | (%) | (g/mol) | (g/mol) | ||
| tBG1 | 2:8 | 1:10 | 12 | 56.8 | 18,988 | 22,469 | 1.06 |
| tBG2 | 3:7 | 1:10 | 12 | 59.6 | 27,704 | 31,654 | 1.07 |
| tBG3 | 4:6 | 1:10 | 12 | 61.3 | 36,581 | 40,788 | 1.22 |
| tBG4 | 5:5 | 1:10 | 2 | 9.8 | 10,625 | 10,023 | 1.05 |
| tBG4 | 5:5 | 1:10 | 4 | 20.5 | 17,722 | 21,172 | 1.09 |
| tBG4 | 5:5 | 1:10 | 8 | 37.4 | 28,930 | 30,181 | 1.17 |
| tBG4 | 5:5 | 1:10 | 12 | 61.5 | 44,914 | 48,131 | 1.24 |
| tBG4 | 5:5 | 1:10 | 24 | 72.7 | 52,342 | 57,895 | 1.30 |
| tBG4 | 5:5 | 1:10 | 48 | 78.0 | 55,857 | 60,583 | 1.37 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, P.; Wu, J.; Shi, X.; Wang, Z.; Song, F.; Liu, S. Synthesis of Thermo-Responsive Block-Graft Copolymer Based on PCL and PEG Analogs, and Preparation of Hydrogel via Click Chemistry. Polymers 2019, 11, 765. https://doi.org/10.3390/polym11050765
Shang P, Wu J, Shi X, Wang Z, Song F, Liu S. Synthesis of Thermo-Responsive Block-Graft Copolymer Based on PCL and PEG Analogs, and Preparation of Hydrogel via Click Chemistry. Polymers. 2019; 11(5):765. https://doi.org/10.3390/polym11050765
Chicago/Turabian StyleShang, Pei, Jie Wu, Xiaoyu Shi, Zhidan Wang, Fei Song, and Shouxin Liu. 2019. "Synthesis of Thermo-Responsive Block-Graft Copolymer Based on PCL and PEG Analogs, and Preparation of Hydrogel via Click Chemistry" Polymers 11, no. 5: 765. https://doi.org/10.3390/polym11050765
APA StyleShang, P., Wu, J., Shi, X., Wang, Z., Song, F., & Liu, S. (2019). Synthesis of Thermo-Responsive Block-Graft Copolymer Based on PCL and PEG Analogs, and Preparation of Hydrogel via Click Chemistry. Polymers, 11(5), 765. https://doi.org/10.3390/polym11050765