Synthesis and Characterization of Wooden Magnetic Activated Carbon Fibers with Hierarchical Pore Structures

Abstract
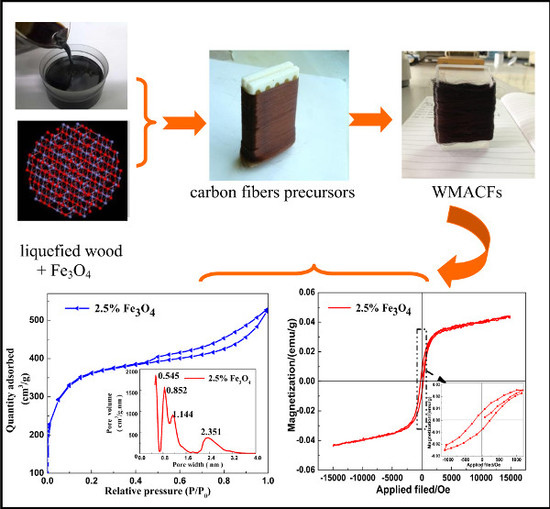
:
1. Introduction
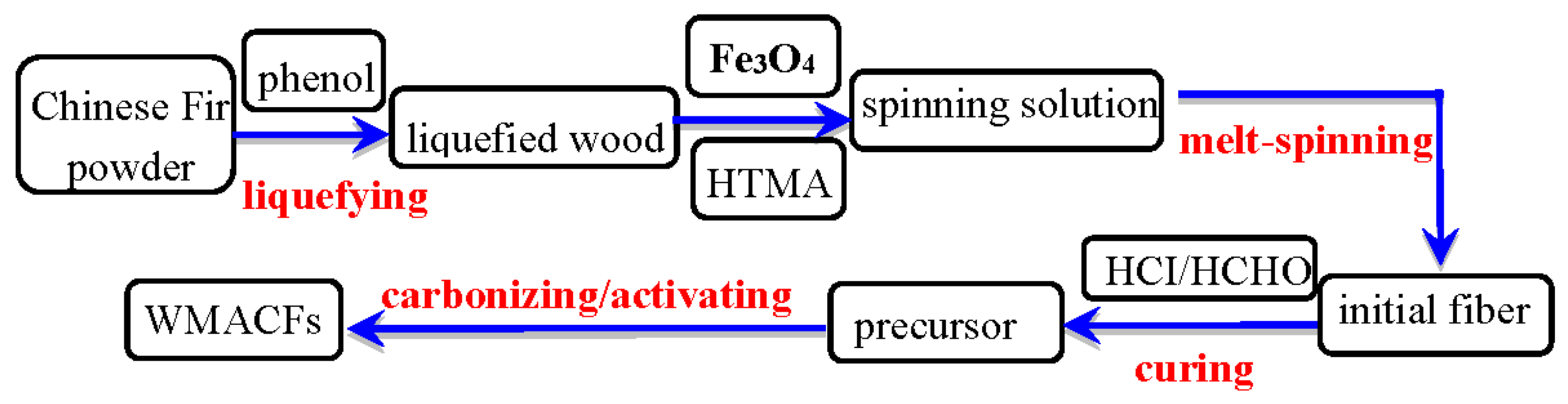
2. Materials and Methods
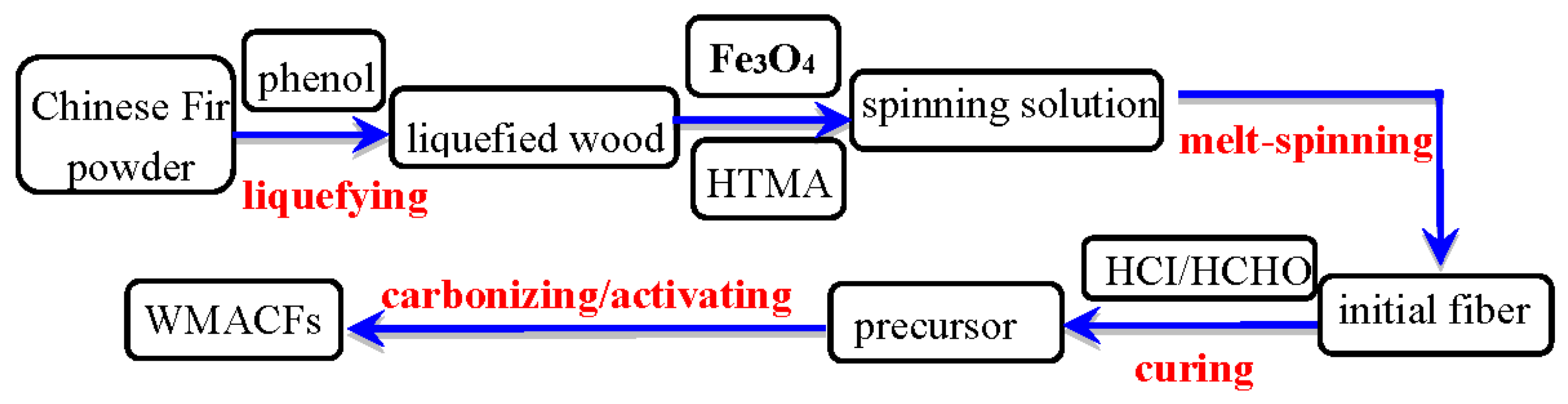
2.1. Samples
2.2. Characterizations
3. Results and Discussion
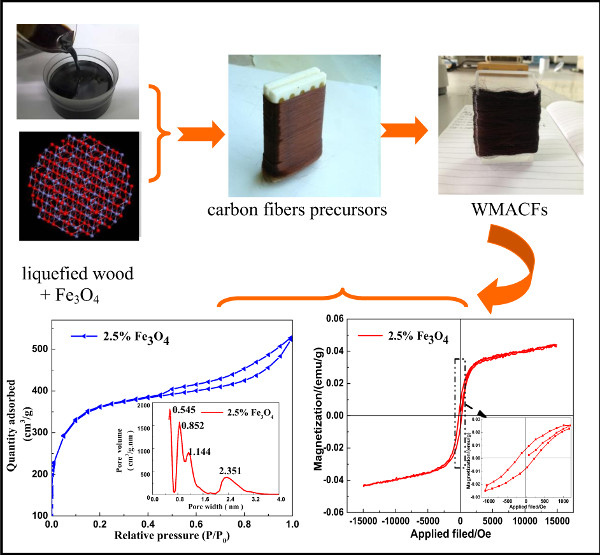
3.1. Morphological Characteristics of the WMACFs
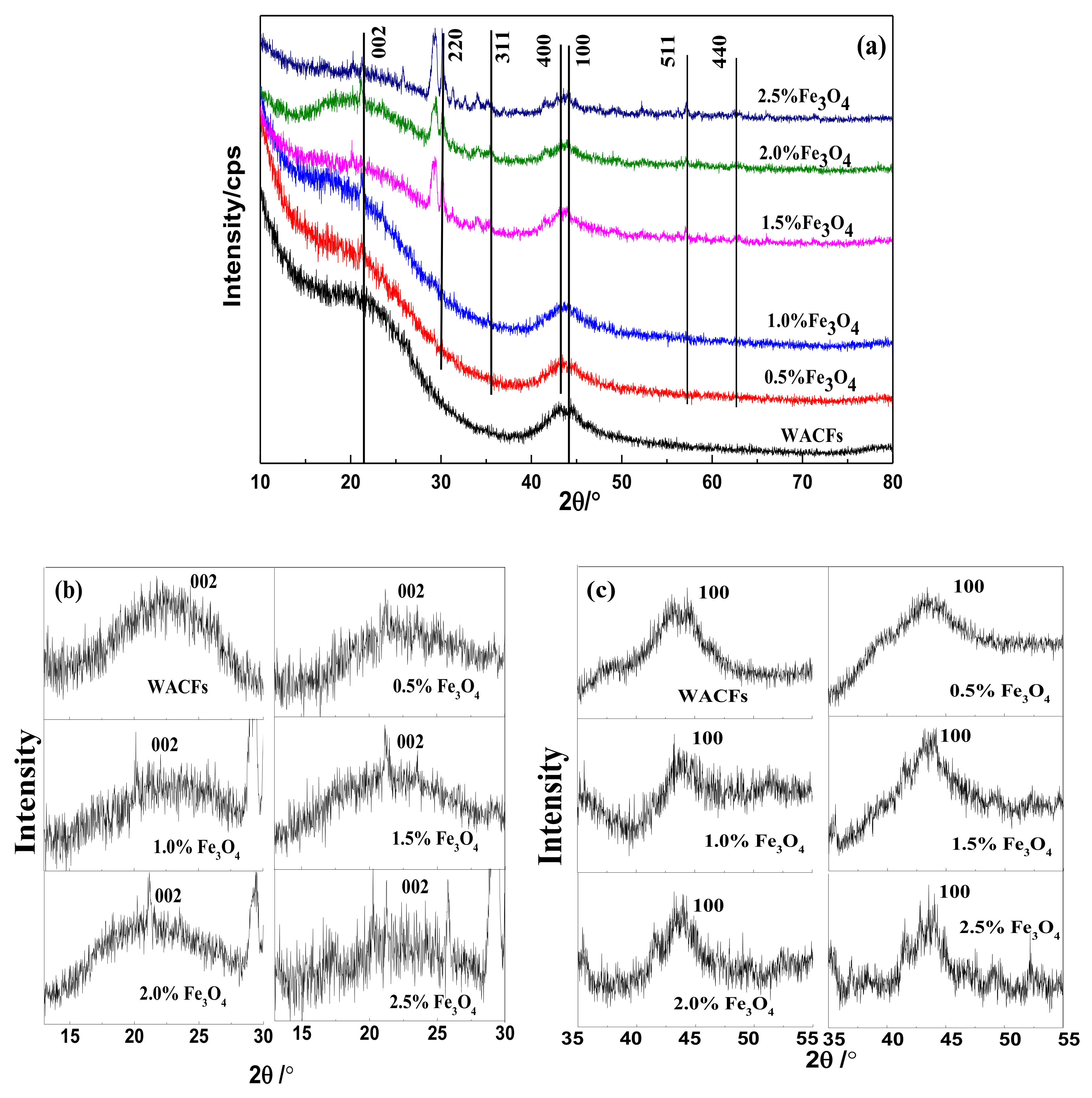
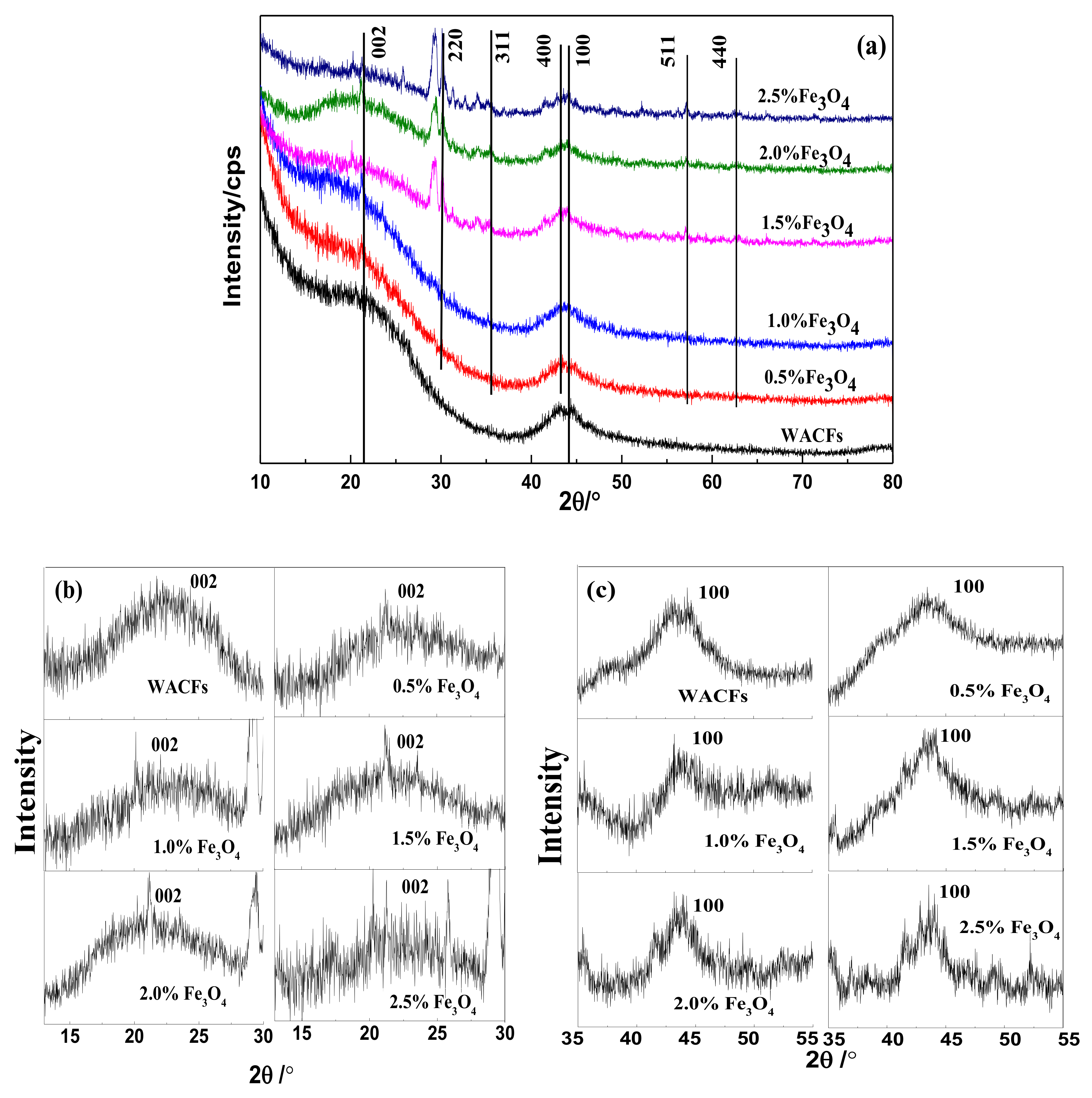
3.2. XRD Analysis of the WMACFs
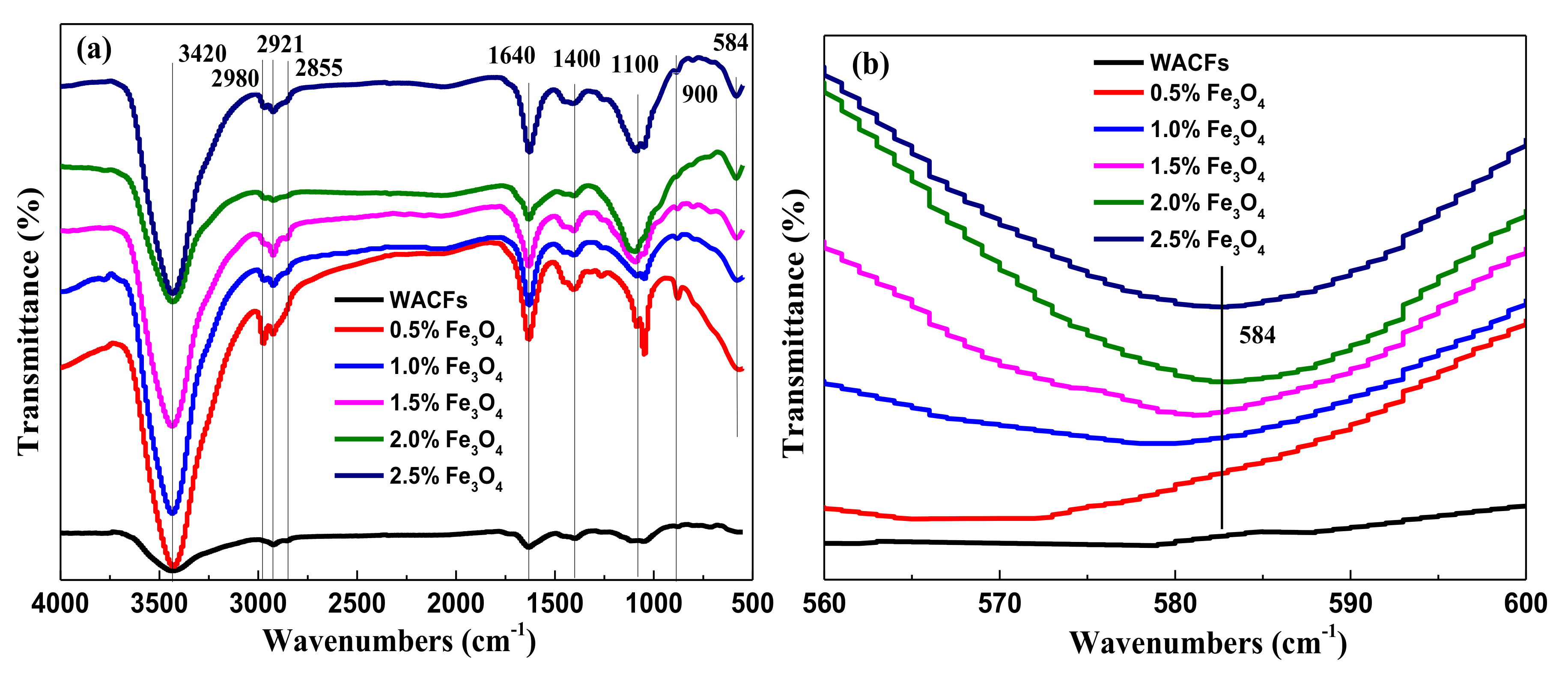
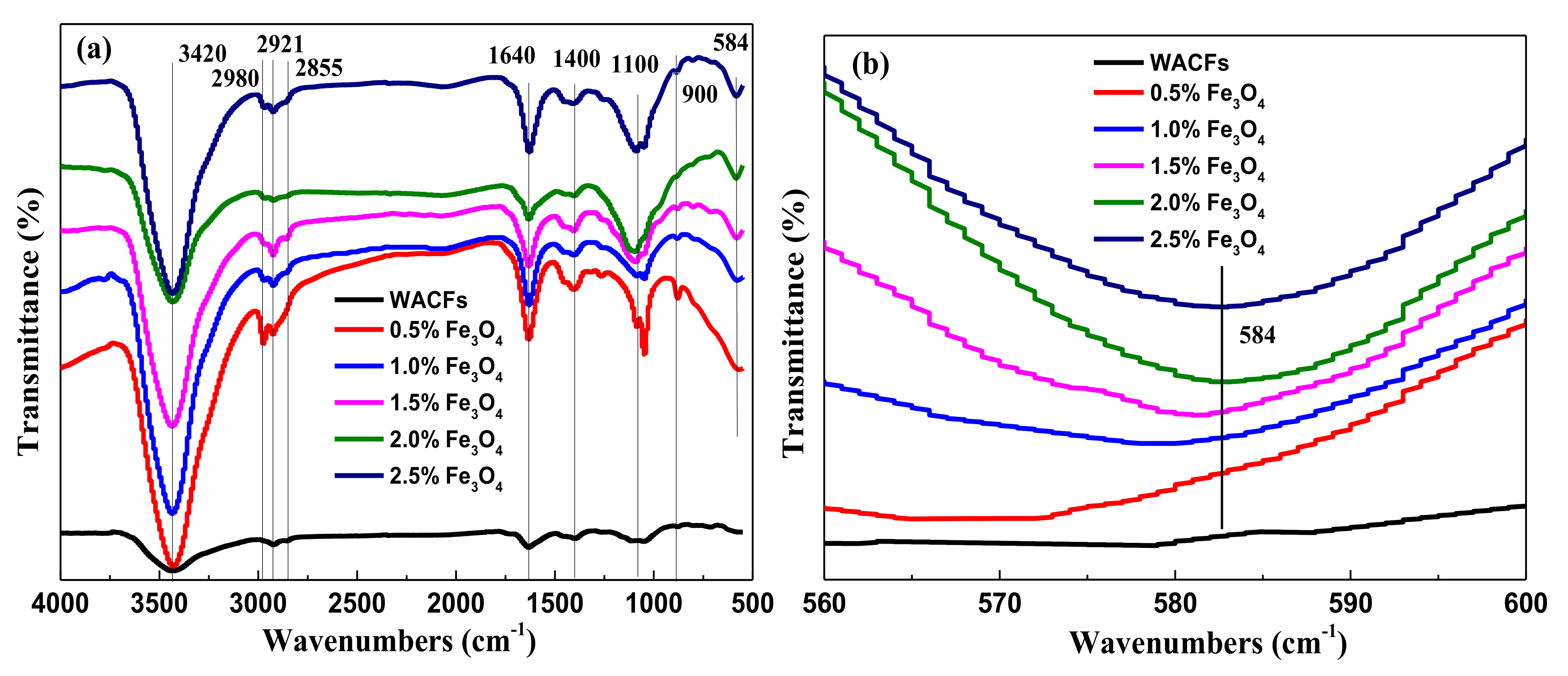
3.3. FTIR Analysis of the WMACFs
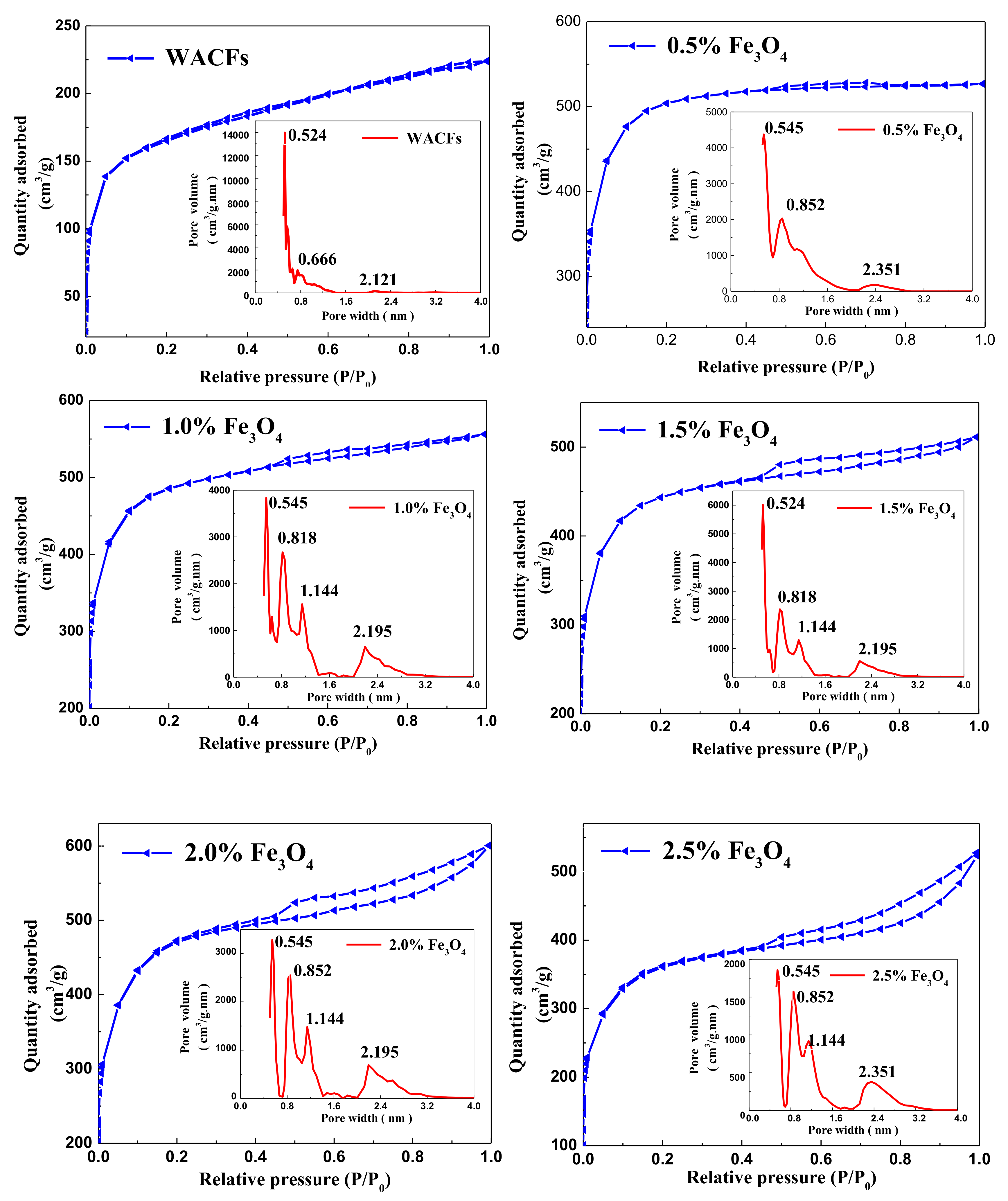
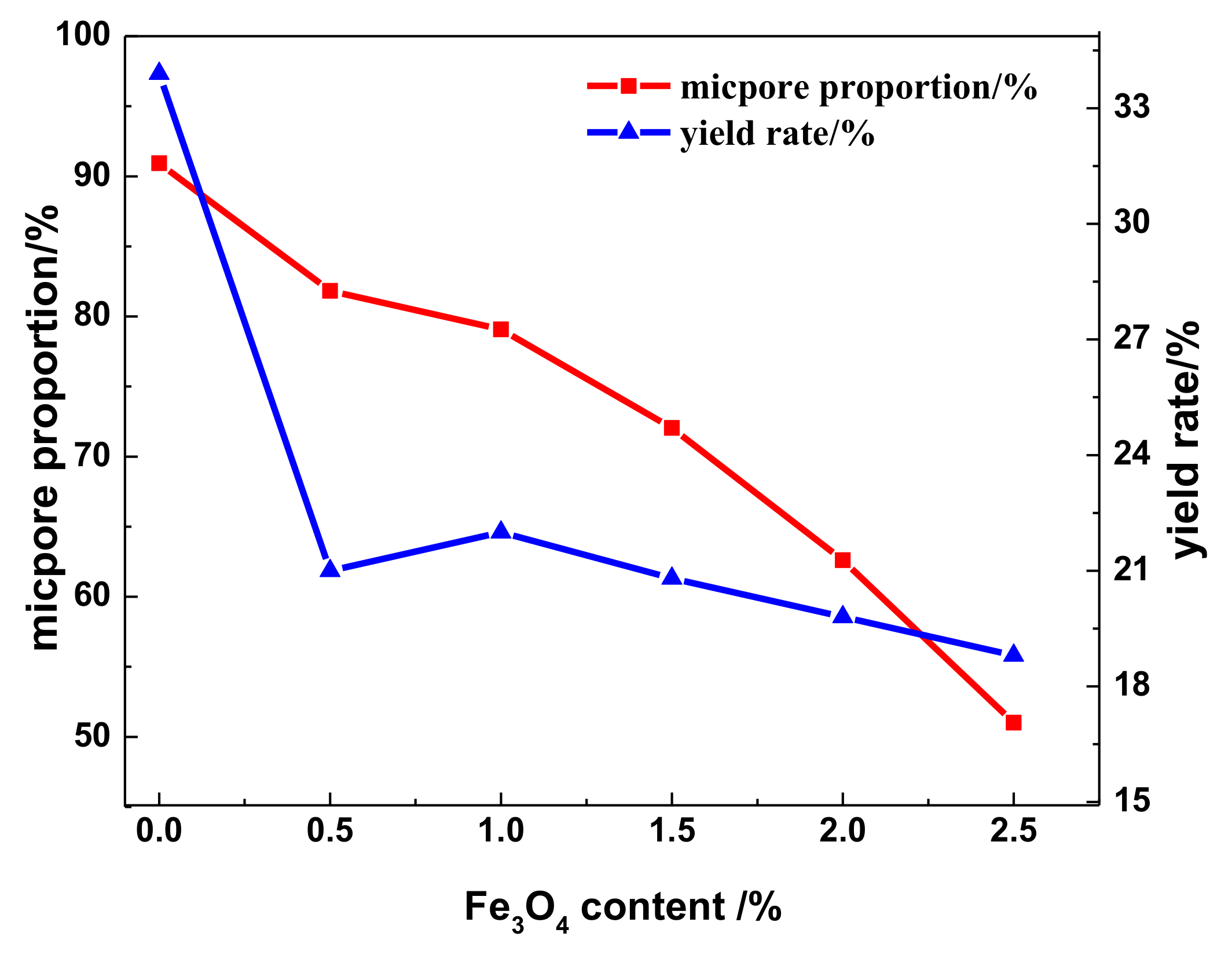
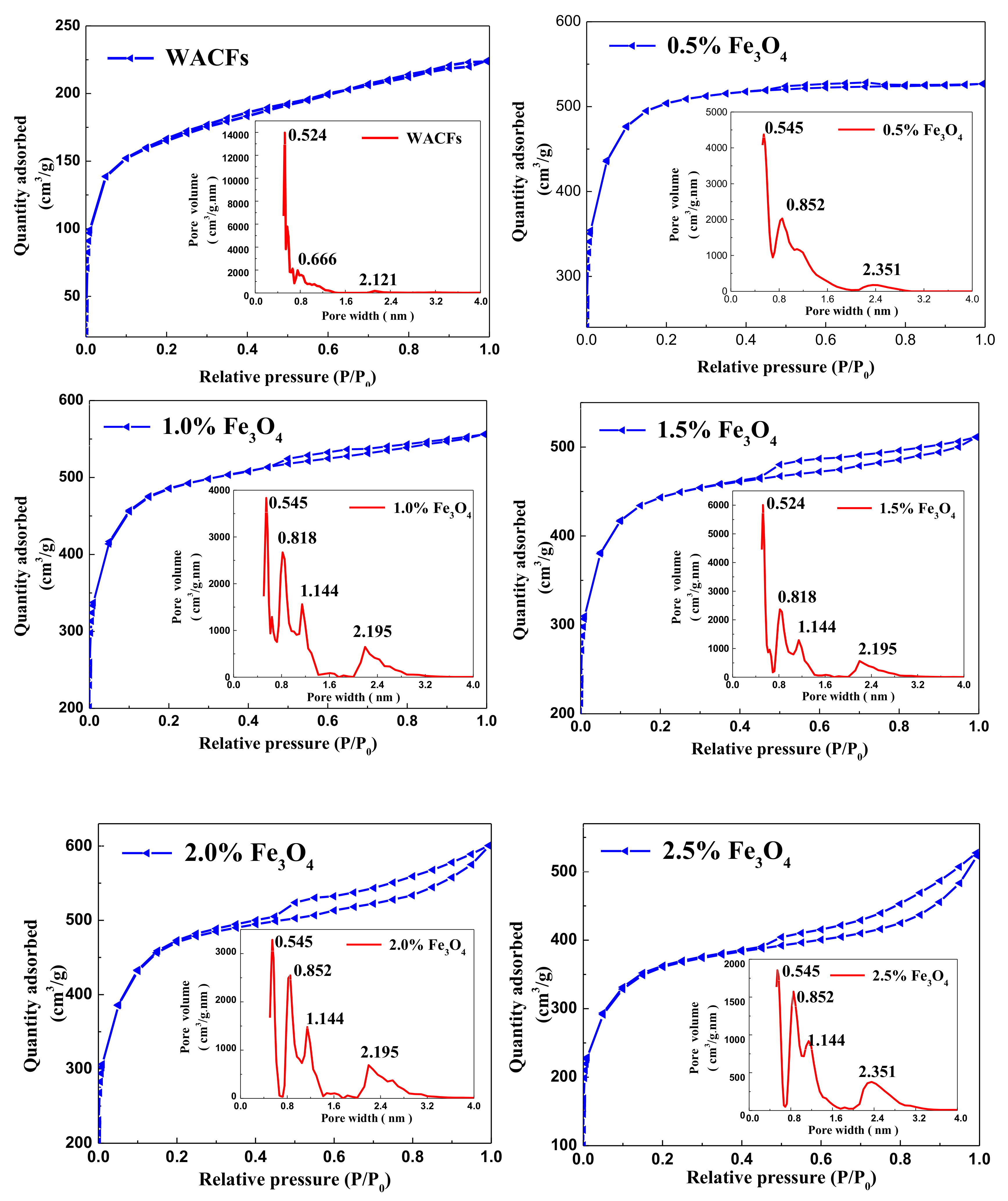
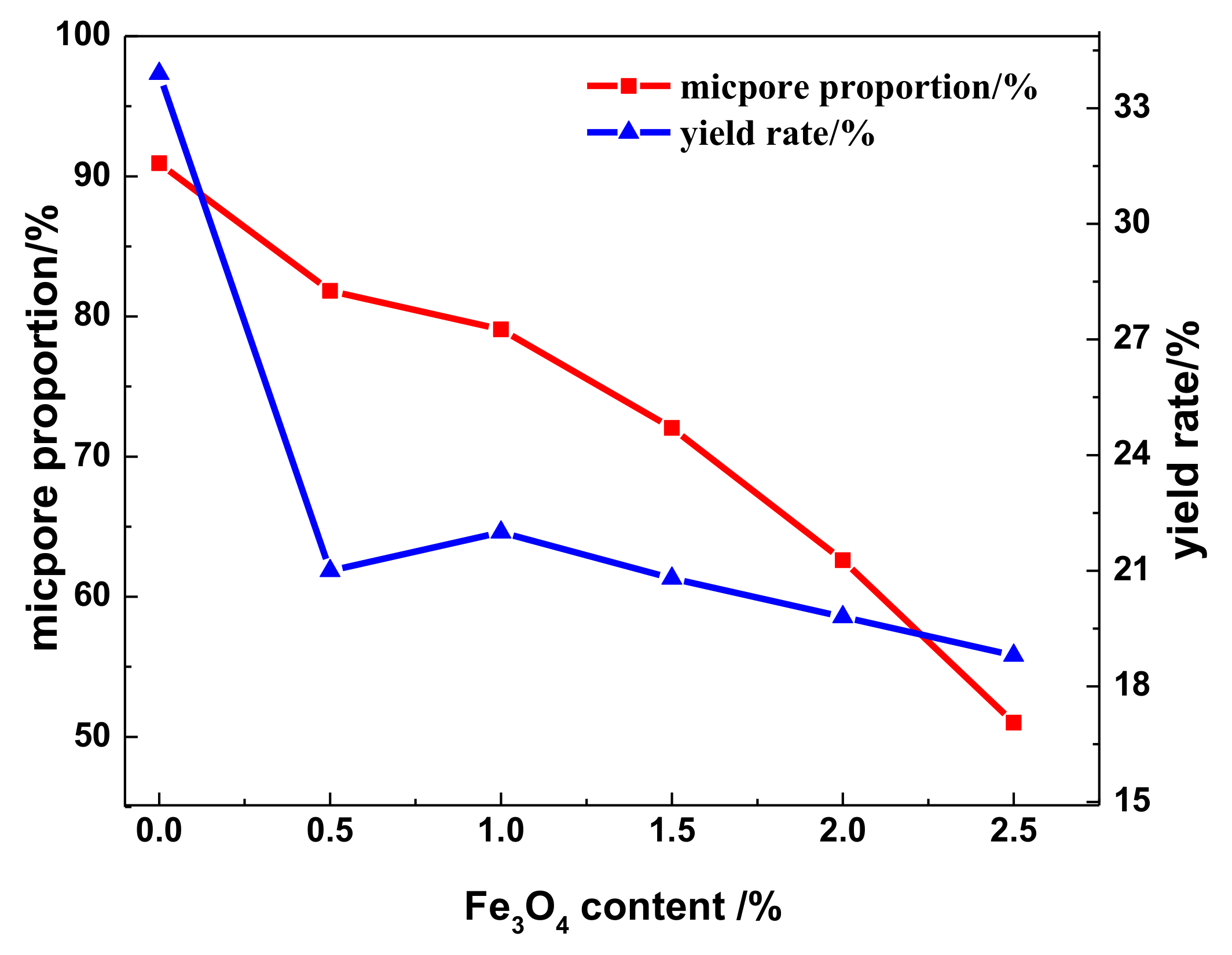
3.4. Adsorption Characteristics, Specific Surface Area, and Pore Distribution of the WMACFs
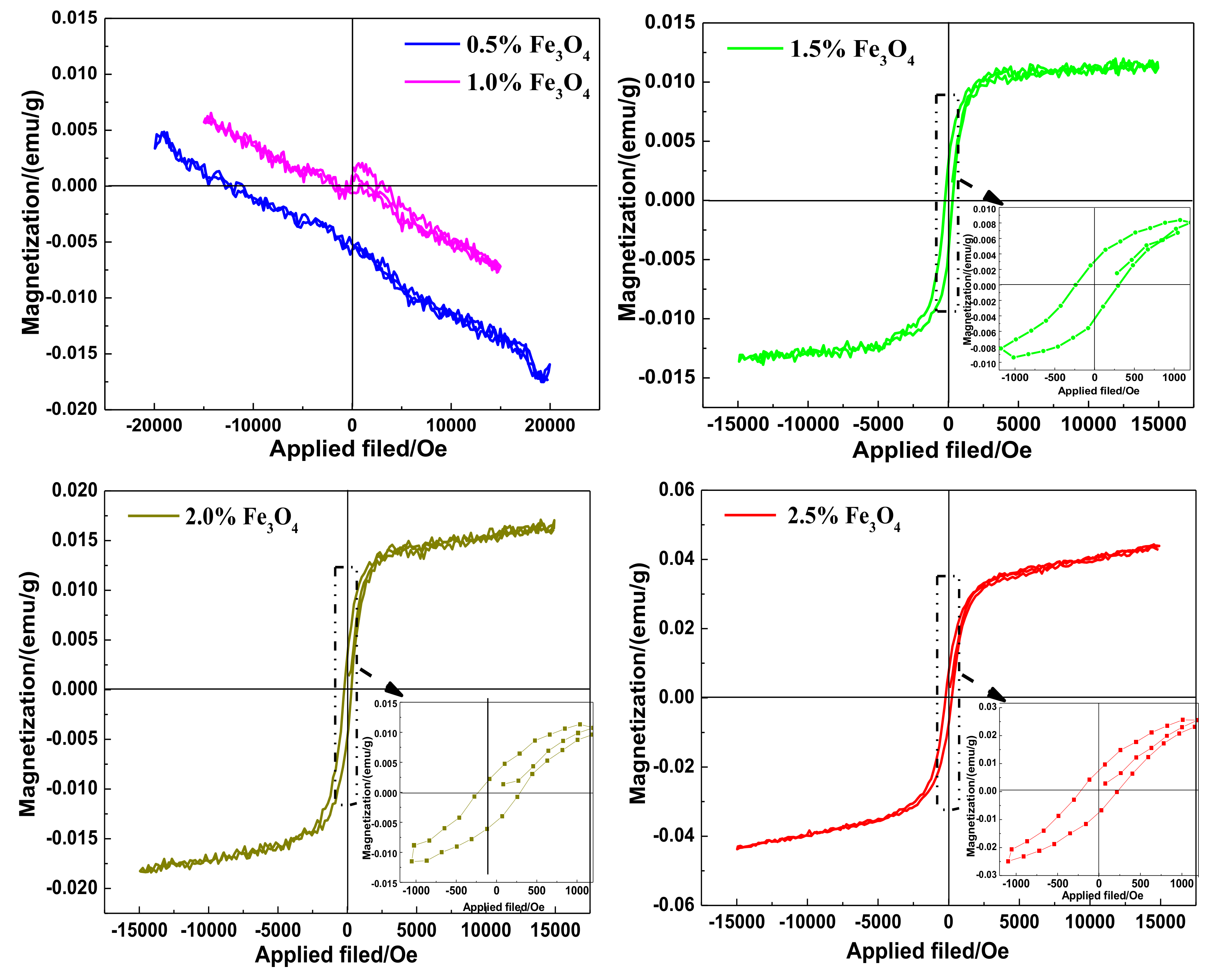
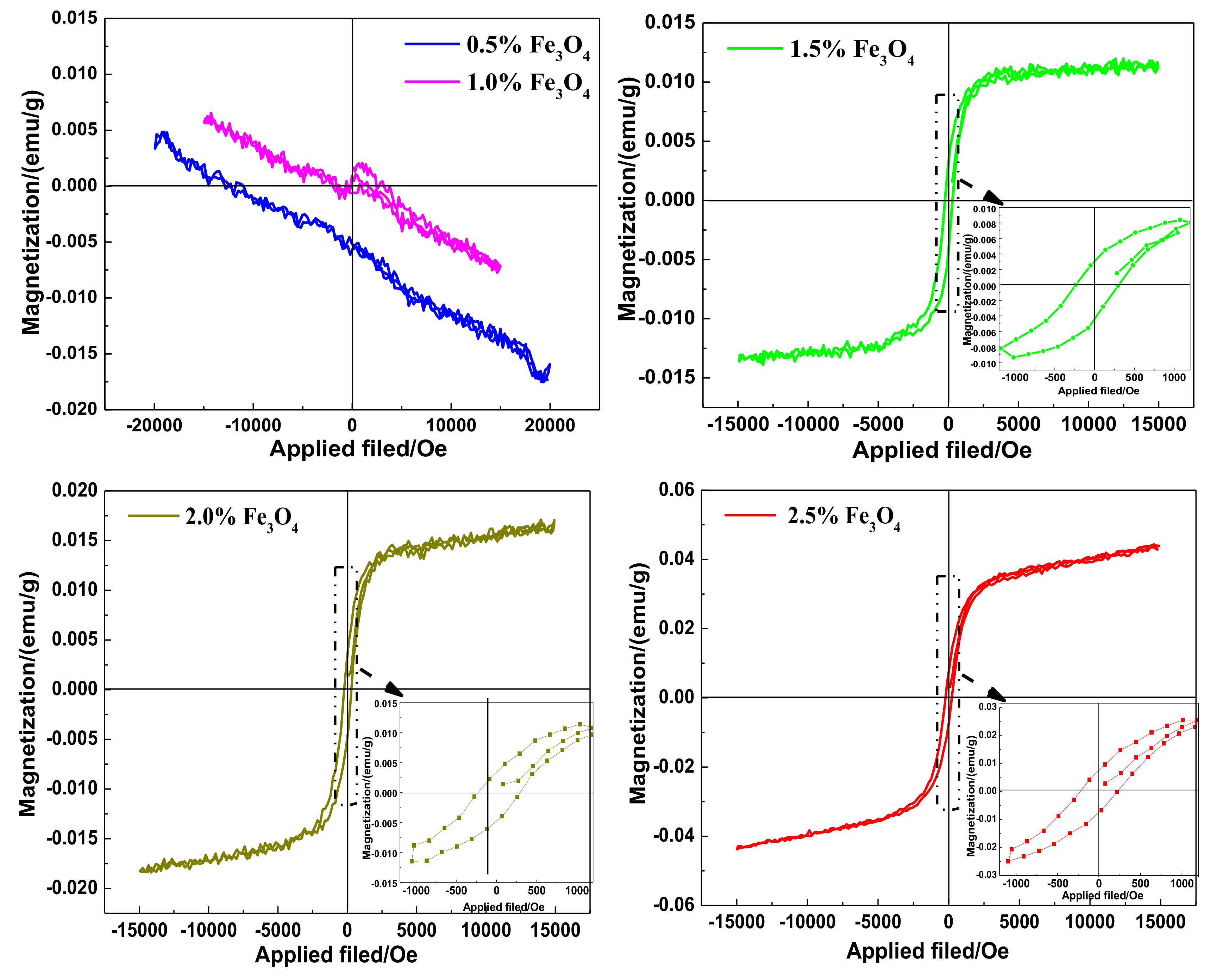
3.5. VSM Analysis of the WMACFs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, D.N.; Ma, X.J. Preparation and characterization of activated carbon fibers from liquefied wood. Cellulose 2013, 20, 1649–1656. [Google Scholar] [CrossRef]
- Huang, Y.X.; Zhao, G.J. A novel method for the production of mesoporous activated carbon fibers from liquefied wood. Mater. Lett. 2016, 178, 190–192. [Google Scholar] [CrossRef]
- Kwiatkowski, M. Analysis of the microporous structure of the low-cost activated carbon fibres obtained from flax and jute cloth. J. Math. Chem. 2017, 55, 1893–1902. [Google Scholar] [CrossRef]
- Yue, Z.R.; Vakili, A. Activated carbon–carbon composites made of pitch-based carbon fibers and phenolic resin for use of adsorbents. J. Mater. Sci. 2017, 52, 12913–12921. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, E.; Zhao, G. Thermal and structure analysis on reaction mechanisms during the preparation of activated carbon fibers by KOH activation from liquefied wood-based fibers. Ind. Crop. Prod. 2015, 69, 447–455. [Google Scholar] [CrossRef]
- Ma, X.J.; Zhang, F.; Wei, L.Q. Effect of wood charcoal contents on the adsorption property, structure, and morphology of mesoporous activated carbon fibers derived from wood liquefaction process. J. Mater. Sci. 2015, 50, 1908–1914. [Google Scholar] [CrossRef]
- Ma, X.J.; Yang, H.; Yu, L.; Chen, Y.; Li, Y. Preparation, surface and pore structure of high surface area activated carbon fibers from bamboo by steam activation. Materials 2014, 7, 4431–4441. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.G.; Zhang, Z.S. Synthesis and characterization of magnetically separable Ag/AgCl-magnetic activated carbon composites for visible light induced photocatalytic detoxification and disinfection. Appl. Catal. B Environ. 2014, 267–278. [Google Scholar] [CrossRef]
- Zhang, S.L.; Tao, L.C.; Jiang, M.; Gou, G.J.; Zhou, Z.W. Single-step synthesis of magnetic activated carbon from peanut shell. Mater. Lett. 2015, 157, 281–284. [Google Scholar] [CrossRef]
- Bai, X.; Qin, C.D.; Huang, X. Voltammetric determination of chloramphenicol using a carbon fiber microelectrode modified with Fe3O4 nanoparticles. Microchim. Acta 2016, 183, 2973–2981. [Google Scholar] [CrossRef]
- Li, C.D.; Lu, J.J.; Li, S.M.; Tong, Y.B.; Ye, B.C. Synthesis of Magnetic Microspheres with Sodium Alginate and Activated Carbon for Removal of Methylene Blue. Materials 2017, 10, 84. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.Q.; Li, A.M.; Zhou, Q.; Shuang, C.D.; Li, Y.; Ma, Y. Utilization of waste cation exchange resin to prepare carbon/iron composites for the adsorption of contaminants in water. J. Ind. Eng. Chem. 2014, 20, 4256–4260. [Google Scholar] [CrossRef]
- Yang, N.; Zhu, S.M.; Zhang, D.; Xu, S. Synthesis and properties of magnetic Fe3O4-activated carbon nanocomposite particles for dye removal. Mater. Lett. 2008, 62, 645–647. [Google Scholar] [CrossRef]
- Majeed, M.I.; Guo, J.; Yan, W.; Tan, B. Preparation of magnetic iron oxide nanoparticles (MIONs) with improved saturation magnetization using multifunctional polymer ligand. Polymers 2016, 8, 392. [Google Scholar] [CrossRef]
- Hai, H.; Grinblat, J.; Sougrati, M.T.; Stievano, L.; Margel, S. Engineering of Iron-Based Magnetic Activated Carbon Fabrics for Environmental Remediation. Materials 2015, 8, 4593. [Google Scholar]
- Chen, H.Y.; Du, Y.; Lu, Q. Microwave-assisted rapid synthesis of Fe2O3/ACF hybrid for high efficient As(V) removal. J. Alloy. Compd. 2016, 674, 399–405. [Google Scholar] [CrossRef]
- Ma, X.J.; Zhao, G.J. Preparation of carbon fibers from liquefied wood. Wood Sci. Technol. 2010, 44, 3–11. [Google Scholar]
- Ma, X.J.; Zhao, G.J. Variations in the microstructure of carbon fibers prepared from liquefied wood during carbonization. J. Appl. Polym. Sci. 2011, 121, 3525–3530. [Google Scholar] [CrossRef]
- Ma, X.J.; Zhang, F.; Zhu, J.Y.; Yu, L.L.; Liu, X.Y. Preparation of highly developed mesoporous activated carbon fiber from liquefied wood using wood charcoal as additive and its adsorption of methylene blue from solution. Bioresour. Technol. 2014, 164, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.J.; Chen, Y. Preparation and characterization of Mn/N co-doped TiO2 loaded on wood-based activated carbon fiber and its visible light photodegradation. Polymers 2015, 7, 1660–1673. [Google Scholar] [CrossRef]
- Foo, K.Y.; Hameed, B.H. Mesoporous activated carbon from wood sawdust by K2CO3 activation using microwave heating. Bioresour. Technol. 2012, 111, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.J.; Frank, C. Recent Advances in Studies of Carbon Fiber Structure. Philos. Trans. 1980, 294, 443–449. [Google Scholar] [CrossRef]
- Cuesta, A.; Dhamelincout, P.; Laureyns, J.; Martinezalonso, A.; Tascon, J.M.D. Comparative performance of X-ray diffraction and Raman microprobe techniques for the study of carbon materials. J. Mater. Chem. 1998, 8, 2875–2879. [Google Scholar] [CrossRef]
- Zou, D.Y.; Zhang, B.H.; Liao, B.G.; Chen, C.G.; Zhang, Q. Study of determining the degree of graphitizing of coke with X-ray diffraction method. J. Chongqing Univ. 1988, 6, 83–93. [Google Scholar]
- Anirudhan, T.S.; Sreekumari, S.S. Adsorptive removal of heavy metal ions from industrial effluents using activated carbon derived from waste coconut buttons. J. Environ. Sci. 2011, 23, 1989–1998. [Google Scholar] [CrossRef]
- Groen, J.C.; Peffer, L.A.; Javier, P.R. Pore size determination in modified micro-and mesoporous materials. Pitfalls and limitations in gas adsorption data analysis. Microporous Mesoporous Mater. 2003, 60, 1–17. [Google Scholar] [CrossRef]
- Elmerraoui, M.; Aoshima, A.M.; Kaneko, K. Micropore Size Distribution of Activated Carbon Fiber Using the Density Functional Theory and Other Methods. Langmuir 2000, 16, 4300–4304. [Google Scholar] [CrossRef]
- Kubo, S.; Yoshida, T.; Kadia, J.F. Surface porosity of lignin/PP blend carbon fibers. J. Wood Chem. Technol. 2007, 27, 257–271. [Google Scholar] [CrossRef]
- Li, G.L.; Jiang, Y.R.; Huang, K.L.; Ding, P.; Yao, L.L. Kinetics of adsorption of Saccharomycescerevisiae mandelated dehydrogenase on magnetic Fe3O4–chitosannanoparticles. Colloids Surf. A 2008, 320, 11–18. [Google Scholar] [CrossRef]
- Dai, C.C.; Wan, J.F.; Shao, J.Q.; Ma, F.W. Hollow activated carbon with unique through-pore structure derived from reed straw for high-performance supercapacitors. Mater. Lett. 2017, 193, 279–282. [Google Scholar] [CrossRef]
- Qiang, C.W.; Xu, J.C.; Zhang, Z.Q.; Tian, L.L.; Xiao, S.T.; Liu, Y.; Xu, P. Magnetic properties and microwave absorption properties of carbon fibers coated by Fe3O4 nanoparticles. J. Alloys Compd. 2010, 506, 93–97. [Google Scholar] [CrossRef]
- Ramesh, T.; Rajalakshmi, N.; Dhathathreyan, K.S. Synthesis and characterization of activated carbon from jutefibers for hydrogen storage. Renew. Energy Environ. Sustain. 2017, 2, 27–36. [Google Scholar] [CrossRef]
- Ge, X.Y.; Wu, Z.S.; Wu, Z.L.; Yan, Y.J.; Cravotto, G.C.; Ye, B.C. Microwave-assisted modification of activated carbon with ammonia for efficient pyrene adsorption. J. Ind. Eng. Chem. 2016, 39, 27–36. [Google Scholar] [CrossRef]
- Jin, G.; Eom, Y.J.; Lee, T.G. Removal of Hg(II) from aquatic invironments using activated carbon impregnated with humic acid. J. Ind. Eng. Chem. 2016, 42, 46–52. [Google Scholar] [CrossRef]
- Lee, Y.S.; Basova, Y.V.; Edie, D.D.; Reid, L.K.; Newcombe, S.R.; Ryu, S.K. Preparation and characterization of trilobal activated carbon fibers. Carbon 2003, 41, 2573–2584. [Google Scholar] [CrossRef]
- Zhang, S.J.; Feng, H.M.; Wang, J.P.; Yu, H.Q. Structure evolution and optimization in the fabrication of PVA-based activated carbon fibers. J. Colloid Interface Sci. 2008, 321, 96–102. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fe3O4 Content | d(002)/nm | Lc(002)/nm | La(100)/nm | Lc/d(002) | g/% |
|---|---|---|---|---|---|
| 0% | 0.4931 | 0.9561 | 3.232 | 1.939 | −17.34 |
| 0.5% | 0.5657 | 0.9350 | 3.027 | 1.653 | −25.78 |
| 1.0% | 0.5491 | 0.9554 | 3.625 | 1.739 | −23.86 |
| 1.5% | 0.4635 | 0.9748 | 3.809 | 2.103 | −13.89 |
| 2.0% | 0.4553 | 1.003 | 4.239 | 2.203 | −12.94 |
| 2.5% | 0.3237 | 1.064 | 4.303 | 3.287 | 2.36 |
| Fe3O4 Content | 0.5% | 1% | 1.5% | 2% | 2.5% |
| Average Crystallite Size | 18.73 nm | 14.22 nm | 11.13 nm | 11.45 nm | 10.58 nm |
| Fe3O4 Content | SBET (m2/g) | Smicro (m2/g) | Smeso (m2/g) | Vtot (m2/g) | Vmicro (m2/g) | Vmeso (m2/g) | MP-Ratio (%) |
|---|---|---|---|---|---|---|---|
| 0% | 1173 | 1090 | 64 | 0.551 | 0.501 | 0.040 | 7 |
| 0.5% | 1894 | 1667 | 203 | 0.858 | 0.702 | 0.142 | 17 |
| 1.0% | 1816 | 1550 | 203 | 0.836 | 0.661 | 0.154 | 18 |
| 1.5% | 1657 | 1380 | 241 | 0.737 | 0.531 | 0.181 | 25 |
| 2.0% | 1716 | 1377 | 299 | 0.896 | 0.561 | 0.300 | 33 |
| 2.5% | 1578 | 1206 | 322 | 0.929 | 0.474 | 0.415 | 45 |
| Fe3O4 Content | 0.5% | 1.0% | 1.5% | 2.0% | 2.5% |
|---|---|---|---|---|---|
| Residual Magnetization (emu/g) | - | - | 0.0037 | 0.0041 | 0.0075 |
| Saturation Magnetization (emu/g) | - | - | 0.0129 | 0.0177 | 0.0440 |
| Coercive Force (Oe) | - | - | 270.4 | 261.3 | 224.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Li, J.; Ren, B.; Li, T.; Ma, X. Synthesis and Characterization of Wooden Magnetic Activated Carbon Fibers with Hierarchical Pore Structures. Polymers 2018, 10, 435. https://doi.org/10.3390/polym10040435
Li D, Li J, Ren B, Li T, Ma X. Synthesis and Characterization of Wooden Magnetic Activated Carbon Fibers with Hierarchical Pore Structures. Polymers. 2018; 10(4):435. https://doi.org/10.3390/polym10040435
Chicago/Turabian StyleLi, Dongna, Jianing Li, Biyun Ren, Tongtong Li, and Xiaojun Ma. 2018. "Synthesis and Characterization of Wooden Magnetic Activated Carbon Fibers with Hierarchical Pore Structures" Polymers 10, no. 4: 435. https://doi.org/10.3390/polym10040435
APA StyleLi, D., Li, J., Ren, B., Li, T., & Ma, X. (2018). Synthesis and Characterization of Wooden Magnetic Activated Carbon Fibers with Hierarchical Pore Structures. Polymers, 10(4), 435. https://doi.org/10.3390/polym10040435