A Methodologic Approach for the Selection of Bio-Resorbable Polymers in the Development of Medical Devices: The Case of Poly(l-lactide-co-ε-caprolactone)

 ,
,  ,
,  and
and
Abstract
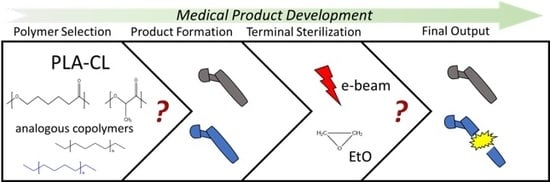
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Terminal Sterilization Procedures
2.2.1. Ethylene Oxide Processing
2.2.2. Electron Beam Processing
2.3. Analytical Methods
2.3.1. Gel Permeation Chromatography
2.3.2. Differential Scanning Calorimetry
2.3.3. X-ray Diffraction
2.3.4. Nuclear Magnetic Resonance (1H-NMR)
3. Results and Discussion
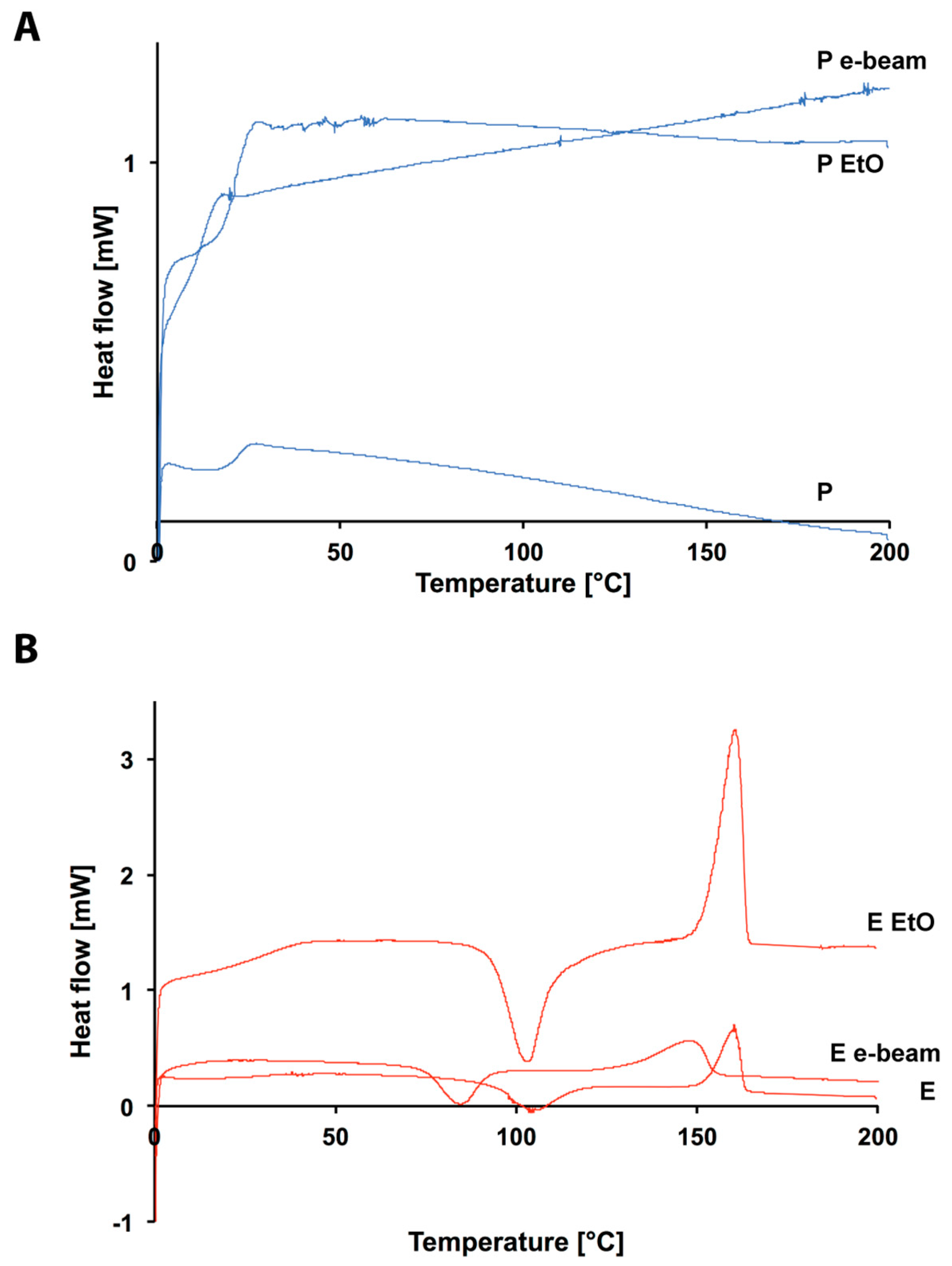
3.1. Prystine Polymer Characterization
3.2. Ethylene Oxide Processing
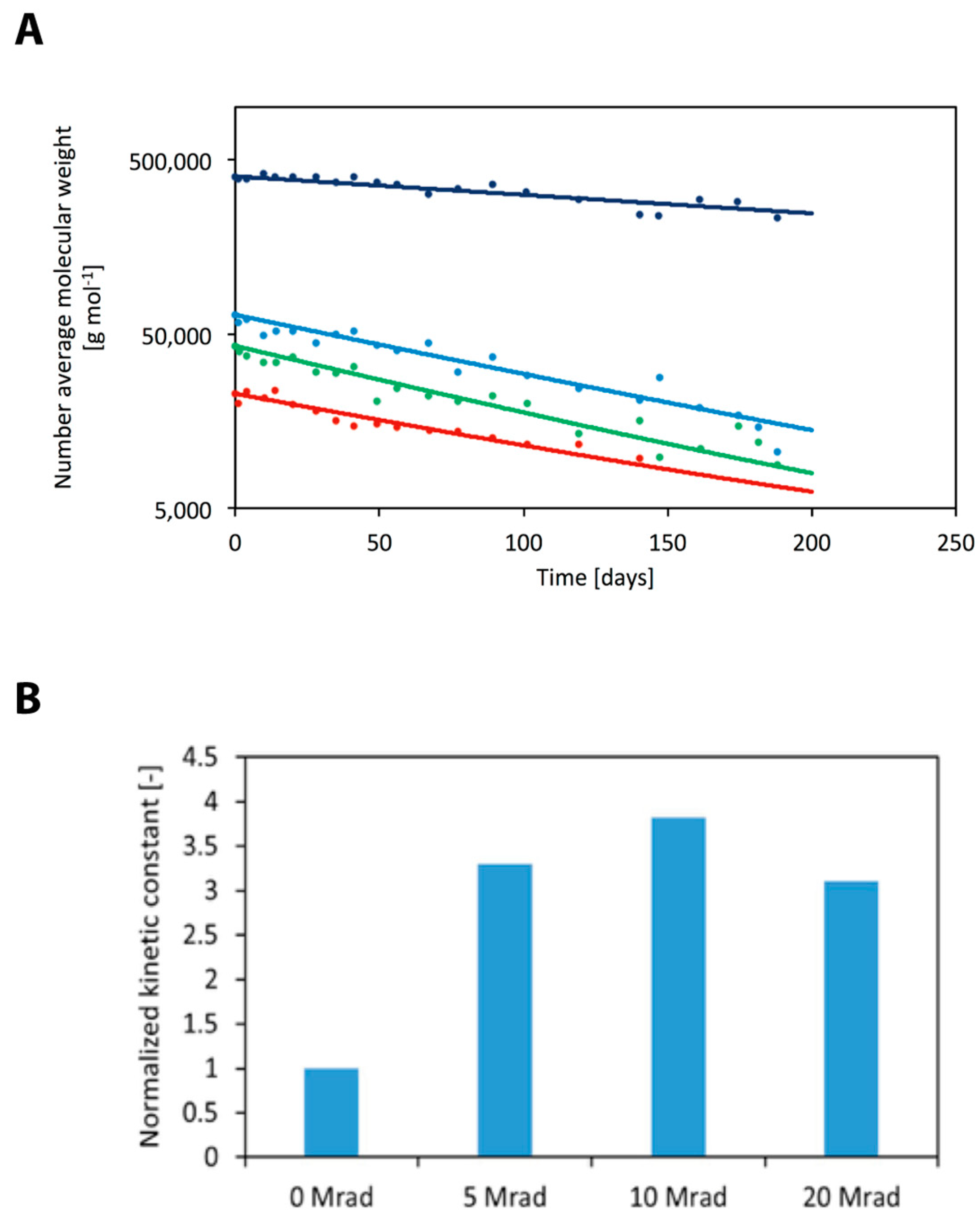
3.3. Electron Beam Processing
3.4. Mathematical Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Perale, G.; Hilborn, J. Bioresorbable Polymers for Biomedical Applications—From Fundamentals to Translational Medicine, 1st ed.; Woodhead Publishing Series in Biomaterials: Sawston, UK, 2016. [Google Scholar]
- Cameron, R.E.; Kamvari-Moghaddam, A. Synthetic Bioresorbable Polymers; Woodhead Publishing Limited: Sawston, UK, 2012; ISBN 9781845699291. [Google Scholar]
- Planell, J.A. Bone Repair Biomaterials; Woodhead Publishing Limited: Sawston, UK, 2009; ISBN 9781845693763. [Google Scholar]
- Armentano, I.; Bitinis, N.; Fortunati, E.; Mattioli, S.; Rescignano, N.; Verdejo, R.; Lopez-Manchado, M.A.; Kenny, J.M. Multifunctional nanostructured PLA materials for packaging and tissue engineering. Prog. Polym. Sci. 2013, 38, 1720–1747. [Google Scholar] [CrossRef]
- Ceccarelli, G.; Presta, R.; Benedetti, L.; Cusella De Angelis, M.G.; Lupi, S.M.; Rodriguez y Baena, R. Emerging Perspectives in Scaffold for Tissue Engineering in Oral Surgery. Stem Cells Int. 2017, 2017, 4585401. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, K.A.; Niederauer, G.G.; Agrawal, C.M. Sterilization, toxicity, biocompatibility and clinical applications of polylactic acid/polyglycolic acid copolymers. Biomaterials 1996, 17, 93–102. [Google Scholar] [CrossRef]
- Casalini, T.; Perale, G. Types of bioresorbable polymers for medical applications. Durab. Reliab. Med. Polym. 2012, 3–29. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Z.; Najeeb, S.; Khurshid, Z.; Verma, V.; Rashid, H.; Glogauer, M. Biodegradable materials for bone repair and tissue engineering applications. Materials 2015, 8, 5744–5794. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.D.; Williams, D.F. The in vivo and in vitro degradation of poly(glycolic acid) suture material as a function of applied strain. Biomaterials 1984, 5, 365–368. [Google Scholar] [CrossRef]
- Prakasam, M.; Locs, J.; Salma-Ancane, K.; Loca, D.; Largeteau, A.; Berzina-Cimdina, L. Biodegradable Materials and Metallic Implants—A Review. J. Funct. Biomater. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Perale, G. Using synthetic bioresorbable polymers for orthopedic tissue regeneration. Durab. Reliab. Med. Polym. 2012, 119–139. [Google Scholar] [CrossRef]
- Colombo, C.; Gatti, S.; Ferrari, R.; Casalini, T.; Cuccato, D.; Morosi, L.; Zucchetti, M.; Moscatelli, D. Self-assembling amphiphilic PEGylated block copolymers obtained through RAFT polymerization for drug-delivery applications. J. Appl. Polym. Sci. 2016, 133, 1–8. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, R.; Cingolani, A.; Moscatelli, D. Solvent effect in PLA-PEG based nanoparticles synthesis through surfactant free polymerization. Macromol. Symp. 2013, 324, 107–113. [Google Scholar] [CrossRef]
- Ferrari, R.; Yu, Y.; Morbidelli, M.; Hutchinson, R.A.; Moscatelli, D. ε-Caprolactone-Based Macromonomers Suitable for Biodegradable Nanoparticles Synthesis Through Free Radical Polymerization. Macromolecules 2011, 44, 9205–9212. [Google Scholar] [CrossRef]
- Pillai, C.K.S.; Sharma, C.P. Review paper: Absorbable polymeric surgical sutures: Chemistry, production, properties, biodegradability, and performance. J. Biomater. Appl. 2010, 25, 291–366. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Freed, L.E.; Vunjak-Novakovic, G.; Biron, R.J.; Eagles, D.B.; Lesnoy, D.C.; Barlow, S.K.; Langer, R. Biodegradable Polymer Scaffolds for Tissue Engineering. Nat. Biotechnol. 1994, 12, 1119–1124. [Google Scholar] [CrossRef]
- Kutz, M. Applied Plastics Engineering Handbook; Elsevier Inc.: New York, NY, USA, 2011. [Google Scholar]
- Lambert, B.J.; Mendelson, T.A.; Craven, M.D. Radiation and Ethylene Oxide Terminal Sterilization Experiences with Drug Eluting Stent Products. AAPS PharmSciTech 2011, 12, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Dearth, C.L.; Keane, T.J.; Carruthers, C.A.; Reing, J.E.; Huleihel, L.; Ranallo, C.A.; Kollar, E.W.; Badylak, S.F. The effect of terminal sterilization on the material properties and in vivo remodeling of a porcine dermal biologic scaffold. Acta Biomater. 2016, 33, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, N.P.; Burgess, D.J. Sterilization of implantable polymer-based medical devices: A review. Int. J. Pharm. 2018, 544, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Mendes, G.C.; Brandão, T.R.; Silva, C.L. Ethylene oxide sterilization of medical devices: A review. Am. J. Infect. Control. 2007, 35, 574–581. [Google Scholar] [CrossRef] [PubMed]
- McKeen, L. Effect of Sterlization Method on Plastics and Elastomers, 3rd ed.; Elsevier Inc.: New York, NY, USA, 2012. [Google Scholar]
- Loo, S.C.J.; Tan, H.T.; Ooi, C.P.; Boey, Y.C.F. Hydrolytic degradation of electron beam irradiated high molecular weight and non-irradiated moderate molecular weight PLLA. Acta Biomater. 2006, 2, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.C.J.; Ooi, C.P.; Boey, Y.C.F. Radiation effects on poly(lactide-co-glycolide) (PLGA) and poly(l-lactide) (PLLA). Polym. Degrad. Stab. 2004, 83, 259–265. [Google Scholar] [CrossRef]
- Loo, S.C.J.; Ooi, C.P.; Boey, Y.C.F. Degradation of poly(lactide-co-glycolide) (PLGA) and poly(l-lactide) (PLLA) by electron beam radiation. Biomaterials 2005, 26, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Pertici, G. The effect of molecular structure on the properties of biomedical polymers. Durab. Reliab. Med. Polym. 2012, 30–48. [Google Scholar] [CrossRef]
- Roato, I.; Belisario, D.C.; Compagno, M.; Verderio, L.; Sighinolfi, A.; Mussano, F.; Genova, T.; Veneziano, F.; Pertici, G.; Perale, G.; et al. Adipose-Derived Stromal Vascular Fraction/Xenohybrid Bone Scaffold: An Alternative Source for Bone Regeneration. Stem Cells Int. 2018, 2018, 4126379. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.; Perale, G.; Milazzo, M.; Moscato, S.; Stefanini, C.; Pertici, G.; Danti, S. Bovine bone matrix/poly(l-lactic-co-ε-caprolactone)/gelatin hybrid scaffold (SmartBone®) for maxillary sinus augmentation: A histologic study on bone regeneration. Int. J. Pharm. 2017, 523, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Pertici, G.; Carinci, F.; Carusi, G.; Epistatus, D.; Villa, T.; Crivelli, F.; Rossi, F.; Perale, G. Composite Polymer-Coated Mineral Scaffolds for Bone Regeneration: From Material Characterization To Human Studies. J. Biol. Regul. Homeost. Agents 2015, 29, 136–148. [Google Scholar] [PubMed]
- Scardi, P.; Leoni, M.; Delhez, R. Line broadening analysis using integral breadth methods: A critical review. J. Appl. Crystallogr. 2004, 37, 381–390. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, B.; Yang, G.; Gauthier, M. Poly(Lactic Acid)-Based Biomaterials: Synthesis, Modification and Applications; INTECH Open Access Publisher: London, UK, 2012. [Google Scholar]
- Cairns, M.L.; Sykes, A.; Dickson, G.R.; Orr, J.F.; Farrar, D.; Dumba, A.; Buchanan, F.J. Through-thickness control of polymer bioresorption via electron beam irradiation. Acta Biomater. 2011, 7, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Morgan, D.A.F.; Forwood, M.R. Sterilization of allograft bone: Is 25 kGy the gold standard for gamma irradiation? Cell Tissue Bank. 2007, 8, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhou, L.; Lv, J. Effects of expandable graphite and modified ammonium polyphosphate on the flame-retardant and mechanical properties of wood flour-polypropylene composites. Polym. Polym. Compos. 2013, 21, 449–456. [Google Scholar] [CrossRef]
- Narkis, M.; Sibony-Chaouat, S.; Siegmann, A.; Shkolnik, S.; Bell, J.P. Irradiation effects on polycaprolactone. Polymer 1985, 26, 50–54. [Google Scholar] [CrossRef]
- Gupta, M.C.; Deshmukh, V.G. Radiation effects on poly(lactic acid). Polymer 1983, 24, 827–830. [Google Scholar] [CrossRef]
- Filipczak, K.; Wozniak, M.; Ulanski, P.; Olah, L.; Przybytniak, G.; Olkowski, R.M.; Lewandowska-Szumiel, M.; Rosiak, J.M. Poly(ε-caprolactone) biomaterial sterilized by E-beam irradiation. Macromol. Biosci. 2006, 6, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, K.; Kriz, D.; Salovey, R.; Narkis, M.; Wallerstein, R. Crosslinking of polycaprolactone in the pre-gelation region. Polym. Eng. Sci. 1988, 28, 1484–1490. [Google Scholar] [CrossRef]
- Oláh, L.; Filipczak, K.; Czvikovszky, T.; Czigány, T.; Borbás, L. Changes of porous poly(ε-caprolactone) bone grafts resulted from e-beam sterilization process. Radiat. Phys. Chem. 2007, 76, 1430–1434. [Google Scholar] [CrossRef]
- Loo, S.C.J.; Ooi, C.P.; Boey, Y.C.F. Influence of electron-beam radiation on the hydrolytic degradation behaviour of poly(lactide-co-glycolide) (PLGA). Biomaterials 2005, 26, 3809–3817. [Google Scholar] [CrossRef] [PubMed]
- Perale, G.; Arosio, P.; Moscatelli, D.; Barri, V.; Müller, M.; Maccagnan, S.; Masi, M. A new model of resorbable device degradation and drug release: Transient 1-dimension diffusional model. J. Control. Release 2009, 136, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Casalini, T.; Rossi, F.; Lazzari, S.; Perale, G.; Masi, M. Mathematical modeling of PLGA microparticles: From polymer degradation to drug release. Mol. Pharm. 2014, 11, 4036–4048. [Google Scholar] [CrossRef] [PubMed]
- Ramkrishna, D. Population Balances—Theory and Applications to Particulate Systems in Engineering; Elsevier Inc.: New York, NY, USA, 2000. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPC | 1H-NMR | DSC | XRD | |||||
|---|---|---|---|---|---|---|---|---|
| # Sample | Mw (Da) | Mn (Da) | PD (−) | CL (% g·gtot−1) | Crystallinity (%) | Crystallinity (%) | FWHM (°) | Lhkl (A) |
| E | 119,600 | 85,660 | 1.12 | 25.4 | 61.1 | 65 | 4.5 | 0.308 |
| P | 172,750 | 123,800 | 1.6 | 25.5 | 36.1 | 35.2 | 7.5 | 0.185 |
| E EtO | 96,000 | 78,700 | 1.2 | 25 | 59.6 | 60 | 4.5 | 0.308 |
| P EtO | 159,400 | 97,300 | 1.64 | 25.7 | 34.3 | 35 | 7.5 | 0.185 |
| E e-beam | 51,800 | 43,660 | 1.3 | 25.6 | 60.6 | 59.2 | 4.5 | 0.317 |
| P e-beam | 133,300 | 64,100 | 2.08 | 25.4 | 35.5 | 32.1 | 7.5 | 0.185 |
| Film thickness (μm) | 55 |
| Mmon (g·mol−1) | 90.08 |
| ρpol | 1.2 |
| DM0 (cm2·s−1) | 10−10 |
| Dolig0 (cm2·s−1) | 10−10 |
| DW0 (cm2·s−1) | 10−8 |
| Radiation Dose (Mrad) | Number Average Molecular Weight (g·mol−1) | Polydispersity (−) | Degradation Constant (cm6·mol−2·s−1) |
|---|---|---|---|
| 0 | 406,000 | 1.60 | 3.85 × 10−5 |
| 5 | 64,700 | 1.68 | 1.27 × 10−4 |
| 10 | 43,200 | 1.73 | 1.47 × 10−4 |
| 20 | 23,100 | 1.76 | 1.21 × 10−4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cingolani, A.; Casalini, T.; Caimi, S.; Klaue, A.; Sponchioni, M.; Rossi, F.; Perale, G. A Methodologic Approach for the Selection of Bio-Resorbable Polymers in the Development of Medical Devices: The Case of Poly(l-lactide-co-ε-caprolactone). Polymers 2018, 10, 851. https://doi.org/10.3390/polym10080851
Cingolani A, Casalini T, Caimi S, Klaue A, Sponchioni M, Rossi F, Perale G. A Methodologic Approach for the Selection of Bio-Resorbable Polymers in the Development of Medical Devices: The Case of Poly(l-lactide-co-ε-caprolactone). Polymers. 2018; 10(8):851. https://doi.org/10.3390/polym10080851
Chicago/Turabian StyleCingolani, Alberto, Tommaso Casalini, Stefano Caimi, Antoine Klaue, Mattia Sponchioni, Filippo Rossi, and Giuseppe Perale. 2018. "A Methodologic Approach for the Selection of Bio-Resorbable Polymers in the Development of Medical Devices: The Case of Poly(l-lactide-co-ε-caprolactone)" Polymers 10, no. 8: 851. https://doi.org/10.3390/polym10080851
APA StyleCingolani, A., Casalini, T., Caimi, S., Klaue, A., Sponchioni, M., Rossi, F., & Perale, G. (2018). A Methodologic Approach for the Selection of Bio-Resorbable Polymers in the Development of Medical Devices: The Case of Poly(l-lactide-co-ε-caprolactone). Polymers, 10(8), 851. https://doi.org/10.3390/polym10080851