DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates

 ,
, 

Abstract
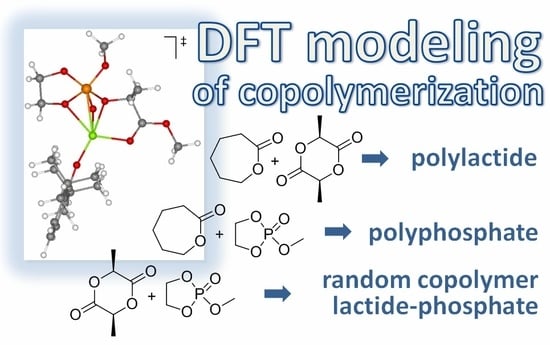
1. Introduction
2. Materials and Methods
2.1. DFT Calculations
2.2. General Experiment Remarks
2.3. Polymerization Experiments
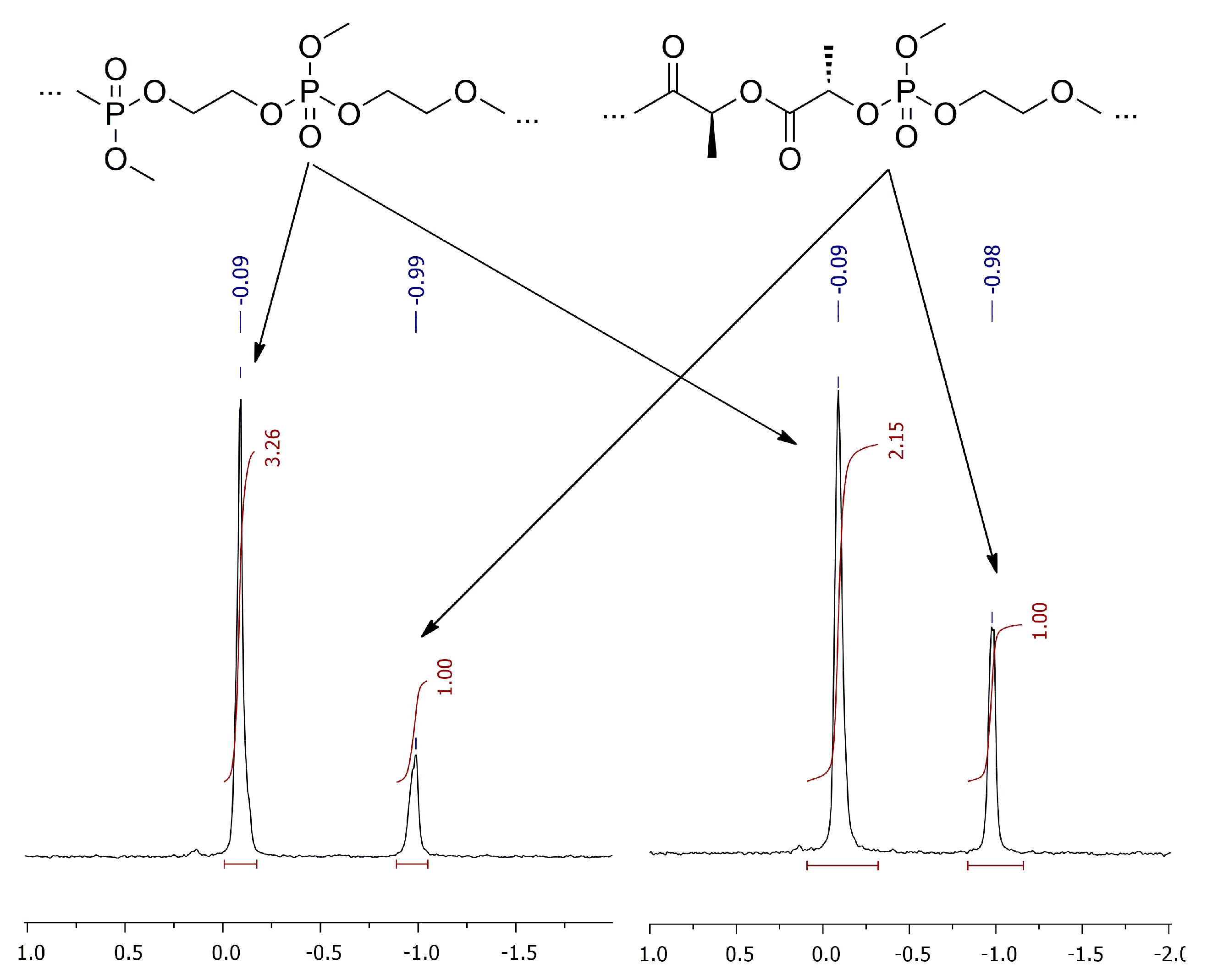
- For PCL, CH2OC=O, δ = 4.2 ppm (monomer) and 4.0 ppm (polymer);
- for PLA, CH(CH3)OC=O, δ = 5.0 ppm (monomer) and 5.1–5.2 ppm (polymer);
- for poly(MeOEP), CH2O, δ = 4.4 ppm (monomer) and 4.2 ppm (polymer).
3. Results
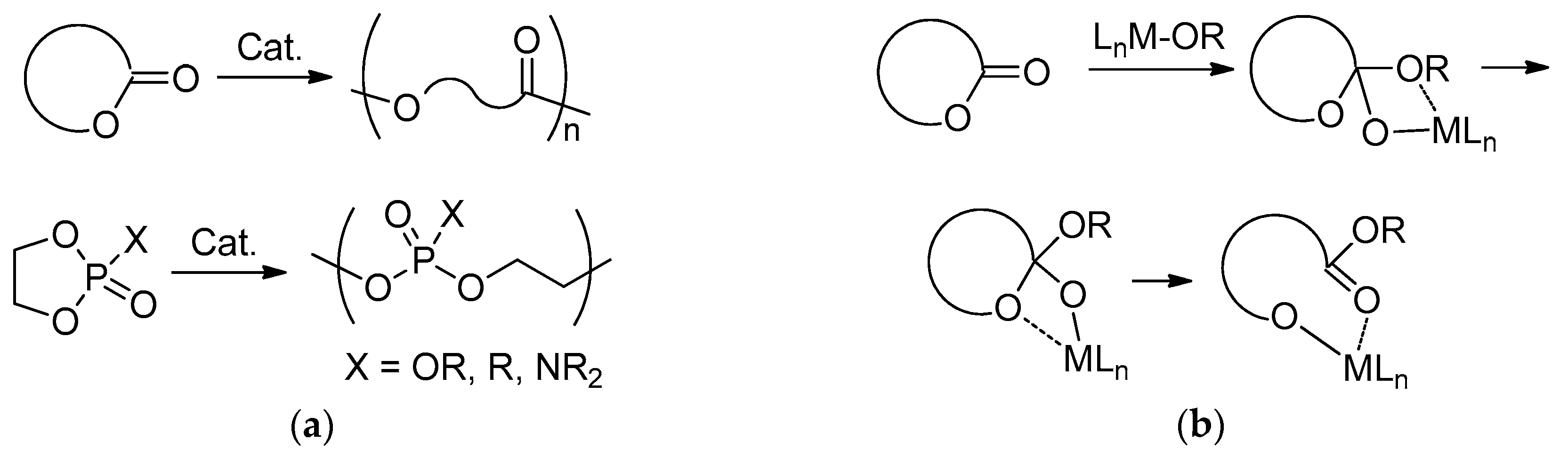
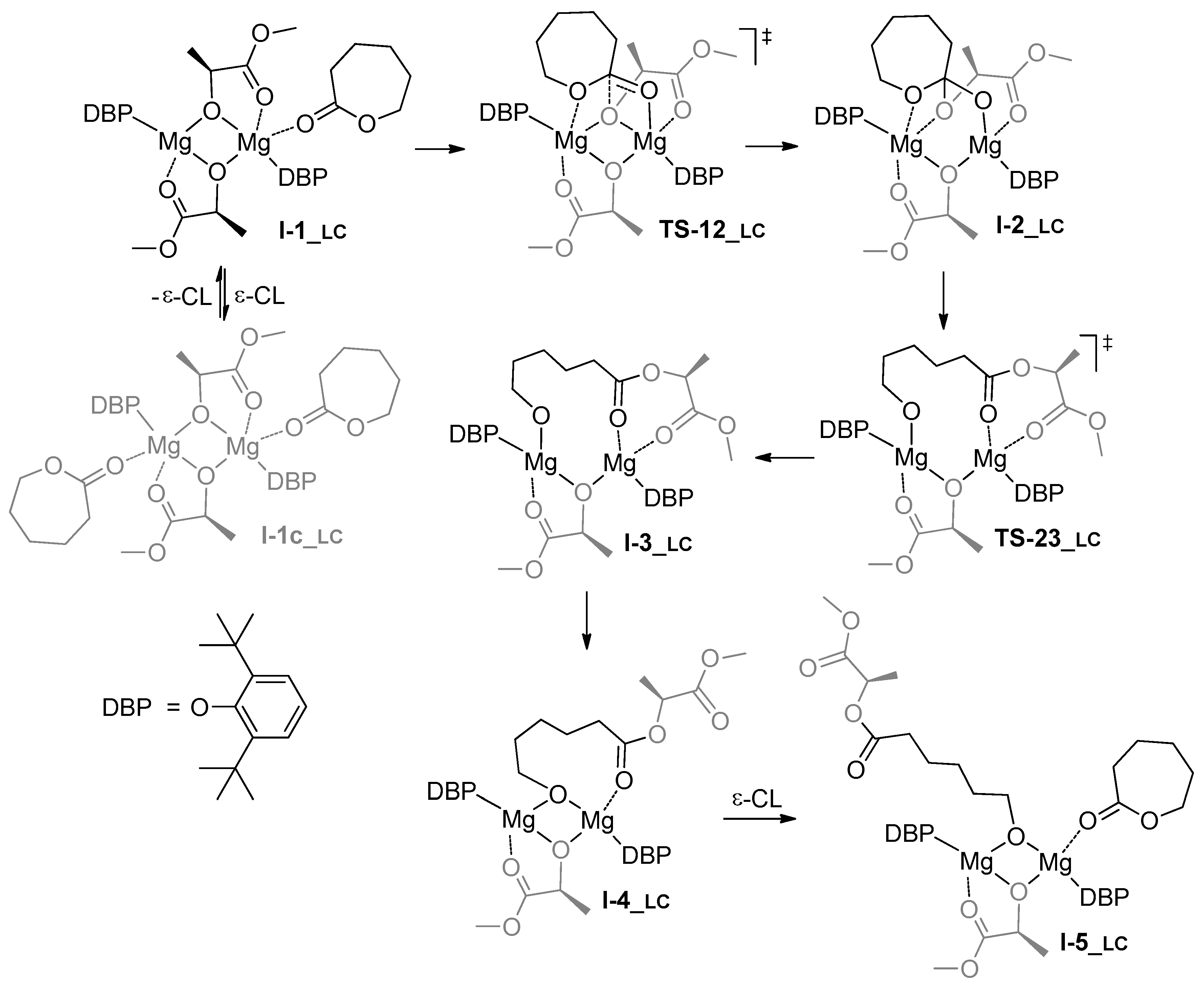
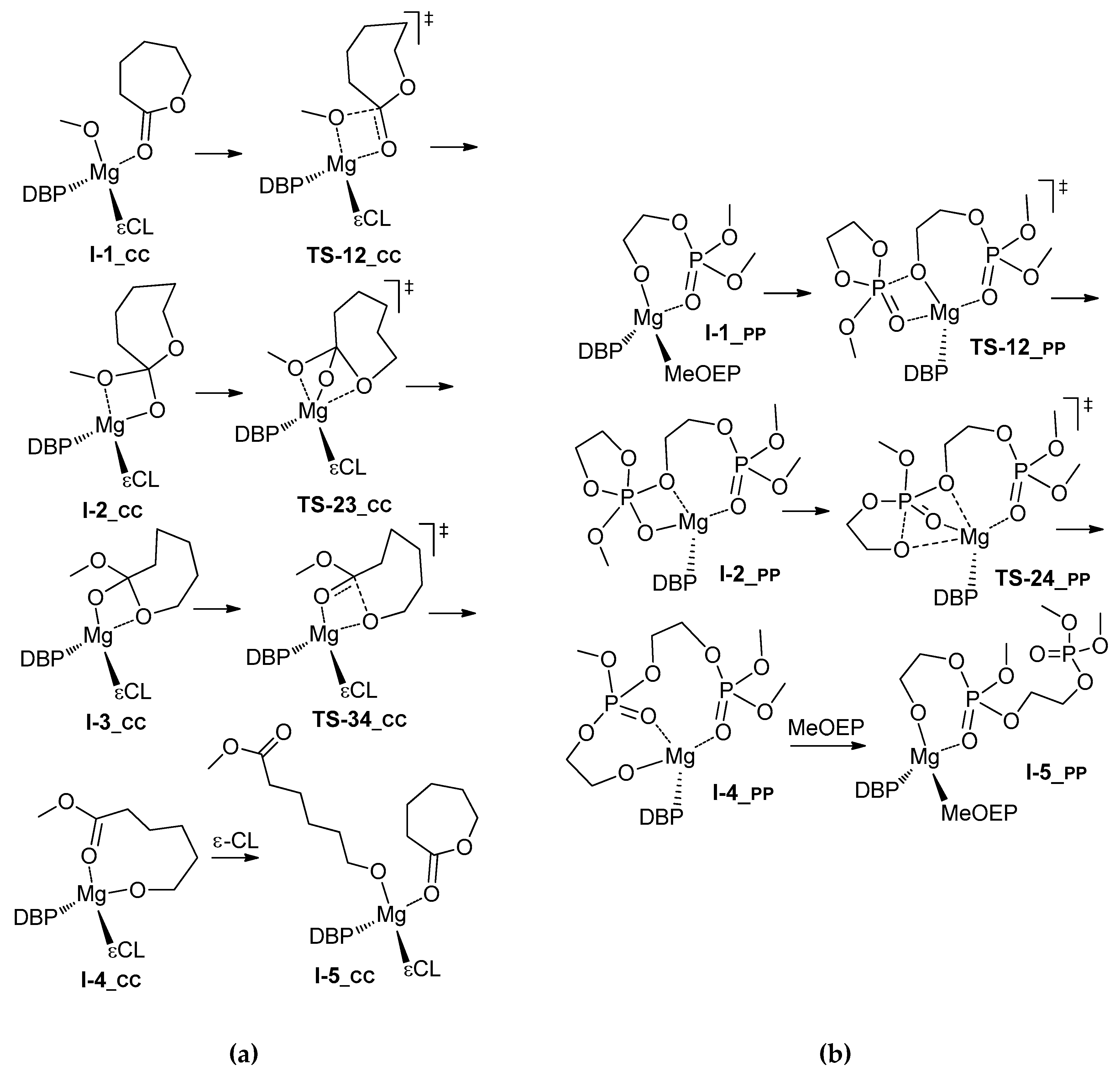
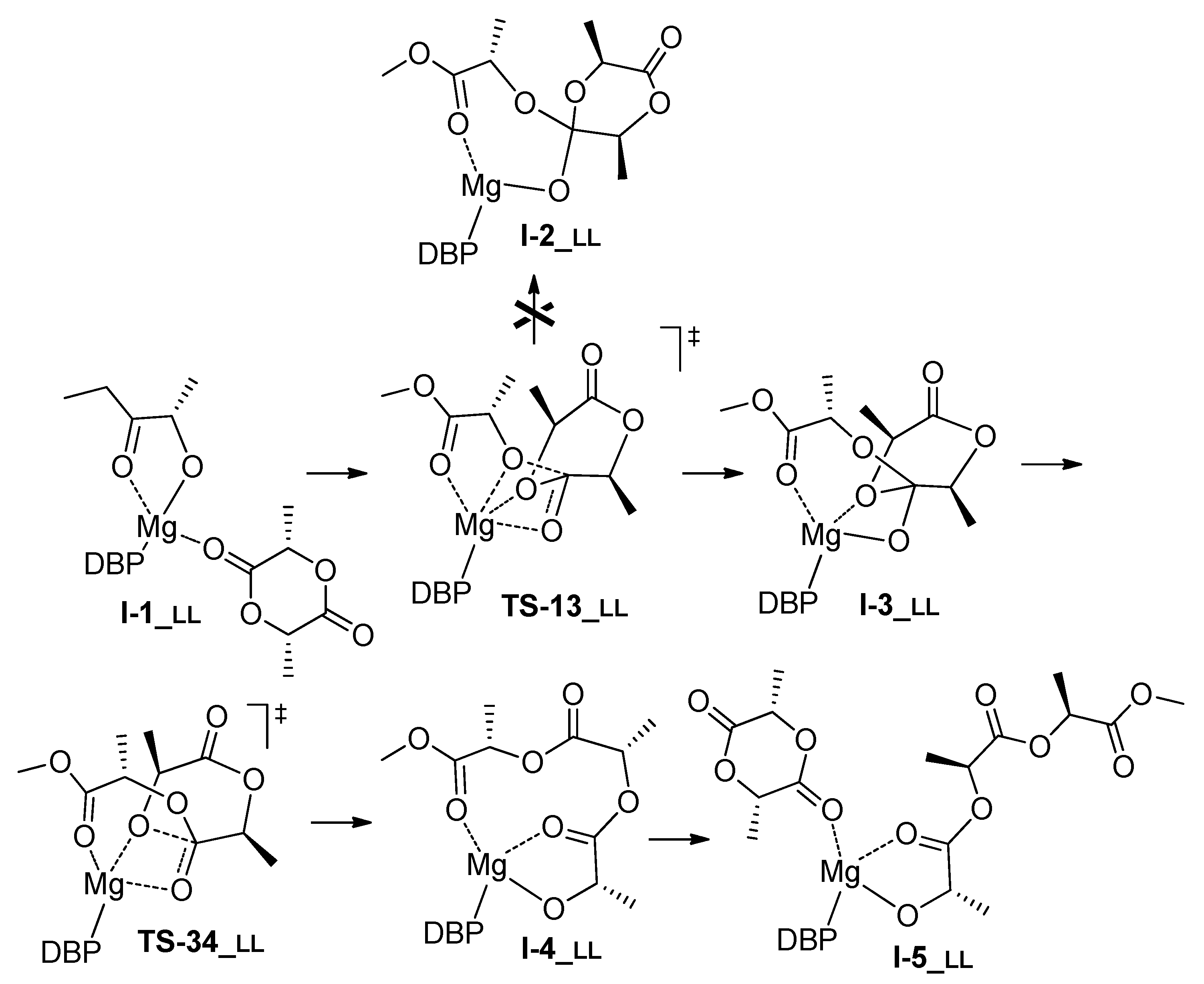
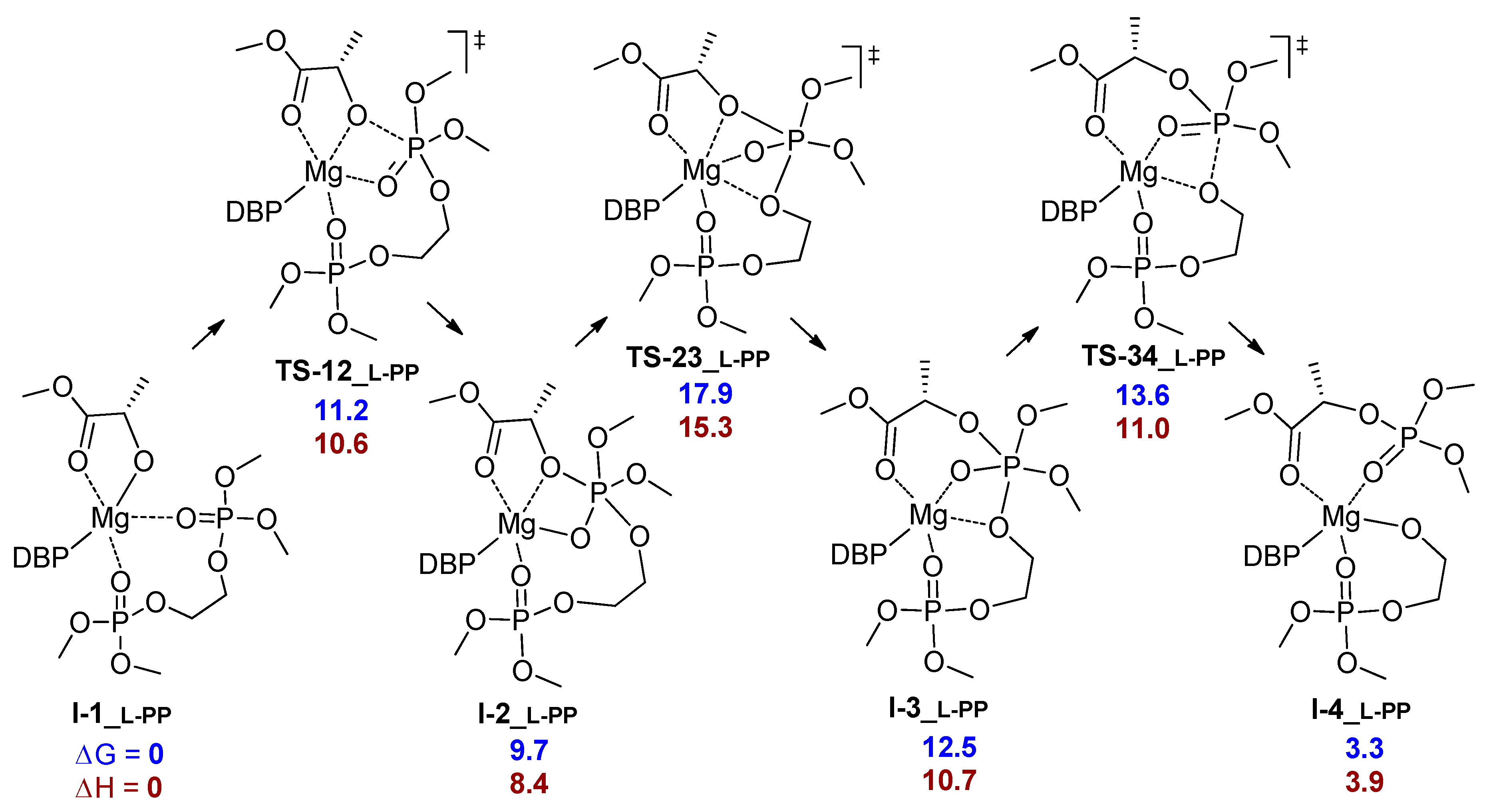
3.1. DFT Modeling of ε-CL/l-LA Copolymerization
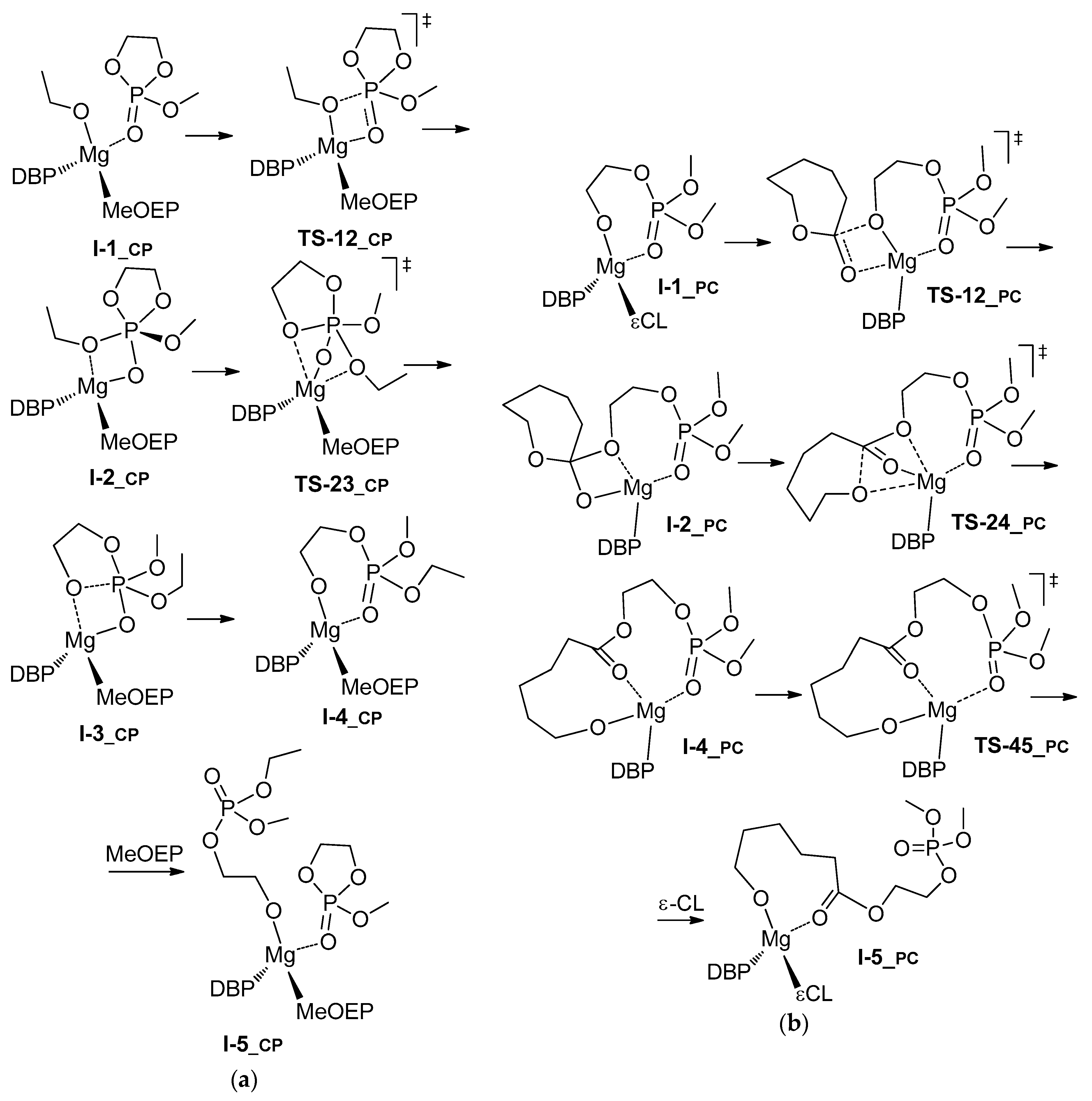
3.2. DFT Modeling of ε-CL/MeOEP Copolymerization
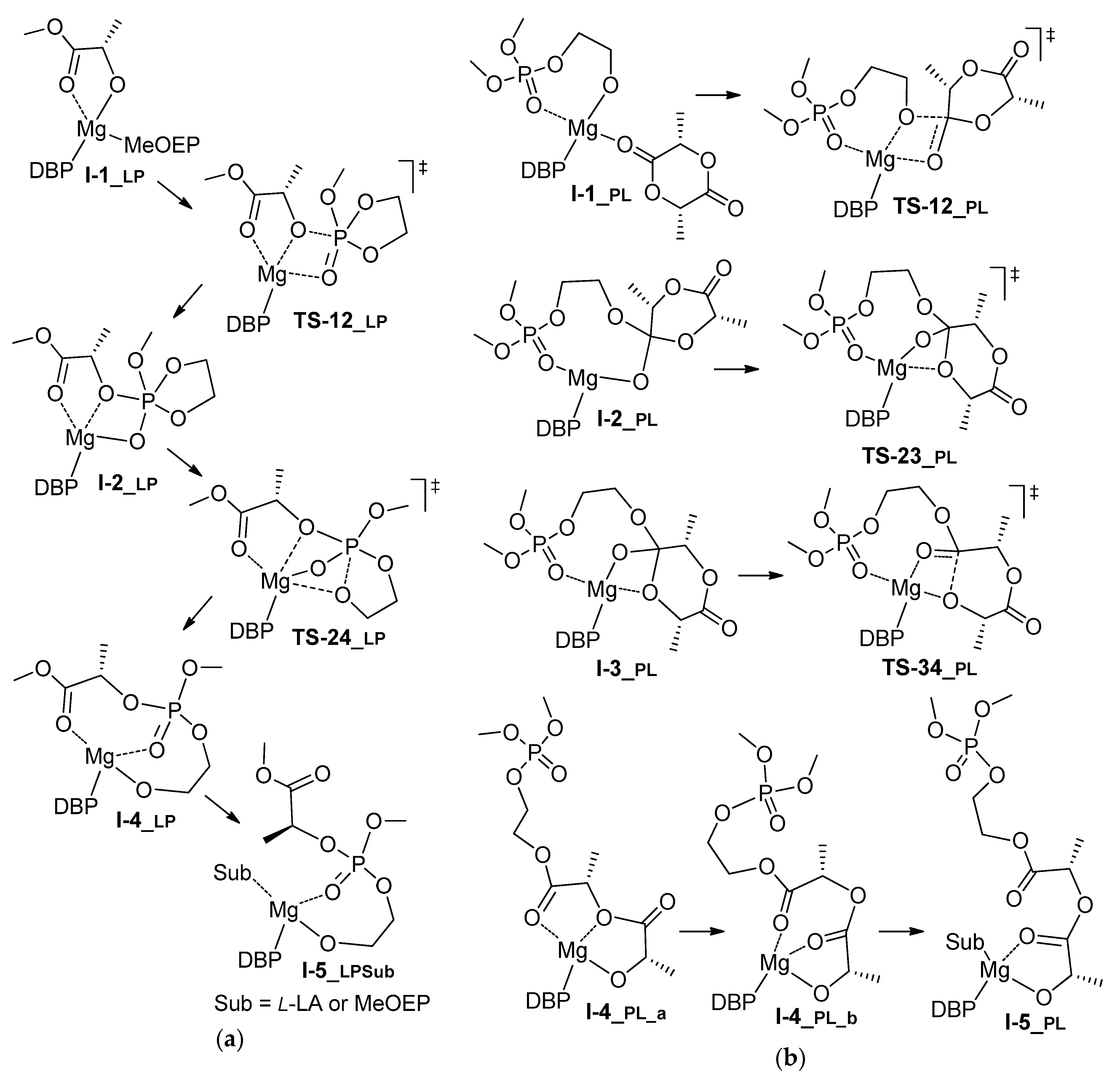
3.3. DFT Modeling of l-LA/MeOEP Copolymerization
3.4. Copolymerization Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef]
- Vert, M. Aliphatic Polyesters: Great Degradable Polymers That Cannot Do Everything. Biomacromolecules 2005, 6, 538–546. [Google Scholar] [CrossRef]
- Naira, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Teo, A.J.T.; Mishra, A.; Park, I.; Kim, Y.-J.; Park, W.-T.; Yoon, Y.-J. Polymeric Biomaterials for Medical Implants and Devices. ACS Biomater. Sci. Eng. 2016, 2, 454–472. [Google Scholar] [CrossRef]
- Sheikh, Z.; Najeeb, S.; Khurshid, Z.; Verma, V.; Rashid, H.; Glogauer, M. Biodegradable Materials for Bone Repair and Tissue Engineering Applications. Materials 2015, 8, 5744–5794. [Google Scholar] [CrossRef]
- Bari, S.S.; Chattarjee, A.; Mishra, S. Biodegradable polymer nanocomposites: An overview. Polym. Rev. 2016, 56, 287–328. [Google Scholar] [CrossRef]
- Klein, R.; Wurm, F.R. Aliphatic Polyethers: Classical Polymers for the 21st Century. Macromol. Rapid Commun. 2015, 36, 1147–1165. [Google Scholar] [CrossRef]
- Rydz, J.; Sikorska, W.; Kyulavska, M.; Christova, D. Polyester-Based (Bio) degradable Polymers as Environmentally Friendly Materials for Sustainable Development. Int. J. Mol. Sci. 2015, 16, 564–596. [Google Scholar] [CrossRef]
- Pang, K.; Kotek, R.; Tonelli, A. Review of conventional and novel polymerization processes for polyesters. Prodr. Polym. Sci. 2006, 31, 10091037. [Google Scholar] [CrossRef]
- Stanford, M.J.; Dove, A.P. Stereocontrolled ring-opening polymerisation of lactide. Chem. Soc. Rev. 2010, 39, 486–494. [Google Scholar] [CrossRef]
- Dijkstra, P.J.; Du, H.; Feijen, J. Single site catalysts for stereoselective ring-opening polymerization of lactides. Polym. Chem. 2011, 2, 520527. [Google Scholar] [CrossRef]
- Okada, M. Chemical synthesis of biodegradable polymers. Prog. Polym. Sci. 2002, 27, 87–133. [Google Scholar] [CrossRef]
- Duda, A. 4.11-ROP of Cyclic Esters. Mechanisms of Ionic and Coordination Processes. In Polymer Science: A Comprehensive Reference; V. 4: Ring-Opening Polymerization and Special Polymerization Processes; Matyjaszewski, K., Möller, M., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2012. [Google Scholar] [CrossRef]
- Wheaton, C.A.; Hayes, P.G.; Ireland, B.J. Complexes of Mg, Ca and Zn as homogeneous catalysts for lactide polymerization. Dalton Trans. 2009, 25, 4832–4846. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Mialon, L.; Abboud, K.A.; Miller, S.A. Comparative Study of Lactide Polymerization with Lithium, Sodium, Magnesium, and Calcium Complexes of BHT. Organometallics 2012, 31, 5252–5261. [Google Scholar] [CrossRef]
- Dagorne, S.; Fliedel, C. Organoaluminum Species in Homogeneous Polymerization Catalysis. In Modern Organoaluminum Reagents. Topics in Organometallic Chemistry; Woodward, S., Dagorne, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; Volume 41, pp. 125–171. ISBN 978-3-642-33672-0. [Google Scholar] [CrossRef]
- Dagorne, S.; Normand, M.; Kirillov, E.; Carpentier, J.-F. Gallium and indium complexes for ring-opening polymerization of cyclic ethers, esters and carbonates. Coord. Chem. Rev. 2013, 257, 1869–1886. [Google Scholar] [CrossRef]
- Kremer, A.B.; Mehrkhodavandi, P. Dinuclear catalysts for the ring opening polymerization of lactide. Coord. Chem. Rev. 2019, 380, 35–57. [Google Scholar] [CrossRef]
- Sauer, A.; Kapelski, A.; Fliedel, C.; Dagorne, S.; Kol, M.; Okuda, J. Structurally well-defined group 4 metal complexes as initiators for the ring-opening polymerization of lactide monomers. Dalton Trans. 2013, 42, 9007–9023. [Google Scholar] [CrossRef]
- Sarazin, Y.; Carpentier, J.-F. Discrete Cationic Complexes for Ring-Opening Polymerization Catalysis of Cyclic Esters and Epoxides. Chem. Rev. 2015, 115, 3564–3614. [Google Scholar] [CrossRef]
- Liu, J.; Ling, J.; Li, X.; Shen, Z. Monomer insertion mechanism of ring-opening polymerization of ɛ-caprolactone with yttrium alkoxide intermediate: A DFT study. J. Mol. Catal. A Chem. 2009, 300, 5964. [Google Scholar] [CrossRef]
- Susperregui, N.; Kramer, M.U.; Okuda, J.; Maron, L. Theoretical Study on the Ring-Opening Polymerization of ε-Caprolactone by [YMeX(THF)5]+ with X = BH4, NMe2. Organometallics 2011, 30, 1326–1333. [Google Scholar] [CrossRef]
- Miranda, M.O.; DePorre, Y.; Vazquez-Lima, H.; Johnson, M.A.; Marell, D.J.; Cramer, C.J.; Tolman, W.B. Understanding the Mechanism of Polymerization of ε-Caprolactone Catalyzed by Aluminum Salen Complexes. Inorg. Chem. 2013, 52, 13692–13701. [Google Scholar] [CrossRef] [PubMed]
- Marlier, E.E.; Macaranas, J.A.; Marell, D.J.; Dunbar, C.R.; Johnson, M.A.; DePorre, Y.; Miranda, M.O.; Neisen, B.D.; Cramer, C.J.; Hillmyer, M.A.; et al. Mechanistic Studies of ε-Caprolactone Polymerization by (salen)AlOR Complexes and a Predictive Model for Cyclic Ester Polymerizations. ACS Catal. 2016, 6, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Lu, W.-Y.; Chang, H.-Y.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y.; Chen, H.-Y. Comparative Study of Aluminum Complexes Bearing N,O- and N,S-Schiff Base in Ring-Opening Polymerization of ε-Caprolactone and l-Lactide. Inorg. Chem. 2015, 54, 1129211298. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Miranda, M.O.; Moscato-Goodpaster, B.; Ajellal, N.; Breyfogle, L.E.; Hermes, E.D.; Schaller, C.P.; Roe, S.E.; Cramer, C.J.; Hillmyer, M.A.; et al. Roles of Monomer Binding and Alkoxide Nucleophilicity in Aluminum-Catalyzed Polymerization of ε-Caprolactone. Macromolecules 2012, 45, 5387–5396. [Google Scholar] [CrossRef]
- Kuzdrowska, M.; Annunziata, L.; Marks, S.; Schmid, M.; Jaffredo, C.G.; Roesky, P.W.; Guillaume, S.M.; Maron, L. Organometallic calcium and strontium borohydrides as initiators for the polymerization of ε-caprolactone and L-lactide: Combined experimental and computational investigations. Dalton Trans. 2013, 42, 9352–9360. [Google Scholar] [CrossRef] [PubMed]
- Jitonnom, J.; Molloy, R.; Punyodom, W.; Meelua, W. Theoretical Studies on Aluminum Trialkoxide-Initiated Lactone Ring-Opening Polymerizations: Roles of Alkoxide Substituent and Monomer Ring Structure. Comput. Theor. Chem. 2016, 1097, 25–32. [Google Scholar] [CrossRef]
- Jędrzkiewicz, D.; Kantorska, D.; Wojtaszak, J.; Ejfler, J.; Szafert, S. DFT calculations as a ligand toolbox for the synthesis of active initiators for ROP of cyclic esters. Dalton Trans. 2017, 46, 4929–4942. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Ivchenko, P.V.; Shlyakhtin, A.V.; Ivanyuk, A.V. Polymerization of Trimethylene Carbonate and Lactones in the Presence of Magnesium Monoionolate: A Comparative Theoretical and Experimental Study. Polym. Sci. Ser. B 2017, 59, 147–156. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Minyaev, M.E.; Churakov, A.V.; Karchevsky, S.G.; Birin, K.P.; Ivchenko, P.V. Mono-BHT heteroleptic magnesium complexes: Synthesis, molecular structure and catalytic behavior in the ring-opening polymerization of cyclic esters. Dalton Trans. 2017, 46, 12132–12146. [Google Scholar] [CrossRef]
- Wei, J.; Riffel, M.N.; Diaconescu, P.L. Redox Control of Aluminum Ring-Opening Polymerization: A Combined Experimental and DFT Investigation. Macromolecules 2017, 50, 1847–1861. [Google Scholar] [CrossRef]
- Li, P.; Xi, Y.; Li, L.; Li, H.; Sun, W.-H.; Lei, M. A DFT study on ring-opening polymerization of ε-caprolactone initiated by Mg and Al complexes. Inorg. Chim. Acta 2018, 477, 34–39. [Google Scholar] [CrossRef]
- Marshall, E.L.; Gibson, V.C.; Rzepa, H.S. A Computational Analysis of the Ring-Opening Polymerization of rac-Lactide Initiated by Single-Site β-Diketiminate Metal Complexes: Defining the Mechanistic Pathway and the Origin of Stereocontrol. J. Am. Chem. Soc. 2005, 127, 6048–6051. [Google Scholar] [CrossRef] [PubMed]
- Tabthong, S.; Nanok, T.; Sumrit, P.; Kongsaeree, P.; Prabpai, S.; Chuawong, P.; Hormnirun, P. Bis (pyrrolidene) Schiff Base Aluminum Complexes as Isoselective-Biased Initiators for the Controlled Ring-Opening Polymerization of rac-Lactide: Experimental and Theoretical Studies. Macromolecules 2015, 48, 6846–6861. [Google Scholar] [CrossRef]
- Dai, Z.; Zhang, J.; Gao, Y.; Tang, N.; Huang, Y.; Wu, J. Synthesis and structures of tridentate β-diketiminato zinc phenoxides as catalysts for immortal ring-opening polymerization of L-lactide. Cat. Sci. Technol. 2013, 3, 32683277. [Google Scholar] [CrossRef]
- Bouyahyi, M.; Ajellal, N.; Kirillov, E.; Thomas, C.M.; Carpentier, J.-F. Exploring Electronic versus Steric Effects in Stereoselective Ring-Opening Polymerization of Lactide and β-Butyrolactone with Amino-alkoxy-bis (phenolate)–Yttrium Complexes. Chem. Eur. J. 2011, 17, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Cheshmedzhieva, D.; Angelova, I.; Ilieva, S.; Georgiev, G.S.; Galabov, B. Initiation of ring-opening polymerization of lactide: The effect of metal alkoxide catalyst. Comput. Theor. Chem. 2012, 995, 8–16. [Google Scholar] [CrossRef]
- Fliedel, C.; Vila-Viçosa, D.; Calhorda, M.J.; Dagorne, S.; Avilés, T. Dinuclear Zinc–N-Heterocyclic Carbene Complexes for Either the Controlled Ring-Opening Polymerization of Lactide or the Controlled Degradation of Polylactide Under Mild Conditions. ChemCatChem 2014, 6, 1357–1367. [Google Scholar] [CrossRef]
- Jędrzkiewicz, D.; Czeluśniak, I.; Wierzejewska, M.; Szafert, S.; Ejfler, J. Well-controlled, zinc-catalyzed synthesis of low molecular weight oligolactides by ring opening reaction. J. Mol. Catal. A Chem. 2015, 396, 155163. [Google Scholar] [CrossRef]
- Roymuhury, S.K.; Chakraborty, D.; Ramkumar, V. Aluminium complexes bearing N,O-aminophenol ligands as efficient catalysts for the ring opening polymerization of lactide. Eur. Polym. J. 2015, 70, 203–214. [Google Scholar] [CrossRef]
- Liu, B.; Roisnel, T.; Maron, L.; Carpentier, J.-F.; Sarazin, Y. Discrete Divalent Rare-Earth Cationic ROP Catalysts: Ligand-Dependent Redox Behavior and Discrepancies with Alkaline-Earth Analogues in a Ligand-Assisted Activated Monomer Mechanism. Chem. Eur. J. 2013, 19, 3986–3994. [Google Scholar] [CrossRef]
- Ivchenko, P.V.; Shlyakhtin, A.V.; Nifant’ev, I.E. Ring-opening polymerization of glycolide and rac-lactide, catalyzed by aryloxy magnesium complexes: DFT study of reaction profile and stereocontrol mechanism. Mendeleev Commun. 2017, 27, 278–280. [Google Scholar] [CrossRef]
- Vogt-Geisse, S.; Matac, R.A.; Toro-Labbé, A. High level potential energy surface and mechanism of Al(CH3)2OCH3-promoted lactone polymerization: Initiation and propagation. Phys. Chem. Chem. Phys. 2017, 19, 8989–8999. [Google Scholar] [CrossRef] [PubMed]
- Chandanabodhi, D.; Nanok, T. A DFT study of the ring-opening polymerization mechanism of l-lactide and ε-caprolactone using aluminium salen-type initiators: Towards an understanding of their reactivities in homo- and copolymerization. Mol. Catal. 2017, 436, 145–156. [Google Scholar] [CrossRef]
- Robert, C.; Schmid, T.E.; Richard, V.; Haquette, P.; Raman, S.K.; Rager, M.-N.; Gauvin, R.M.; Morin, Y.; Trivelli, X.; Guérineau, V.; et al. Mechanistic Aspects of the Polymerization of Lactide Using a Highly Efficient Aluminum(III) Catalytic System. J. Am. Chem. Soc. 2017, 139, 6217–6225. [Google Scholar] [CrossRef] [PubMed]
- Stasiw, D.E.; Luke, A.M.; Rosen, T.; League, A.B.; Mandal, M.; Neisen, B.D.; Cramer, C.J.; Kol, M.; Tolman, W.B. Mechanism of the Polymerization of rac-Lactide by Fast Zinc Alkoxide Catalysts. Inorg. Chem. 2017, 56, 14366–14372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mehmood, A.; Zhao, Y.; Qu, J.; Luo, Y. Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst. Inorganics 2017, 5, 46. [Google Scholar] [CrossRef]
- del Rosal, I.; Brignou, P.; Guillaume, S.M.; Carpentier, J.-F.; Maron, L. DFT investigations on the ring-opening polymerization of cyclic carbonates catalyzed by zinc-{β-diiminate} complexes. Polym. Chem. 2011, 2, 2564–2573. [Google Scholar] [CrossRef]
- del Rosal, I.; Brignou, P.; Guillaume, S.M.; Carpentier, J.-F.; Maron, L. DFT investigations on the ring-opening polymerization of substituted cyclic carbonates catalyzed by zinc-{β-diketiminate} complexes. Polym. Chem. 2015, 6, 3336–3352. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Shlyakhtin, A.; Kosarev, M.; Karchevsky, S.; Ivchenko, P. Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data. Polymers 2018, 10, 1105. [Google Scholar] [CrossRef]
- Wen, J.; Kim, G.J.A.; Leong, K.W. Poly (d,l-lactide–co-ethyl ethylene phosphate)s as new drug carriers. J. Control. Release 2006, 92, 39–48. [Google Scholar] [CrossRef]
- Florczak, M.; Duda, A. Effect of the Configuration of the Active Center on Comonomer Reactivities: The Case of ε-Caprolactone/l,l-Lactide Copolymerization. Angew. Chem. Int. Ed. 2008, 47, 9088–9091. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Akita, A.; Ishii, R.; Mizuno, M. Random Copolymerization of ε-Caprolactone with Lactide Using a Homosalen−Al Complex. J. Am. Chem. Soc. 2010, 132, 1750–1751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, H. Exploitation of dinuclear salan aluminum complexes for versatile copolymerization of ε-caprolactone and L-lactide. Chem. Commun. 2012, 6729–6731. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lamberti, M.; Pappalardo, D.; Pellecchia, C. Random Copolymerization of ε-Caprolactone and Lactides Promoted by Pyrrolylpyridylamido Aluminum Complexes. Macromolecules 2012, 45, 8614–8620. [Google Scholar] [CrossRef]
- Li, L.; Liu, B.; Liu, D.; Wu, C.; Li, S.; Liu, B.; Cui, D. Copolymerization of ε-Caprolactone and l-Lactide Catalyzed by Multinuclear Aluminum Complexes: An Immortal Approach. Organometallics 2014, 33, 6474–6480. [Google Scholar] [CrossRef]
- Kan, C.; Ma, H. Copolymerization of L-lactide and ε-caprolactone catalyzed by mono-and dinuclear salen aluminum complexes bearing bulky 6,6′-dimethylbipheyl-bridge: Random and tapered copolymer. RSC Adv. 2016, 6, 47402–47409. [Google Scholar] [CrossRef]
- Pilone, A.; De Maio, N.; Press, K.; Venditto, V.; Pappalardo, D.; Mazzeo, M.; Pellecchia, C.; Kol, M.; Lamberti, M. Ring-opening homo- and co-polymerization of lactides and ε-caprolactone by salalen aluminum complexes. Dalton Trans. 2015, 44, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Wang, B.; Zhang, Y.-P.; Li, Y.-S. Bimetallic aluminum complexes with cyclic β-ketiminato ligands: The cooperative effect improves their capability in polymerization of lactide and ε-caprolactone. Polym. Chem. 2016, 7, 5819–5827. [Google Scholar] [CrossRef]
- Yang, J.; Sun, Z.; Duan, R.; Li, L.; Pang, X.; Chen, X. Copolymer of lactide and ε-caprolactone catalyzed by bimetallic Schiff base aluminum complexes. Sci. China Chem. 2016, 59, 1384–1389. [Google Scholar] [CrossRef]
- Shi, T.; Luo, W.; Liu, S.; Li, Z. Controlled random copolymerization of rac-lactide and ɛ-caprolactone by well-designed phenoxyimine Al complexes. J. Polym. Sci. Part. A Polym. Chem. 2018, 56, 611–617. [Google Scholar] [CrossRef]
- Beament, J.; Wolf, T.; Markwart, J.C.; Wurm, F.R.; Jones, M.D.; Buchard, A. Copolymerization of Cyclic Phosphonate and Lactide: Synthetic Strategies toward Control of Amphiphilic Microstructure. Macromolecules 2019, 52, 1220–1226. [Google Scholar] [CrossRef]
- Honrado, M.; Otero, A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Garcés, A.; Lara-Sánchez, A.; Rodríguez, A.M. Copolymerization of Cyclic Esters Controlled by Chiral NNO-Scorpionate Zinc Initiators. Organometallics 2016, 35, 189–197. [Google Scholar] [CrossRef]
- Lin, L.; Xu, Y.; Wang, S.; Xiao, M.; Meng, Y. Ring-opening polymerization of l-lactide and ε-caprolactone catalyzed by versatile tri-zinc complex: Synthesis of biodegradable polyester with gradient sequence structure. Eur. Polym. J. 2016, 74, 109–119. [Google Scholar] [CrossRef]
- Dakshinamoorthy, D.; Peruch, F. Block and random copolymerization of ε-caprolactone, L-, and rac-lactide using titanium complex derived from aminodiol ligand. J. Polym. Sci. Part. A Polym. Chem. 2012, 50, 2161–2171. [Google Scholar] [CrossRef]
- Piskun, Y.A.; Vasilenko, I.V.; Zaitsev, K.V.; Karlov, S.S.; Zaitseva, G.S.; Gaponik, L.V.; Kostjuk, S.V. Controlled homo and copolymerization of ε-caprolactone and d, l-lactide in the presence of TiIV complexes. Russ. Chem. Bull. 2015, 64, 181–188. [Google Scholar] [CrossRef]
- Lapenta, R.; Mazzeo, M.; Grisi, F. Monoamidinate titanium complexes: Highly active catalysts for the polymerization and copolymerization of L-lactide and ε-caprolactone. RSC Adv. 2015, 5, 87635–87644. [Google Scholar] [CrossRef]
- Gilmour, D.J.; Webster, R.L.; Perrya, M.R.; Schafer, L.L. Titanium pyridonates for the homo- and copolymerization of rac-lactide and ε-caprolactone. Dalton Trans. 2015, 44, 12411–12419. [Google Scholar] [CrossRef]
- Pappuru, S.; Chakraborty, D.; Sundar, J.V.; Roymuhury, S.K.; Ramkumar, V.; Subramanian, V.; Chand, D.K. Group 4 complexes of salicylbenzoxazole ligands as effective catalysts for the ring-opening polymerization of lactides, epoxides and copolymerization of ε-caprolactone with L-lactide. Polymer 2016, 102, 231–247. [Google Scholar] [CrossRef]
- Della Monica, F.; Luciano, E.; Buonerba, A.; Grassi, A.; Milione, S.; Capacchione, C. Poly (lactide-co-ε-caprolactone) copolymers prepared using bis-thioetherphenolate group 4 metal complexes: Synthesis, characterization and morphology. RSC Adv. 2014, 4, 51262–51267. [Google Scholar] [CrossRef]
- Wen, J.; Zhuo, R.-X. Preparation and characterization of poly (d,l-lactide-co-ethylene methyl phosphate). Polym. Int. 1998, 47, 503–509. [Google Scholar] [CrossRef]
- Wang, L.; Kefalidis, C.E.; Sinbandhit, S.; Dorcet, V.; Carpentier, J.-F.; Maron, L.; Sarazin, Y. Heteroleptic Tin (II) Initiators for the Ring-Opening (Co) Polymerization of Lactide and Trimethylene Carbonate: Mechanistic Insights from Experiments and Computations. Chem. Eur. J. 2013, 19, 13463–13478. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.-J.; Lai, P.-S.; Chen, J.-Y.; Hsu, S.C.N.; Peng, W.-D.; Ou, S.-W.; Lai, Y.-C.; Chen, Y.-J.; Chung, H.; Chen, Y.; et al. ε-Caprolactone polymerization under air by the biocatalyst: Magnesium 2,6-di-tert-butyl-4-methylphenoxide. J. Polym. Sci. Part. A Polym. Chem. 2012, 50, 2697–2704. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hopkins, S.A.; Wright, P.M.; Dove, A.P. ‘Immortal’ ring-opening polymerization of ω-pentadecalactone by Mg (BHT)2(THF)2. Polym. Chem. 2014, 5, 2691–2694. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hopkins, S.A.; Wright, P.M.; Dove, A.P. Synthesis of ω-Pentadecalactone Copolymers with Independently Tunable Thermal and Degradation Behavior. Macromolecules 2015, 48, 950–958. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Ivchenko, P.V.; Borisov, R.S.; Churakov, A.V. Monomeric and dimeric magnesium mono-BHT complexes as effective ROP catalysts. Catal. Commun. 2016, 87, 106–111. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Controlled ring-opening polymerisation of cyclic phosphates, phosphonates and phosphoramidates catalysed by hereroleptic BHT-alkoxy magnesium complexes. Polym. Chem. 2017, 8, 6806–6816. [Google Scholar] [CrossRef]
- Minyaev, M.E.; Nifant’ev, I.E.; Shlyakhtin, A.V.; Ivchenko, P.V.; Lyssenko, K.A. Phenoxide and alkoxide complexes of Mg, Al and Zn and their use for ring-opening polymerization of ε-caprolactone with initiators of different nature. Acta Cryst. C Struct. Chem. 2018, C74, 548–557. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Synthesis and ring-opening polymerization of glycidyl ethylene phosphate with a formation of linear and branched polyphosphates. Mendeleev Commun. 2018, 28, 155–157. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Tavtorkin, A.N.; Minyaev, M.E.; Kosarev, M.A.; Ivchenko, P.V. Synthesis in aqueous media of poly(ethylene phosphoric acids) by mild thermolysis of homopolymers and block copolymers based on tert-butyl ethylene phosphate. Eur. Polym. J. 2018, 106, 249–256. [Google Scholar] [CrossRef]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision, A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part, I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation 91 Energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Kosarev, M.A.; Gavrilov, D.E.; Komarov, P.D.; Ilyin, S.O.; Karchevsky, S.G.; Ivchenko, P.V. Mechanistic study of transesterification in TBD-catalyzed ring-opening polymerization of methyl ethylene phosphate. Eur. Polym. J. 2019, 118, 393–403. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Kosarev, M.A.; Komarov, P.D.; Karchevsky, S.G.; Ivchenko, P.V. Data for theoretical study of the mechanisms of ring-opening polymerization of methyl ethylene phosphate. Data Brief. 2019, 26, 104431. [Google Scholar] [CrossRef] [PubMed]
- Fliedel, C.; Mameri, S.; Dagorne, S.; Avilés, T. Controlled ring-opening polymerization of trimethylene carbonate and access to PTMC-PLA block copolymers mediated by well-defined N-heterocyclic carbene zinc alkoxides. Appl. Organomet. Chem. 2014, 28, 504–511. [Google Scholar] [CrossRef]
- Minyaev, M.E.; Churakov, A.V.; Nifant’ev, I.E. Structural diversity of polynuclear MgxOy cores in magnesium phenoxide complexes. Acta Cryst. C Struct. Chem. 2017, 73, 854–861. [Google Scholar] [CrossRef]












{kind=link}
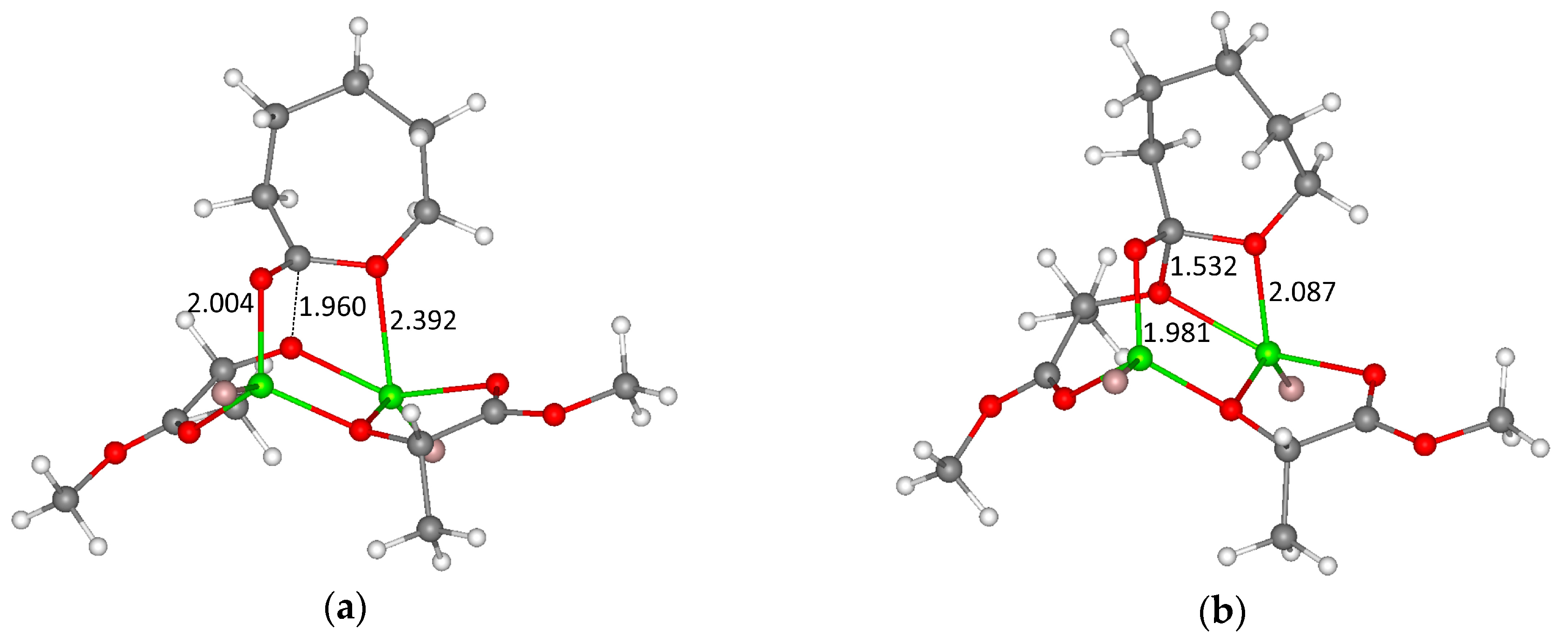{kind=link}
{kind=link}
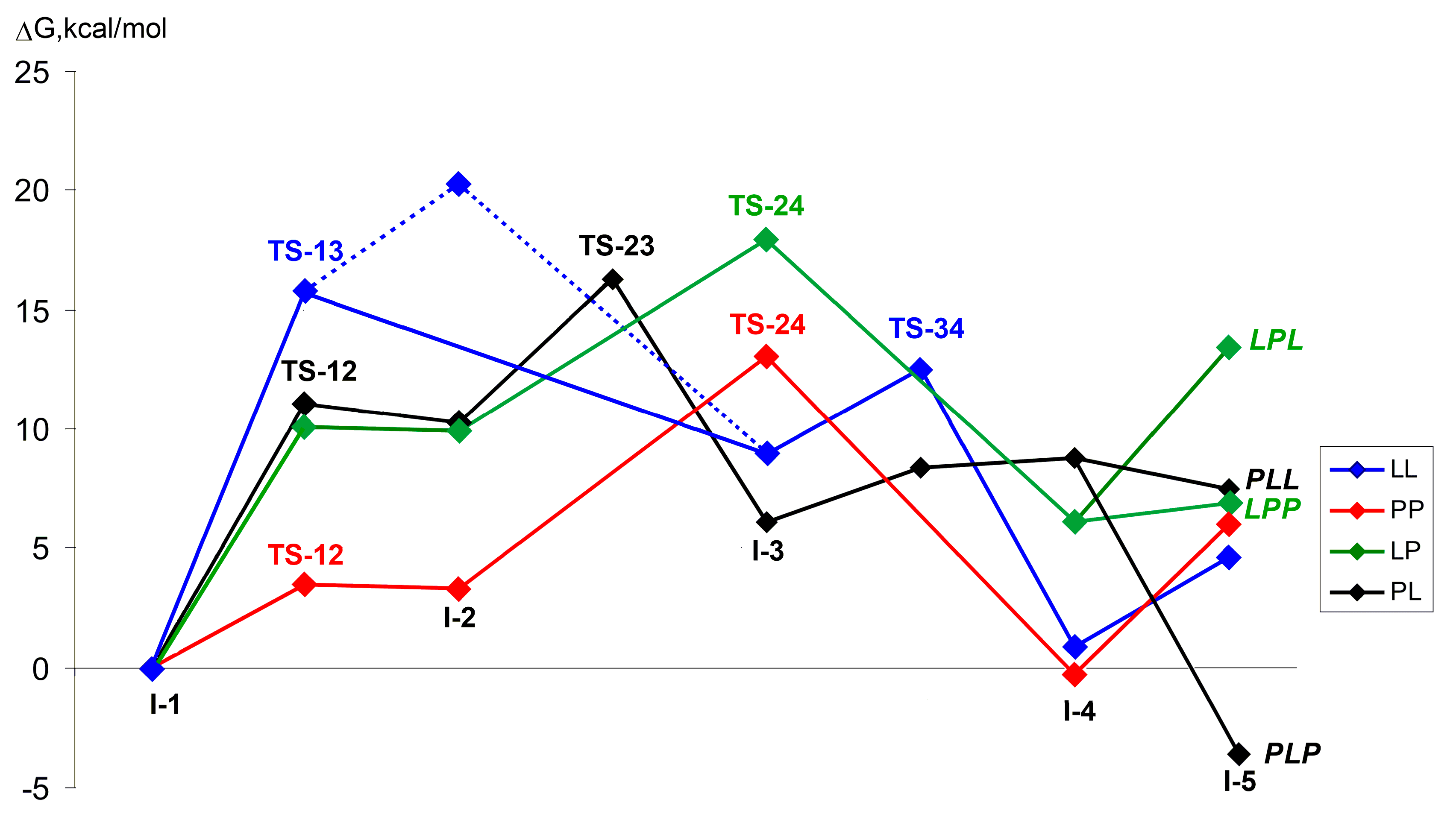{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comonomer Sequence | I-1 | TS-12 | I-2 | TS-23 | I-3 or TS-24 | TS-34 | I-4 | TS-45 | I-5 | |
|---|---|---|---|---|---|---|---|---|---|---|
| PP | G | 0.0 | 3.5 | 3.3 | n.d. | 13.1 2 | n.d. | –0.3 | 13.7 | 6.0 |
| H | 0.0 | 1.5 | 2.0 | n.d. | 9.1 2 | n.d. | –2.1 | 13.0 | –3.9 | |
| CC | G | 0.0 | 14.8 | 8.7 | 13.5 | 4.1 | 14.9 | 2.3 | 13.2 | 0.2 |
| H | 0.0 | 12.8 | 5.7 | 9.0 | 0.3 | 11.2 | 0.8 | –0.9 | –8.9 | |
| CP | G | 0.0 | 6.6 | –2.8 | 8.5 | –3.7 | n.d. | –7.0 | 4.6 | –2.3 |
| H | 0.0 | 3.5 | –6.0 | 3.6 | –7.1 | n.d. | –8.9 | –13.3 | –12.4 | |
| PC | G | 0.0 | 15.4 | 8.5 | n.d. 3 | 11.3 2 | n.d. | 10.6 | 23.2 | 14.7 |
| H | 0.0 | 13.0 | 7.5 | n.d. | 9.2 2 | n.d. | 9.1 | 21.9 | 6.5 | |
| Comonomer Sequence | I-1 | TS-12 | I-2 | TS-23 | I-3 or TS-24 | TS-34 | I-4 | I-5 | |
|---|---|---|---|---|---|---|---|---|---|
| LL | G | 0.0 | 15.8 2 | 20.3 | n.d. | 8.9 | 12.5 | 0.9 | 4.6 |
| H | 0.0 | 10.1 2 | 15.7 | n.d. | 2.9 | 7.5 | –3.1 | –7.4 | |
| PP | G | 0.0 | 3.5 | 3.3 | n.d. | 13.1 2 | n.d. | –0.3 | 6.0 |
| H | 0.0 | 1.5 | 2.0 | n.d. | 9.1 2 | n.d. | –2.1 | –3.9 | |
| LP | G | 0.0 | 10.1 | 9.9 | n.d. | 17.9 2 | n.d. | 6.1 | 13.4/6.9 4 |
| H | 0.0 | 6.9 | 5.3 | n.d. | 13.5 2 | n.d. | 4.2 | 1.1/–6.1 4 | |
| PL | G | 0.0 | 11.0 | 10.3 | 16.3 | 6.1 | 8.4 | 8.8 | 7.5/–3.7 4 |
| H | 0.0 | 8.8 | 8.6 | 13.3 | 4.1 | 5.6 | 12.1 | –2.0/–13.3 4 | |
| Run | Mon1 | Mon2 | Reaction time, min | Reaction T, °C | Conv., Mon1/ Mon2, % | Mntheo ×103 a) | MnNMR ×103 b) | MnSEC ×103 | ÐM |
|---|---|---|---|---|---|---|---|---|---|
| c1 | ε-CL | – | 10 | 5 | 54 | 6.3 | 6.8 | 6.4 c) | 1.18 c) |
| c2 | l-LA | – | 10 | 5 | 21 | 3.1 | 4.6 | 8.1 c) | 1.17 c) |
| c3 | MeOEP | – | 10 | 5 | >99 | 13.9 | 13.7 | 9.8 d) | 1.35 d) |
| 1a | ε-CL | l-LA | 10 | 5 | 0/86 | 6.3 | 7.0 | 8.5 c) | 1.31 c) |
| 1b | ε-CL | l-LA | 1800 | 20 | 0/100 | 7.3 | 10.3 | 10.6 c) | 1.28 c) |
| 2a | ε-CL | MeOEP | 10 | 5 | 0/98 | 6.9 | 9.3 | 11.6 d) | 1.32 d) |
| 2b | ε-CL | MeOEP | 2880 | 20 | 28/100 | 8.6 | 14.1 | 15.1 d) | 1.48 d) |
| 3a | l-LA | MeOEP | 20 | 5 | 89/100 | 13.5 | 13.7 | 16.9 d) | 1.21 d) |
| 3b | l-LA | MeOEP | 60 | 5 | 100/100 | 14.2 | 16.7 | 19.4 d) | 1.25 d) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nifant’ev, I.; Shlyakhtin, A.; Kosarev, M.; Gavrilov, D.; Karchevsky, S.; Ivchenko, P. DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates. Polymers 2019, 11, 1641. https://doi.org/10.3390/polym11101641
Nifant’ev I, Shlyakhtin A, Kosarev M, Gavrilov D, Karchevsky S, Ivchenko P. DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates. Polymers. 2019; 11(10):1641. https://doi.org/10.3390/polym11101641
Chicago/Turabian StyleNifant’ev, Ilya, Andrey Shlyakhtin, Maxim Kosarev, Dmitry Gavrilov, Stanislav Karchevsky, and Pavel Ivchenko. 2019. "DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates" Polymers 11, no. 10: 1641. https://doi.org/10.3390/polym11101641
APA StyleNifant’ev, I., Shlyakhtin, A., Kosarev, M., Gavrilov, D., Karchevsky, S., & Ivchenko, P. (2019). DFT Visualization and Experimental Evidence of BHT-Mg-Catalyzed Copolymerization of Lactides, Lactones and Ethylene Phosphates. Polymers, 11(10), 1641. https://doi.org/10.3390/polym11101641