Self-Association Behavior of Cell Membrane-Inspired Amphiphilic Random Copolymers in Water



Abstract
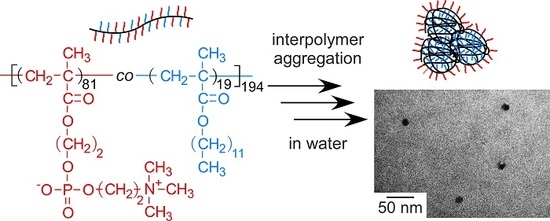
:
1. Introduction
2. Experimental Section
2.1. Materials
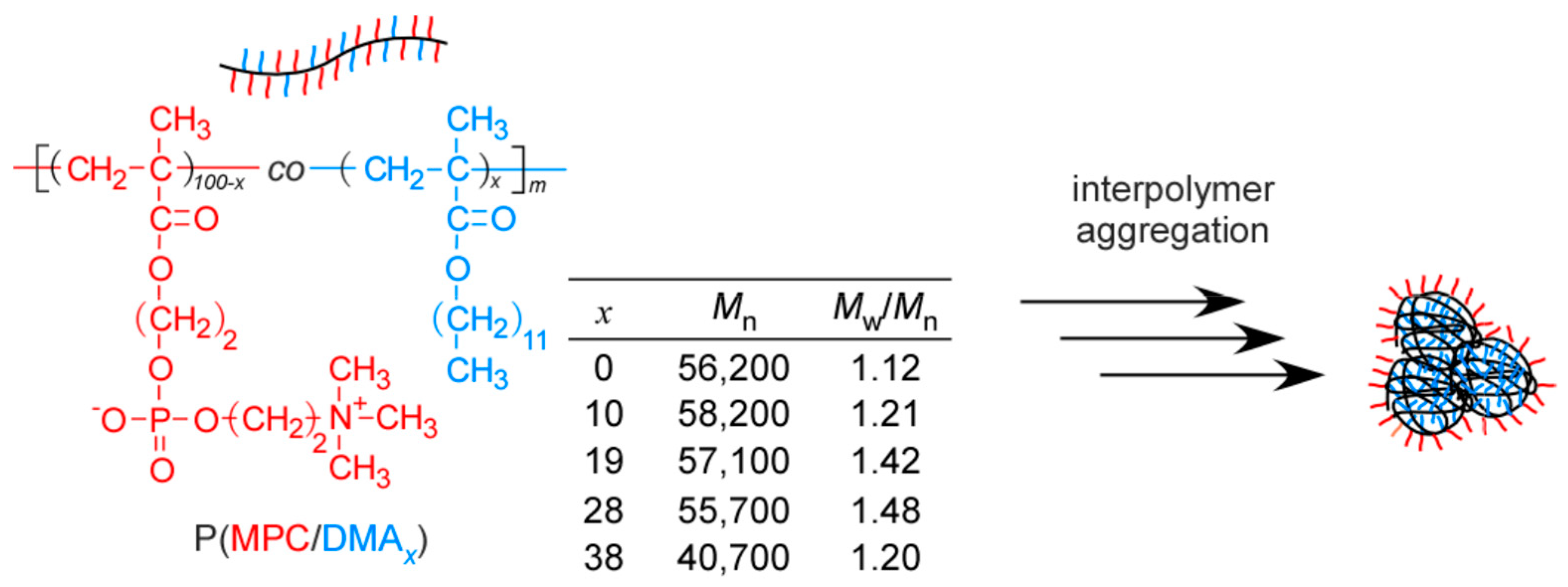
2.2. Preparation of P(MPC/DMAx)
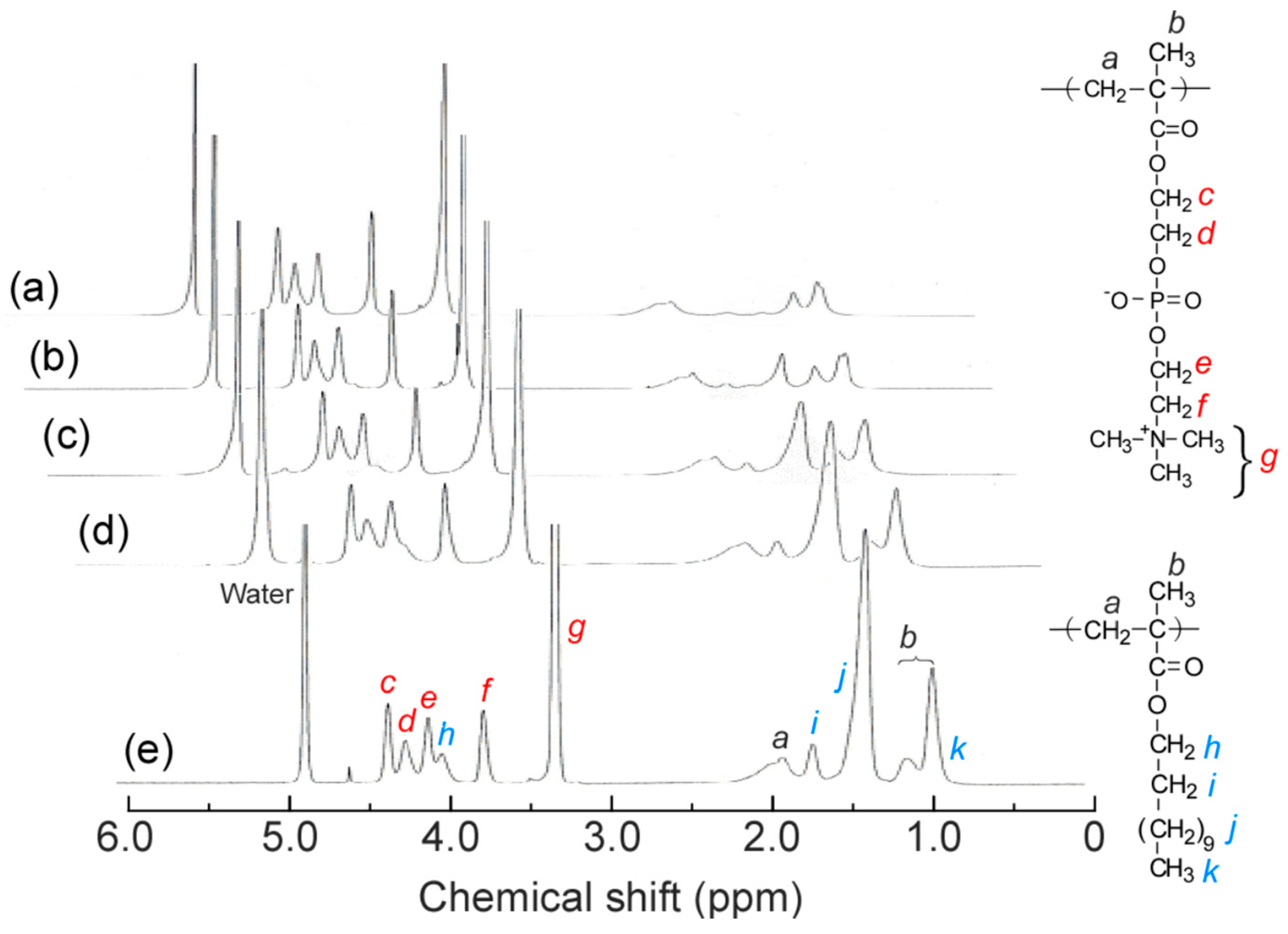
2.3. Characterization of Polymers and Their Aggregates
3. Results and Discussion
3.1. Preparation of P(MPC/DMAx)
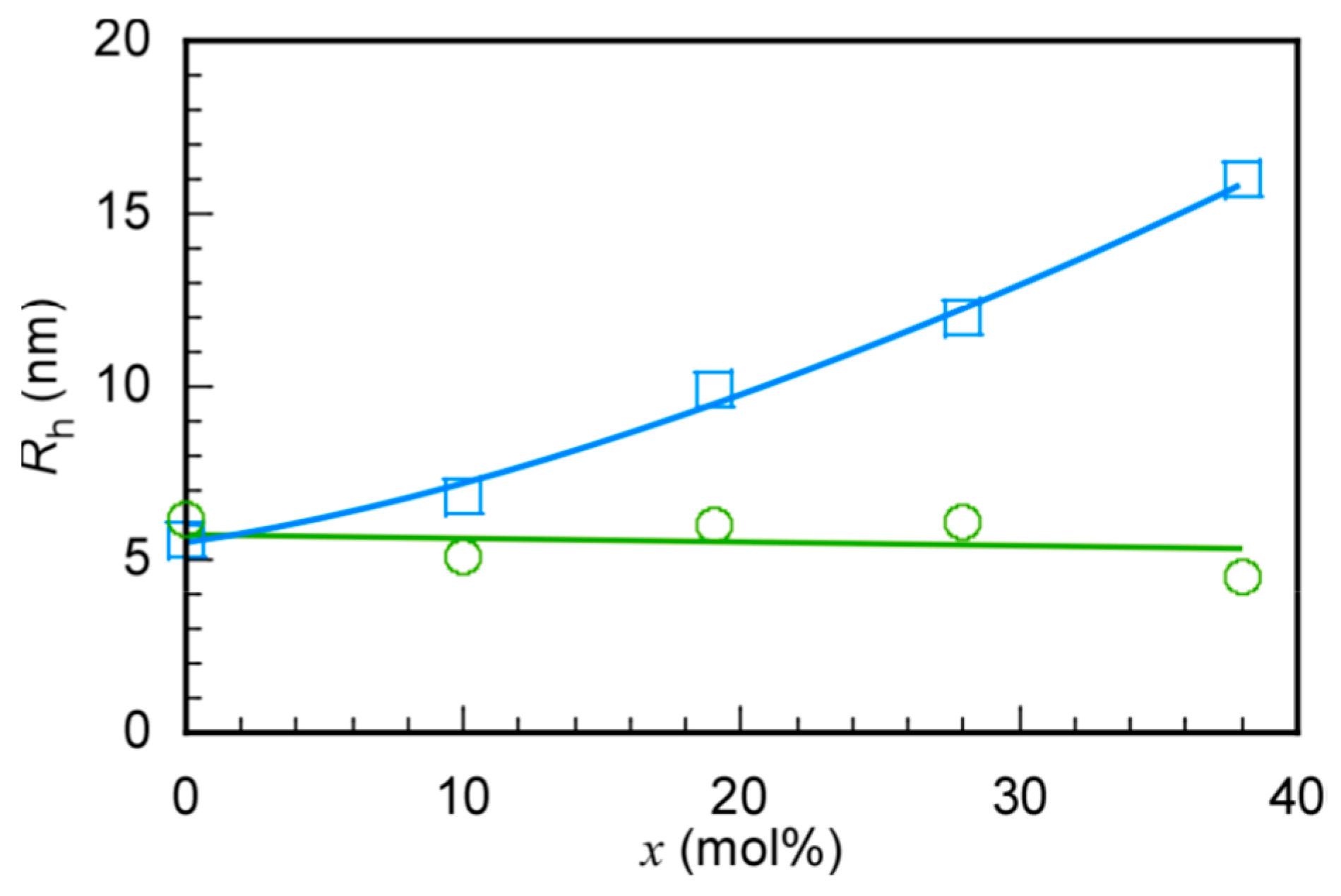
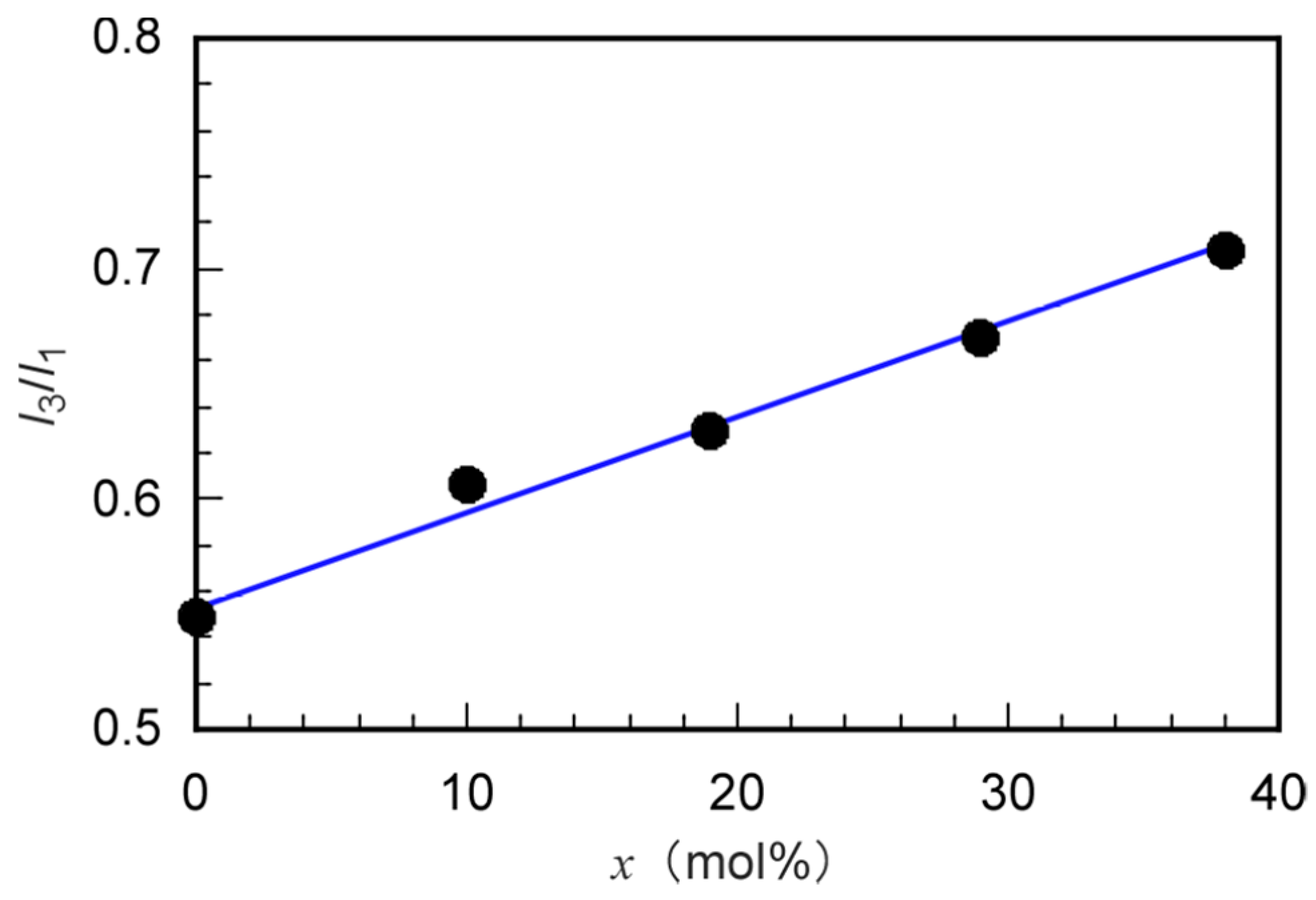
3.2. Self-Association Behavior of P(MPC/DMAx)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Osaki, T.; Takeuchi, S. Artificial cell membrane systems for biosensing applications. Anal. Chem. 2017, 89, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.A.; Chapman, D. Biomembrane surfaces as models for polymer design: The potential for haemocompatibility. Biomaterials 1984, 5, 135–142. [Google Scholar] [CrossRef]
- Bangham, A.D.; Horne, B.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668. [Google Scholar] [CrossRef]
- Regen, S.L.; Czech, B.; Singh, A. Polymerized vesicles. J. Am. Chem. Soc. 1980, 102, 6638–6640. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ishihara, K. Cell membrane-inspired phospholipid polymers for developing medical devices with excellent biointerface. Sci. Technol. Adv. Mater. 2012, 13, 064101. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Oshida, H.; Kurita, K.; Ishihara, K.; Nakabayashi, N. Preparation of 2-methacryloyloxyethly phosphorylcholine copolymers with alkyl methacrylates and their blood compatibility. Polym. J. 1992, 24, 1259–1269. [Google Scholar] [CrossRef]
- Ma, Y.; Tang, Y.; Billingham, N.C.; Armes, S.P.; Lewis, A.L.; Lloyd, A.W.; Salvage, J.P. Well-defined biocompatible block copolymers via atom transfer radical polymerization of 2-methacryloyloxyethyl phosphorylcholine in protic media. Macromolecules 2003, 36, 3475–3484. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Akiyoshi, K. Design of biodegradable amphiphilic polymers: Well-defined amphiphilic polyphosphates with hydrophilic graft chains via ATRP. Macromolecules 2004, 37, 7637–7642. [Google Scholar] [CrossRef]
- Samanta, D.; McRae, S.; Cooper, B.; Hu, Y.; Emrick, T.; Pratt, J.; Charles, S.A. End-functionalized phosphorylcholine methacrylates and their use in protein conjugation. Biomacromolecules 2008, 9, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Sakakida, M.; Ichinose, K.; Umeda, T.; Uehara, M.; Kajiwara, K.; Miyata, T.; Shichiri, M.; Ishihara, K.; Nakabayashi, N. Development of a ferrocene-mediated needle-type glucose sensor covered with newly designed biocompatible membrane, 2-nethacryloyloxyethyl phosphorylcholine-co-n-butyl methacrylate. Med. Prog. Tech. 1995, 21, 91–103. [Google Scholar]
- Akkahat, P.; Kiatkamjornwong, S.; Yusa, S.; Hoven, V.P.; Iwasaki, Y. Development of a novel antifouling platform for biosensing probe immobilization from methacryloyloxyethyl phosphorylcholine containing copolymer brushes. Langmuir 2012, 28, 5872–5881. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, K.; Takai, M.; Ishihara, K. Stabilization of three-dimensional nanostructures biointerface constructed with phospholipid polymer and immobilized antibody for highly sensitive immunoassay. Colloid Surf. B Biointerfaces 2010, 77, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Matsuno, R.; Konno, T.; Takai, M.; Ishihara, K. Artificial cell membrane-covered nanoparticles embedding quantum dots as stable and highly sensitive fluorescence bioimaging probes. Biomacromolecules 2008, 9, 3252–3257. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Chen, W.; Liu, Y.; Tsukamoto, Y.; Inoue, Y. Cytocompatible and multifunctional polymeric nanoparticles for transportation of bioactive molecules into and in cells. Sci. Technol. Adv. Mater. 2016, 17, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; McCormick, C.L. Water-soluble copolymers. 49. Effect of the distribution of the hydrophobic cationic monomer dimethyldodecyl (2-acrylamidoethyl) ammonium bromide on the solution behavior of associating acrylamide copolymers. Macromolecules 1993, 26, 6121–6126. [Google Scholar] [CrossRef]
- Morishima, Y.; Nomura, S.; Ikeda, T.; Seki, M.; Kamachi, M. Characterization of unimolecular micelles of random copolymers of sodium 2-(acrylamido)-2-methylpropanesulfonate and methacrylamides bearing bulky hydrophobic substituents. Macromolecules 1995, 28, 2874–2881. [Google Scholar] [CrossRef]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and single-chain folding of amphiphilic random copolymers in water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Hattori, G.; Takenaka, M.; Sawamoto, M.; Terashima, T. Nanostructured materials via the pendant self-assembly of amphiphilic crystalline random copolymers. J. Am. Chem. Soc. 2018, 140, 8376–8379. [Google Scholar] [CrossRef]
- Ishihara, K.; Iwasaki, Y.; Nakabayashi, M. Polymeric lipid nanosphere constituted of poly(2-methacryloyloxyethyl phosphorylcholine-co-n-butyl methacrylate. Polym. J. 1999, 31, 1231–1236. [Google Scholar] [CrossRef]
- Ishihara, K.; Mu, M.; Konno, T. Water-soluble and amphiphilic phospholipid polymers having 2-methacryloyloxyethyl phosphorylcholine unit for solubilization of bioactive compounds. J. Biomater. Sci. Polym. Ed. 2018, 29, 844–862. [Google Scholar] [CrossRef]
- Ishihara, K.; Ueda, T.; Nakabayashi, N. Preparation of phospholipid polymers and their properties as polymer hydrogel membranes. Polym. J. 1990, 22, 335–360. [Google Scholar] [CrossRef]
- Mitsukami, Y.; Donovan, M.S.; Lowe, A.B.; McCormick, C.L. Water-soluble polymers. 81. Direct synthesis of hydrophilic styrenic-based homopolymers and block copolymers in aqueous solution via RAFT. Macromolecules 2001, 34, 2248–2256. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–746. [Google Scholar] [CrossRef]
- Haas, C.; Drenth, J.; Wilson, W.W. Relation between the solubility of proteins in aqueous solutions and the second virial coefficient of the solution. J. Phys. Chem. B 1999, 103, 2808–2811. [Google Scholar] [CrossRef]
- Akcasu, A.Z.; Han, C.C. Molecular weight and temperature dependence of polymer dimensions in solution. Macromolecule 1979, 12, 276–280. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Thomas, J.K. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. [Google Scholar] [CrossRef]
- Sakaki, S.; Iwasaki, Y.; Nakabayashi, N.; Ishihara, K. Water-soluble 2-methacryloyloxyethyl phosphorylcholine copolymer as a novel synthetic blocking reagent in immunoassay system. Polym. J. 2000, 32, 637–641. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x (mol %) | Conversion (%) | Mn(theory) × 10−4 | Mn(NMR) × 10−4 | DP(NMR) | Mn(GPC) × 10−4 | Mw/Mn |
|---|---|---|---|---|---|---|
| 0 | 94.7 | 5.62 | 5.91 | 200 | 2.93 | 1.12 |
| 10 | 99.7 | 5.82 | 5.78 | 197 | 2.81 | 1.21 |
| 19 | 99.5 | 5.71 | 5.60 | 194 | 3.27 | 1.42 |
| 28 | 99.8 | 5.65 | 5.57 | 195 | 3.14 | 1.48 |
| 38 | 74.2 | 4.07 | 5.21 | 185 | 2.57 | 1.20 |
| x | Mwa × 10−5 (g/mol) | Rga (nm) | Rhb (nm) | Rg/Rh | RTEMc (nm) | Naggd | A2a × 10−5 (cm3·g−2·mol) |
|---|---|---|---|---|---|---|---|
| 0 | 0.60 | 8.08 | 5.6 | 1.44 | - | 1.1 | 27.1 |
| 10 | 2.49 | 7.90 | 6.80 | 1.16 | 5.45 | 3.6 | 26.9 |
| 19 | 4.47 | 11.3 | 9.90 | 1.14 | 6.94 | 5.2 | 2.11 |
| 28 | 7.82 | 21.0 | 11.8 | 1.78 | 7.58 | 8.4 | 1.59 |
| 38 | 8.76 | 45.9 | 17.6 | 2.61 | 14.0 | 13.7 | 1.40 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohshio, M.; Ishihara, K.; Yusa, S.-i. Self-Association Behavior of Cell Membrane-Inspired Amphiphilic Random Copolymers in Water. Polymers 2019, 11, 327. https://doi.org/10.3390/polym11020327
Ohshio M, Ishihara K, Yusa S-i. Self-Association Behavior of Cell Membrane-Inspired Amphiphilic Random Copolymers in Water. Polymers. 2019; 11(2):327. https://doi.org/10.3390/polym11020327
Chicago/Turabian StyleOhshio, Maho, Kazuhiko Ishihara, and Shin-ichi Yusa. 2019. "Self-Association Behavior of Cell Membrane-Inspired Amphiphilic Random Copolymers in Water" Polymers 11, no. 2: 327. https://doi.org/10.3390/polym11020327
APA StyleOhshio, M., Ishihara, K., & Yusa, S.-i. (2019). Self-Association Behavior of Cell Membrane-Inspired Amphiphilic Random Copolymers in Water. Polymers, 11(2), 327. https://doi.org/10.3390/polym11020327