Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends



Abstract
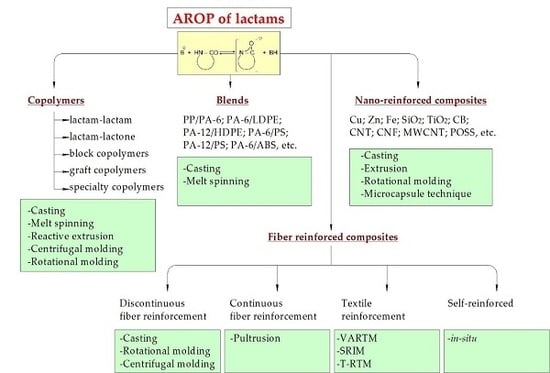
1. Introduction
2. Homopolymers
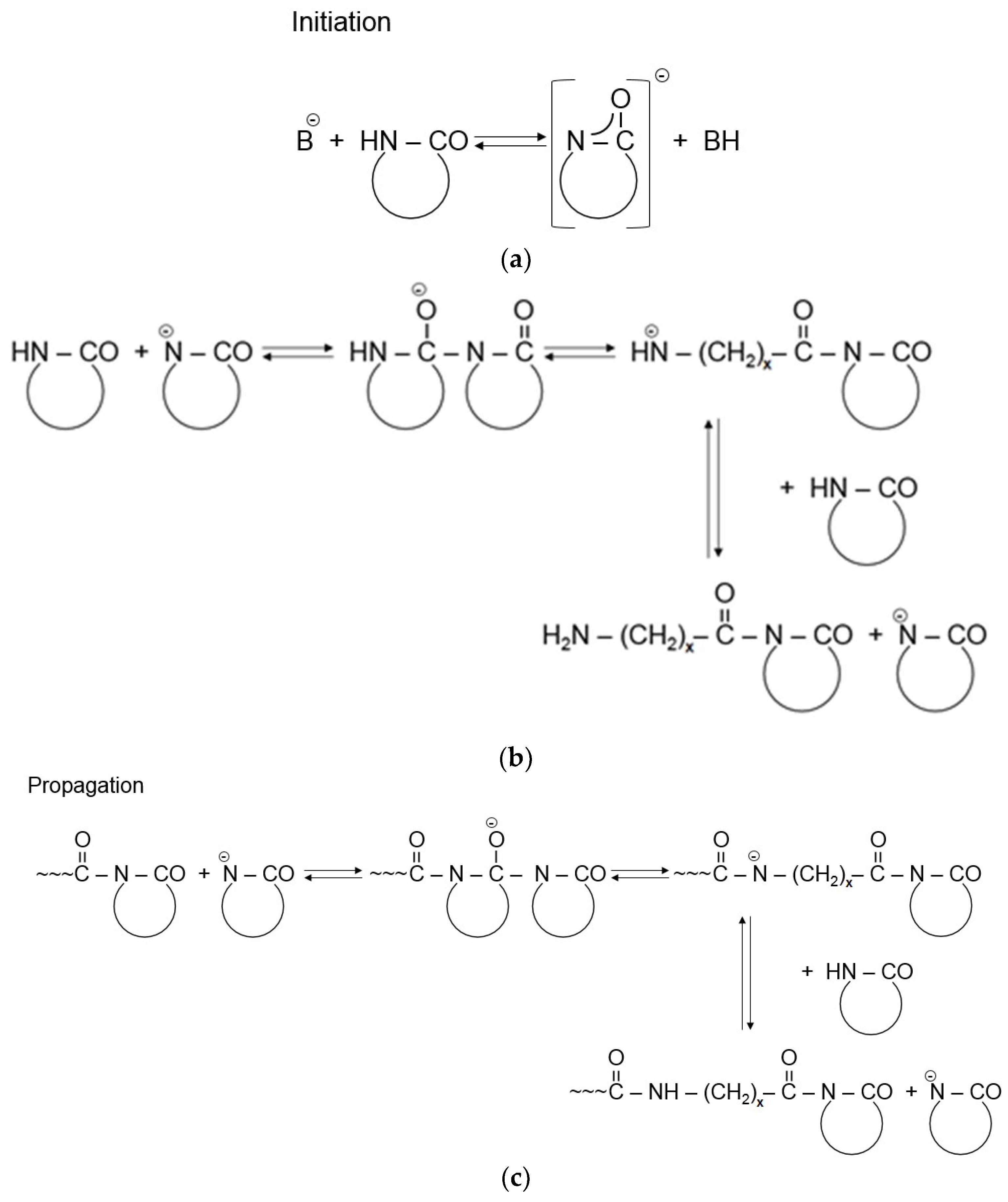
2.1. Chemistry
2.2. Properties
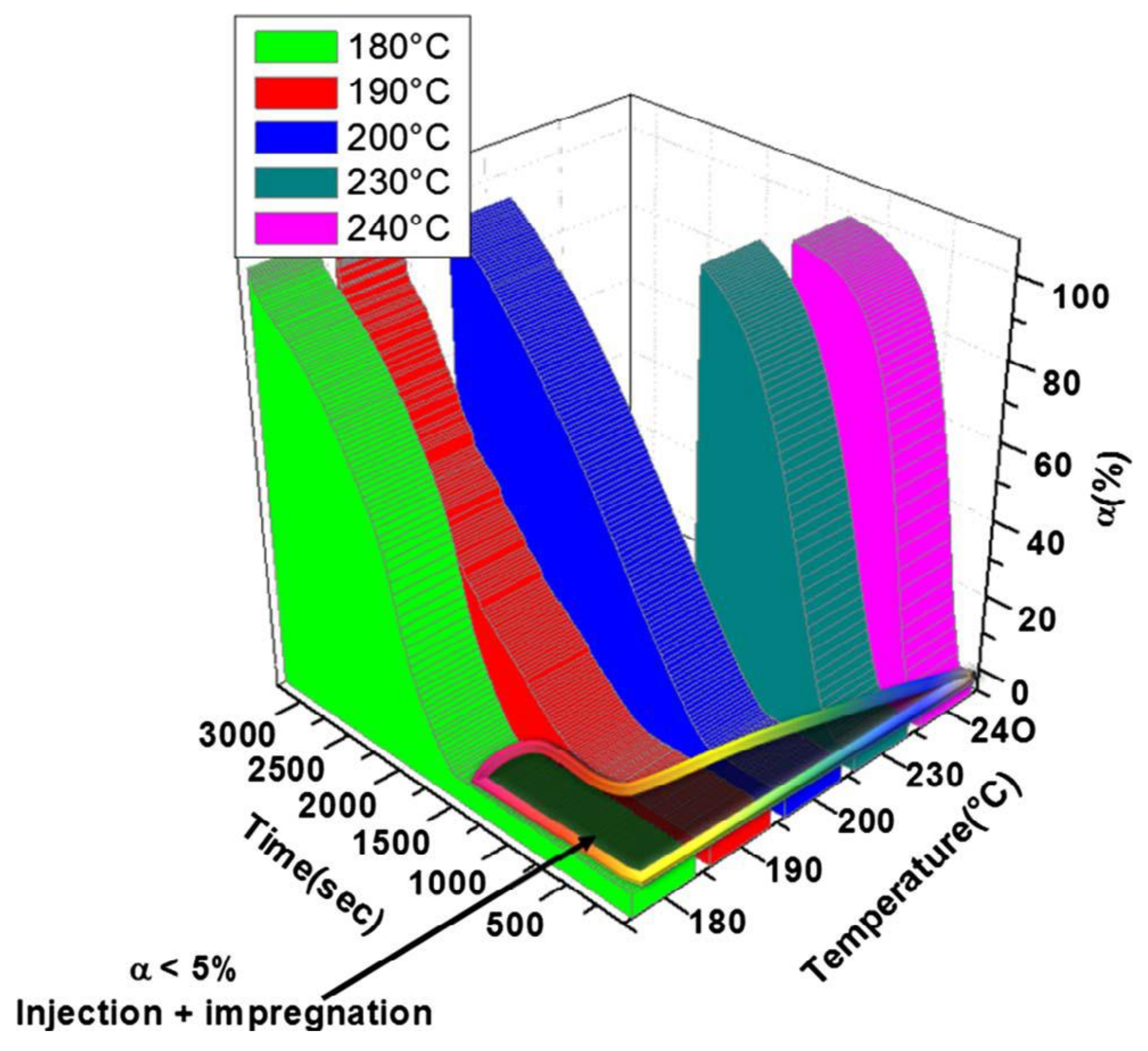
2.3. Manufacturing
3. Copolymers
3.1. Chemical and Structural Aspects
3.1.1. Lactam-Lactam Copolymers
3.1.2. Lactam-Lactone Copolymers
3.1.3. Block Copolymers
3.1.4. Graft Copolymers
3.1.5. Specialty Copolymers
3.2. Manufacturing
4. Blends
5. Nanocomposites
6. Composites
6.1. Discontinuous Fiber Reinforced
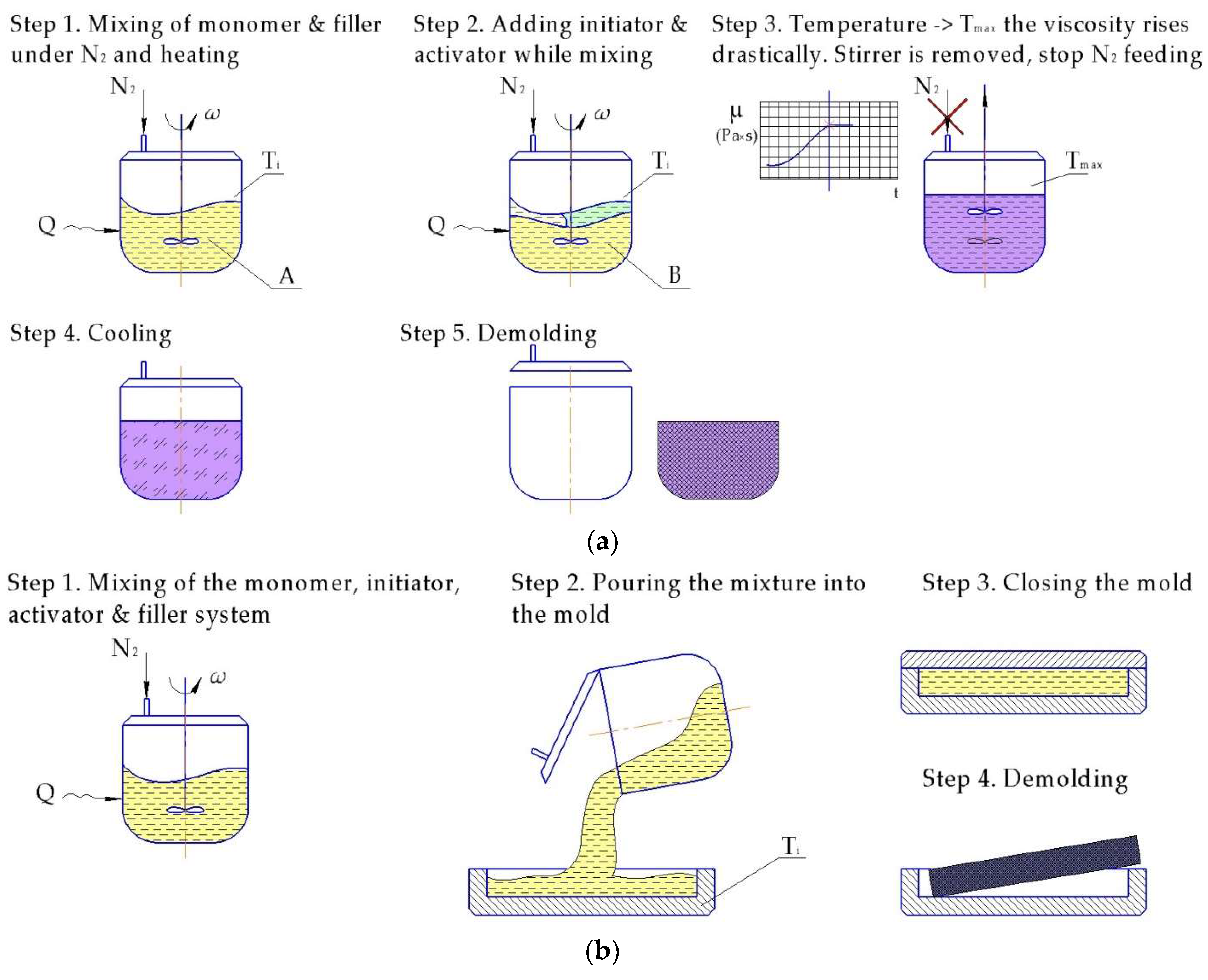
6.1.1. Casting
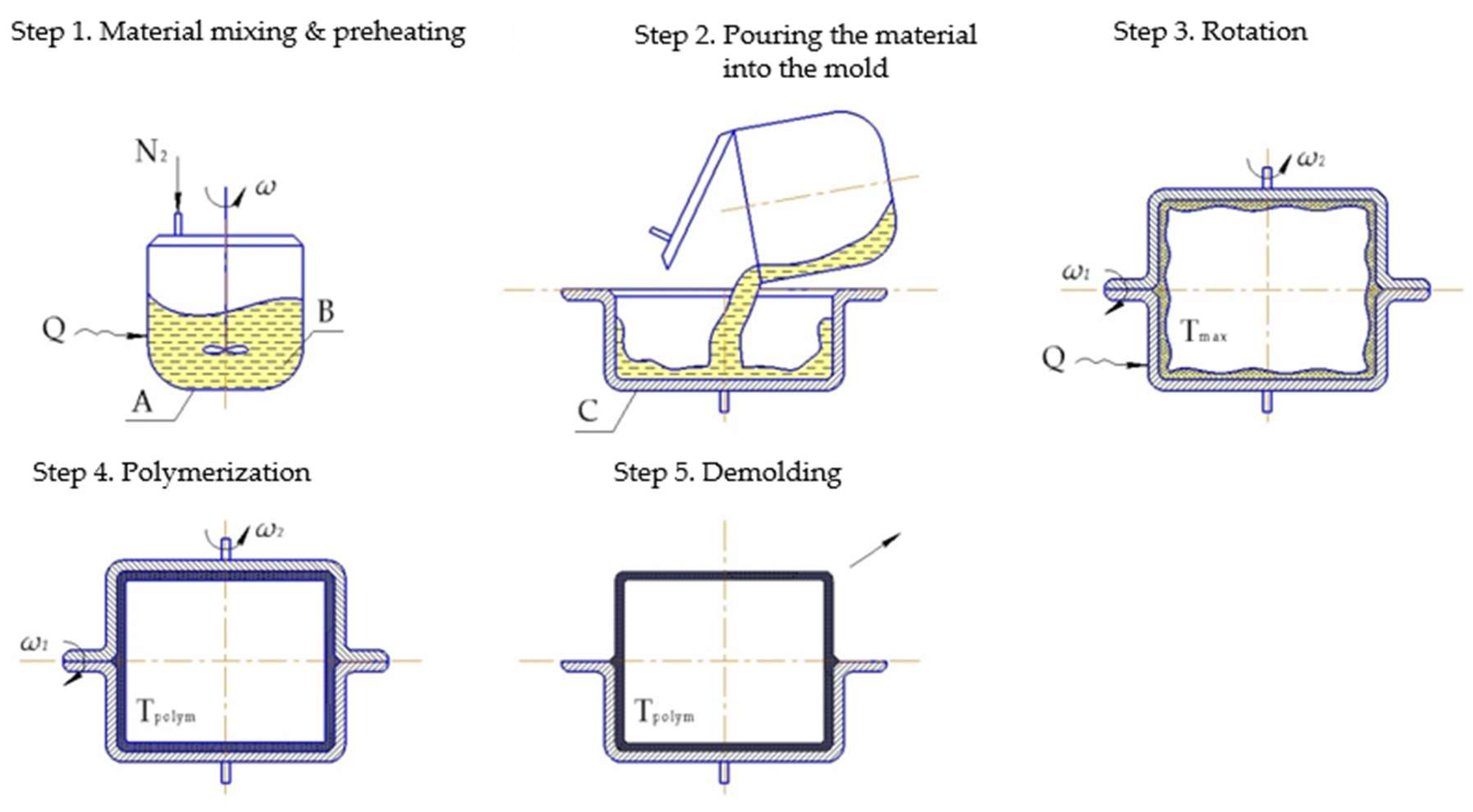
6.1.2. Rotation Molding
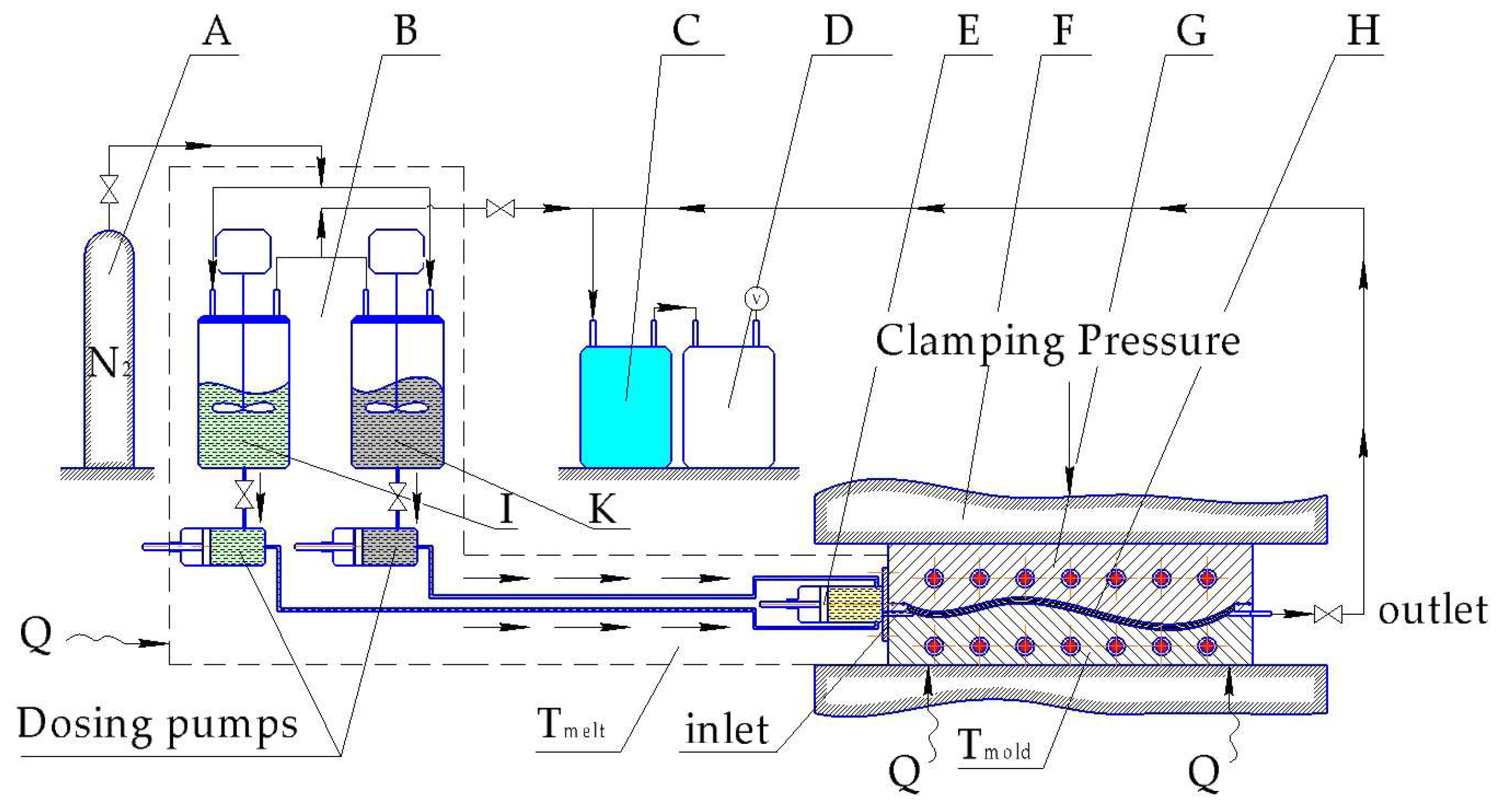
6.2. Continuous Fiber Reinforced.
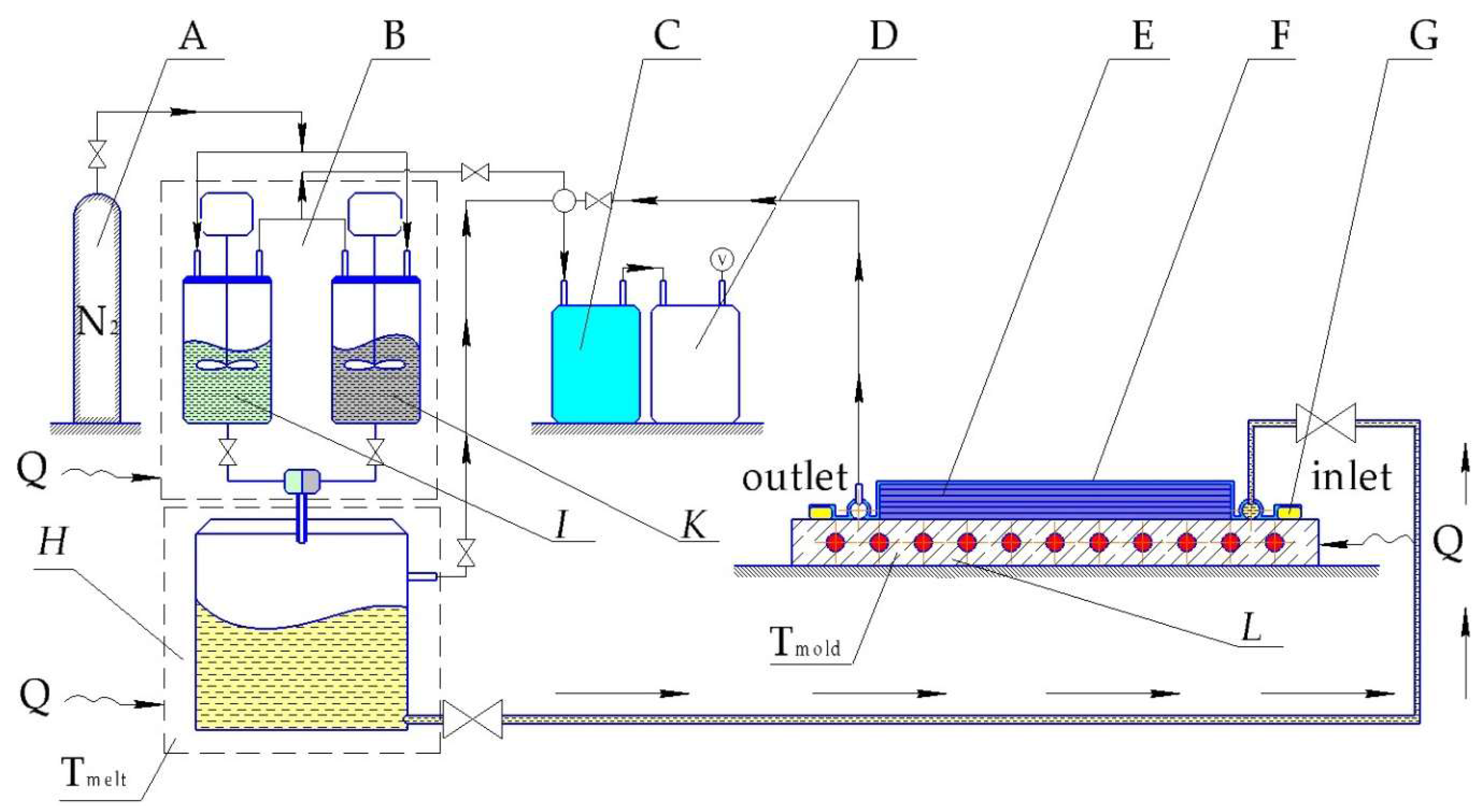
Pultrusion
6.3. Textile Reinforced
6.4. Single-Polyamide Comosites
7. Outlook and Future Trends
- -
- CL remains the preferred monomer for AROP and in the future. The present commercially available initiators and activators will be used in the near future, and attempts will be made to reduce their “environmental” (i.e., humidity) sensitivity. Solvent-borne, liquid initiators and activators may likely be preferred. Search for effective latent initiators/activators will be under the spotlight of research in academia targeting the use of a one-component system that is prone to “polymerization on demand”. The copolymerization strategy will further focus on the toughness improvement of the related PA-6-based (block) copolymers. Besides the traditional block segments (polyether- and polyester-based diols) others, like polycaprolactone, polylactic acid etc. may be incorporated in order to enhance the renewable content and support biodegradability. The in situ blending via AROP will hardly achieve industrial breakthrough.
- -
- Vigorous development can be predicted for PA-6-based nanocomposites produced through AROP thereby making use of the “grafting from” (i.e., transforming the surface of the nanofillers into a suitable “nanoactivator” for grafting the CL chains) approach. This development will target the production of new tribological compounds, containing novel carbonaceous nanofillers, which will be most likely produced still by casting. The toughness of such nanocomposites will be a key factor and thus the related works will be supported by extensive modeling [198]. According to our view, novel and adapted manufacturing methods will be the real future drivers of the development of thermoplastic composites with AROP-produced matrix. Additive manufacturing via ink jetting should be mentioned among the emerging novel techniques. Among the “adapted” techniques a bright future can be predicted for thermoplastic reaction injection pultrusion (TRI-pultrusion), thermoplastic resin transfer molding (T-RTM), and other liquid composite molding procedures. This claim is based not only on the straightforward recyclability of the related composite parts, but also on other beneficial design- and post-processing-related features, such as part integration, overmolding (with and without additional reinforcements), surface coating and finishing, and welding. The related research and developments works will run parallel with extensive modeling (especially via finite element codes) studies. The potential of PA-6-based single-polymer (self-reinforced) composites has been strongly underestimated, therefore in this field interesting developments may be expected.
Acknowledgments
Conflicts of Interest
Abbreviations
| 1H-NMR | proton nuclear magnetic resonance |
| ABS | acrylonitrile-butadiene-styrene |
| AE | acoustic emission |
| AFM | atomic force microscopy |
| AM | additive manufacturing |
| AROP | anionic (activated) ring-opening polymerization |
| C1 | Bruggolen C1 (initiator: CLMgBr) |
| C10 | Bruggolen C10 (initiator: NaCL) |
| C20 | Bruggolen C20P (activator: hexamethylene-1.6-dicarbomoylcaprolactam) |
| CB | carbon black |
| CBT | cyclic butylene terephthalate |
| CF | carbon fiber |
| CL | ε-caprolactam |
| CLMgBr | ε-caprolactam magnesium bromide |
| CNC | nanocrystalline cellulose |
| CNF | carbon nanofibers |
| CNT | carbon nanotubes |
| CTT | conversion-temperature transformation |
| DMA | dynamic mechanical analysis |
| DMF | dimethylformamide |
| DOC | degree of conversion |
| dpolym | length of the polymerization part of the pultrusion die |
| DSC | differential scanning calorimetry |
| E | Young’s modulus |
| Fpul | pulling force |
| FR | flow rate |
| FTIR | Fourier-transform infrared spectroscopy |
| G | shear modulus |
| GF | glass fiber |
| GO | graphene oxide |
| GPC | gel permeation chromatography |
| gsm | gram per square meter (surface weight) |
| HDT | heat distortion temperature |
| HMDI | hexamethylene di-isocyanate |
| HSIMT | high speed impact-bending test |
| ILSS | interlaminar shear strength |
| IS | impact strength |
| LCM | liquid composite molding |
| LDPE | low density polyethylene |
| LDPE-g-MA | LDPE grafted by maleic anhydride |
| LL | ω-laurolactam |
| LP-RTM | low pressure resin transfer molding |
| MA | maleic anhydride |
| MC | microcapsulation |
| MMA | methyl methacrylate |
| MMT | montmorillonite clay |
| Mn | number average MW |
| Mv | viscosity average MW |
| Mw | weight average MW |
| MW | molecular weight |
| MWCNT | multiwall carbon nanotube |
| NaCL | sodium caprolactamate |
| NBC | nylon block copolymer |
| NBR | acrylonitrile-butadiene rubber |
| NMP | N-methyl-2-pyrrolidone |
| NMR | nuclear magnetic resonance |
| OMMT | organophilic-modified MMT clay |
| PA | polyamide |
| PA-12 | polyamide-12 |
| PA-6 | polyamide-6 |
| PA-6,6 | polyamide-6,6 |
| PBT | polybutylene terephthalate |
| PCL | polycaprolactone |
| Pclamp | clamping pressure |
| PDMS | polydimethylsiloxane |
| phr | parts per hundred parts of resin |
| PI | polyimide |
| PIC | phenyl isocyanate (also as cyclic trimer) |
| Pinj | injection pressure |
| PMMA | polymethyl methacrylate |
| POSS | polyhedral oligomeric silsequioxane |
| PP | polypropylene |
| Pp | process pressure |
| PPE | polyphenylene ether |
| PPG | polypropyleneglycol |
| PP-g-MA | PP grafted by maleic anhydride |
| PP-g-PA-6 | PP grafted by PA-6 |
| PS | polystyrene |
| R/r | mold major and minor radius |
| RH | relative humidity |
| RIM | reaction injection molding |
| RISP | reaction-induced phase separation |
| RMC | residual monomer content |
| RT | room temperature |
| RTM | resin transfer molding |
| SAN | styrene-acrylonitrile |
| SAXS | small angle X-ray scattering |
| SCF | short carbon fibers |
| SEBS | styrene-ethylene-butylene-styrene |
| SEBS-g-MA | SEBS grafted by maleic anhydride |
| SEC | size exclusion chromatography |
| SEM | scanning electron microscopy |
| SMA | styrene-maleic anhydride |
| SPC | self-reinforced composite |
| SRIM | structural reaction injection molding |
| SWCNT | single-walled carbon nanotubes |
| T0 | initial temperature |
| Tc | crystallization temperature |
| Tc | crystallization temperature |
| Tcool | cooling temperature |
| tcycle | cycle time |
| TDI | toluene 2,4-diisocyanate |
| TEM | transmission electron microscopy |
| Tf | fusion temperature |
| Tg | glass transition temperature |
| TGA | thermogravimetric analysis |
| tinj | injection time |
| Tm | melting temperature |
| Tmax | process maximum temperature |
| Tmold | mold temperature |
| TPC | thermoplastic composites |
| tpolym | polymerization time |
| Tproc | processing temperature |
| TPU | thermoplastic polyurethane |
| Trc | recrystallization temperature |
| TRI-pultrusion | thermoplastic reaction injection pultrusion |
| TTT | time-temperature-transformation |
| VC | void content |
| Vf | fiber (reinforcement) volume content |
| Vpul | pultrusion line speed |
| WAXS | wide angle X-ray scattering |
| Wf | fiber (reinforcement) weight fraction |
| XC | degree of crystallinity |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
| ΔHf | heat of fusion |
| εR | elongation at break |
| η | intrinsic viscosity |
| μ | viscosity |
| ρ | density |
| σ- | ultimate tensile strength |
| ω | rotation speed of the rotors |
References
- Roda, J. Polyamides. In Handbook of Ring-Opening Polymerization; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 165–195. [Google Scholar]
- Joyce, R.M.; Ritter, D.M. Process for Making Polymeric Materials. U.S. Patent 2,251,519, 5 August 1941. [Google Scholar]
- Su, W.-F. Ring-opening polymerization. In Principles of Polymer Design and Synthesis; Springer: Berlin, Heidelberg, 2013; pp. 267–299. [Google Scholar]
- Šebenda, J. Lactam polymerization. J. Macromol. Sci. Chem. 1972, 6, 1145–1199. [Google Scholar] [CrossRef]
- Reimschuessel, H.K. Nylon 6. Chemistry and mechanisms. J. Polym. Sci. Macromol. Rev. 1977, 12, 65–139. [Google Scholar] [CrossRef]
- Endo, T.; Sudo, A. Development and application of novel ring-opening polymerizations to functional networked polymers. J. Polym. Sci. Part A 2009, 47, 4847–4858. [Google Scholar] [CrossRef]
- Zhao, J.; Hadjichristidis, N.; Gnanou, Y. Phosphazene-promoted anionic polymerization. Polimery 2014, 59, 49–59. [Google Scholar] [CrossRef]
- Zhang, J.; Gellman, S.H.; Stahl, S.S. Kinetics of anionic ring-opening polymerization of variously substituted β-lactams: Homopolymerization and copolymerization. Macromolecules 2010, 43, 5618–5626. [Google Scholar] [CrossRef]
- Keul, H.; Höcker, H. Expected and unexpected reactions in ring-opening (co)polymerization. Macromol. Rapid Commun. 2000, 21, 869–883. [Google Scholar] [CrossRef]
- Ricco, L.; Casazza, E.; Mineo, P.; Russo, S.; Scamporrino, E. Nature of a low molar mass peak in anionic poly(ε-caprolactam). Main aspects of its formation. Macromolecules 2008, 41, 3904–3911. [Google Scholar] [CrossRef]
- Bernat, P.; Hladka, O.; Fismanova, M.; Roda, J.; Brozek, J. Polymerization of lactams. 98: Influence of water on the non-activated polymerization of ε-caprolactam. Eur. Polym. J. 2008, 44, 32–41. [Google Scholar] [CrossRef]
- Piskun, Y.A.; Vasilenko, I.V.; Gaponik, L.V.; Kostjuk, S.V. Activated anionic ring-opening polymerization of ε-caprolactam with magnesium di(ε-caprolactamate) as initiator: Effect of magnesium halides. Polym. Bull. 2012, 68, 1501–1513. [Google Scholar] [CrossRef]
- Kříž, J.; Stehlíček, J.; Dybal, J.; Hauer, J. Study of the propagation centre in the anionic polymerization of lactams, 2. Model study of the interaction between an activated monomer and N-acyllactam in dimethyl sulfoxide. Macromol. Chem. Phys. 1996, 197, 483–495. [Google Scholar] [CrossRef]
- Luisier, A.; Bourban, P.-E.; Månson, J.-A.E. Time-temperature-transformation diagram for reactive processing of polyamide 12. J. Appl. Polym. Sci. 2001, 81, 963–972. [Google Scholar] [CrossRef]
- Russo, S.; Maniscalco, S.; Ricco, L. Some new perspectives of anionic polyamide 6 (apa 6) synthesis. Polym. Adv. Technol. 2015, 26, 851–854. [Google Scholar] [CrossRef]
- Chang, W.L.; Frisch, K.C.; Ashida, K. Anionic polymerization of star-shaped nylon 6 with a trifunctional initiator. J. Polym. Sci. Part A 1989, 27, 3637–3649. [Google Scholar] [CrossRef]
- Zhu, N.; Gong, H.; Han, W.; Zeng, W.-B.; Wang, H.-X.; Fang, Z.; Li, X.; Zhang, K.; Li, Z.-J.; Guo, K. Synthesis and characterization of star-branched polyamide 6 via anionic ring-opening polymerization with N,N′,N″-trimesoyltricaprolactam as a multifunctional activator. Chin. Chem. Lett. 2015, 26, 1389–1392. [Google Scholar] [CrossRef]
- Mateva, R.; Dencheva, N. On the behavior of organophosphorus lactam derivatives during anionic polymerization of ε-caprolactam. J. Polym. Sci. Part A 1992, 30, 1449–1462. [Google Scholar] [CrossRef]
- Naumann, S.; Epple, S.; Bonten, C.; Buchmeiser, M.R. Polymerization of ε-caprolactam by latent precatalysts based on protected n-heterocyclic carbenes. ACS Macro Lett. 2013, 2, 609–612. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Szafner, A. Organosilicon crown compounds in the anionic polymerization of ε-caprolactam. Makromol. Chem. 1977, 179, 519–522. [Google Scholar] [CrossRef]
- Zabegaeva, O.N.; Volkova, T.V.; Shaplov, A.S.; Lozinskaya, E.I.; Afonicheva, O.V.; Buzin, M.I.; Sinitsyna, O.V.; Vygodskii, Y.S. Anionic ring-opening polymerization of ε-caprolactam in the presence of an ionic liquid. In Unique Properties of Polymers and Composites: Pure and Applied Science Today and Tomorrow; Hauppauge: New York, NY, USA, 2012; Volume 2, pp. 91–105. [Google Scholar]
- Zhang, C.-L.; Feng, L.-F.; Hu, G.-H. Anionic polymerization of lactams: A comparative study on various methods of measuring the conversion of ε-caprolactam to polyamide 6. J. Appl. Polym. Sci. 2006, 101, 1972–1981. [Google Scholar] [CrossRef]
- Mendichi, R.; Russo, S.; Ricco, L.; Giacometti Schieroni, A. Hexafluoroisopropanol as size exclusion chromatography mobile phase for polyamide 6. J. Sep. Sci. 2004, 27, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Wollny, A.; Nitz, H.; Faulhammer, H.; Hoogen, N.; Mülhaupt, R. In situ formation and compounding of polyamide 12 by reactive extrusion. J. Appl. Polym. Sci. 2003, 90, 344–351. [Google Scholar] [CrossRef]
- Luisier, A.; Bourban, P.-E.; Månson, J.-A.E. Initiation mechanisms of an anionic ring-opening polymerization of lactam-12. J. Polym. Sci. Part A 2002, 40, 3406–3415. [Google Scholar] [CrossRef]
- Abt, T.; Sánchez-Soto, M. A review of the recent advances in cyclic butylene terephthalate technology and its composites. Crit. Rev. Solid State Mater. Sci. 2017, 42, 173–217. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Kiss, L. Attempts of separation of the polymerization and crystallization processes by means of dsc thermograms of activated anionic polymerization of ε-caprolactam. Makromol. Chem. 1979, 180, 1593–1597. [Google Scholar] [CrossRef]
- Khodabakhshi, K.; Gilbert, M.; Fathi, S.; Dickens, P. Anionic polymerisation of caprolactam at the small-scale via dsc investigations. J. Therm. Anal. Calorim. 2014, 115, 383–391. [Google Scholar] [CrossRef]
- Vicard, C.; De Almeida, O.; Cantarel, A.; Bernhart, G. Experimental study of polymerization and crystallization kinetics of polyamide 6 obtained by anionic ring opening polymerization of ε-caprolactam. Polymer 2017, 132, 88–97. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Shang, P.P.; Mohd Ishak, Z.A.; Rösch, M. Melting and crystallization of in-situ polymerized cyclic butylene terephthalates with and without organoclay: A modulated dsc study. eXPRESS Polym. Lett. 2007, 1, 60–68. [Google Scholar] [CrossRef]
- Faridirad, F.; Ahmadi, S.; Barmar, M.D.; Naderi, G. Statistical analysis of the catalyst and activator level on in situ polymerization of laurolactam during reactive melt blending. J. Thermodyn. Catal. 2016, 7, 7. [Google Scholar]
- Russo, S.; Maniscalco, S.; Moretti, P.; Ricco, L. Fast-activated anionic polymerization of ε-caprolactam in the bulk under quasi-adiabatic conditions: Comparison of different kinetic models. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4474–4480. [Google Scholar] [CrossRef]
- Kamal, M.R.; Sourour, S. Kinetics and thermal characterization of thermoset cure. Polym. Eng. Sci. 1973, 13, 59–64. [Google Scholar] [CrossRef]
- Harsch, M.; Karger-Kocsis, J.; Holst, M. Influence of fillers and additives on the cure kinetics of an epoxy/anhydride resin. Eur. Polym. J. 2007, 43, 1168–1178. [Google Scholar] [CrossRef]
- Teuwen, J.J.E.; van Geenen, A.A.; Bersee, H.E.N. Novel reaction kinetic model for anionic polyamide-6. Macromol. Mater. Eng. 2013, 298, 163–173. [Google Scholar] [CrossRef]
- Malkin, A.Y.; Frolov, V.G.; Ivanova, A.N.; Andrianova, Z.S. The nonisothermal anionic polymerization of caprolactam. Polym. Sci. USSR 1979, 21, 691–700. [Google Scholar] [CrossRef]
- Nagy, J.; Reith, L.; Fischlschweiger, M.; Steinbichler, G. Modelling the influence of flow phenomena on the polymerization of ε-caprolactam. Chem. Eng. Sci. 2014, 111, 85–93. [Google Scholar] [CrossRef]
- Simon, S.L.; Gillham, J.K. Thermosetting cure diagrams: Calculation and application. J. Appl. Polym. Sci. 1994, 53, 709–727. [Google Scholar] [CrossRef]
- Maazouz, A.; Lamnawar, K.; Dkier, M. Chemorheological study and in-situ monitoring of pa6 anionic-ring polymerization for rtm processing control. Composites 2018, 107, 235–247. [Google Scholar] [CrossRef]
- Harsch, M.; Karger-Kocsis, J.; Apostolov, A.A. Crystallization-induced shrinkage, crystalline, and thermomechanical properties of in situ polymerized cyclic butylene terephthalate. J. Appl. Polym. Sci. 2008, 108, 1455–1461. [Google Scholar] [CrossRef]
- Khodabakhshi, K.; Gilbert, M.; Dickens, P.; Hague, R. Optimizing conditions for anionic polymerization of caprolactam for inkjetting. Adv. Polym. Technol. 2010, 29, 226–236. [Google Scholar] [CrossRef]
- Khodabakhshi, K.; Gilbert, M.; Dickens, P. Monitoring of small-scale anionic polymerization of caprolactam; a method to be used in an additive manufacturing process. Polym. Adv. Technol. 2013, 24, 503–510. [Google Scholar] [CrossRef]
- Rahim, T.N.A.T.; Abdullah, A.M.; Akil, H.M.; Mohamad, D.; Rajion, Z.A. The improvement of mechanical and thermal properties of polyamide 12 3d printed parts by fused deposition modelling. eXPRESS Polym. Lett. 2017, 11, 963–982. [Google Scholar] [CrossRef]
- Ogila, K.O.; Shao, M.; Yang, W.; Tan, J. Rotational molding: A review of the models and materials. eXPRESS Polym. Lett. 2017, 11, 778–798. [Google Scholar] [CrossRef]
- Rusu, G.; Ueda, K.; Rusu, E.; Rusu, M. Polyamides from lactams by centrifugal molding via anionic ring-opening polymerization. Polymer 2001, 42, 5669–5678. [Google Scholar] [CrossRef]
- Barhoumi, N.; Maazouz, A.; Jaziri, M.; Abdelhedi, R. Polyamide from lactams by reactive rotational molding via anionic ring-opening polymerization: Optimization of processing parameters. eXPRESS Polym. Lett. 2013, 7, 76–87. [Google Scholar] [CrossRef]
- Michaeli, W.; Greefenstein, A.; Berghaus, U. Twin-screw extruders for reactive extrusion. Polym. Eng. Sci. 1995, 35, 1485–1504. [Google Scholar] [CrossRef]
- Kim, I.; White, J.L. Continuous polymerization of ω-lauryl lactam in an intermeshing corotating twin-screw extruder. J. Appl. Polym. Sci. 2005, 97, 1605–1620. [Google Scholar] [CrossRef]
- Kye, H.; White, J.L. Continuous polymerization of caprolactam in a modular intermeshing corotating twin screw extruder integrated with continuous melt spinning of polyamide 6 fiber: Influence of screw design and process conditions. J. Appl. Polym. Sci. 1994, 52, 1249–1262. [Google Scholar] [CrossRef]
- Wu, L.; Jia, Y.; Sun, S.; Zhang, G.; Zhao, G.; An, L. Numerical simulation of reactive extrusion processes of pa6. J. Appl. Polym. Sci. 2007, 103, 2331–2336. [Google Scholar] [CrossRef]
- Verfahren zur Herstellung eines Polyamids durch anionische Polymerisation. German Patent DE 102011007134 A1, 11 October 2012.
- Ricco, L.; Monticelli, O.; Russo, S.; Paglianti, A.; Mariani, A. Fast-activated anionic polymerization of ε-caprolactam in suspension, 1. Role of the continuous phase on characteristics and properties of powdered pa6. Macromol. Chem. Phys. 2002, 203, 1436–1444. [Google Scholar] [CrossRef]
- Crespy, D.; Landfester, K. Anionic polymerization of ε-caprolactam in miniemulsion: Synthesis and characterization of polyamide-6 nanoparticles. Macromol 2005, 38, 6882–6887. [Google Scholar] [CrossRef]
- Mallakpour, S.; Behranvand, V. Polymeric nanoparticles: Recent development in synthesis and application. eXPRESS Polym. Lett. 2016, 10, 895–913. [Google Scholar] [CrossRef]
- Budín, J.; Roda, J.; Brožek, J.; Kříř, J. Anionic copolymerization of ε-caprolactam with ω-laurolactam. Macromol. Symp. 2006, 240, 78–82. [Google Scholar] [CrossRef]
- Ricco, L.; Russo, S.; Orefice, G.; Riva, F. Caprolactam-laurolactam copolymers: Fast activated anionic synthesis, thermal properties and structural investigations. Macromol. Chem. Phys. 2001, 202, 2114–2121. [Google Scholar] [CrossRef]
- Naumann, S.; Schmidt, F.G.; Speiser, M.; Böhl, M.; Epple, S.; Bonten, C.; Buchmeiser, M.R. Anionic ring-opening homo- and copolymerization of lactams by latent, protected N-heterocyclic carbenes for the preparation of PA 12 and PA 6/12. Macromolecules 2013, 46, 8426–8433. [Google Scholar] [CrossRef]
- Vygodskii, Y.S.; Volkova, T.V.; Pashkova, O.N.; Batalova, T.L.; Dubovik, I.I.; Chekulaeva, L.A.; Garbuzova, I.A. Anionic polymerization of ε-caprolactam and its copolymerization with w-dodecanelactam in the presence of aromatic polyimides. Polym. Sci. Ser. A 2006, 48, 557–562. [Google Scholar] [CrossRef]
- Merna, J.; Chromkova, D.; Brozek, J.; Roda, J. Polymerization of lactams: 97. Anionic polymerization of ε-caprolactam activated by esters. Eur. Polym. J. 2006, 42, 1569–1580. [Google Scholar] [CrossRef]
- Kim, I.; White, J.L. Reactive copolymerization of various monomers based on lactams and lactones in a twin-screw extruder. J. Appl. Polym. Sci. 2005, 96, 1875–1887. [Google Scholar] [CrossRef]
- Kim, B.J.; White, J.L. Continuous polymerization of lactam-lactone block copolymers in a twin-screw extruder. J. Appl. Polym. Sci. 2003, 88, 1429–1437. [Google Scholar] [CrossRef]
- Fang, X.; Hutcheon, R.; Scola, D.A. Microwave syntheses of poly(ε-caprolactam-co-ε-caprolactone). J. Polym. Sci. Part A 2000, 38, 1379–1390. [Google Scholar] [CrossRef]
- Klitschke, J.; Bergmann, K. Composition for Anionic Lactam Polymerization. U.S. Patent 9,290,621 B2, 22 March 2016. [Google Scholar]
- Ulrich, H. Speciality monomers. In Reaction Polymers; Gum, W.F., Riese, W., Ulrich, H., Eds.; Hanser Publishers: Munchen, Germany, 1992; pp. 217–220. [Google Scholar]
- Van Geenen, A.; Bongers, J.M. Process for Preparing an Acyl-Lactam Compound. Canadian Patent CA1252089 A, 4 April 1989. [Google Scholar]
- Lozano-González, M.J.; González-De Los Santos, E.A.; Johnson, A.F.; Tsui, S.W. Improvement of dimensional stability of nylon-6 block copolymer using phenolic resin by reaction injection molding. J. Appl. Polym. Sci. 1998, 70, 1811–1816. [Google Scholar] [CrossRef]
- Estrada-Monje, A.; Navarro-Rodríguez, D. Nylon 6-polyesteramide block copolymers (NBC) and NBC/Phenolic resin composites, 1. synthesis, thermal and mechanical properties. Macromol. Mater. Eng. 2004, 289, 933–941. [Google Scholar] [CrossRef]
- Gabbert, J.D.; Garner, A.Y.; Hedrick, R.M. Acid Halide and Acyllactam Functional Materials and the Process for the Preparation of Nylon Block Polymers Therewith. Canadian Patent CA1255322 A, 6 June 1989. [Google Scholar]
- Xu, S.; Ye, L. Monomer casting nylon-6-b-polyether amine copolymers: Synthesis and properties. Composites 2015, 79, 170–181. [Google Scholar] [CrossRef]
- Xiang, M.; Xu, S.; Li, C.; Ye, L. Monomer casting nylon-6-b-polyether amine copolymers: Synthesis and antistatic property. Polym. Eng. Sci. 2016, 56, 817–828. [Google Scholar] [CrossRef]
- Kim, K.J.; Hong, D.S.; Tripathy, A.R.; Kyu, T. Toughening and phase separation behavior of nylon 6-PEG block copolymers and in situ nylon 6-PEG blend via in situ anionic polymerization. J. Appl. Polym. Sci. 1999, 73, 1285–1303. [Google Scholar] [CrossRef]
- Kim, I.; White, J.L. Anionic copolymerization of lauryl lactam and polycaprolactone for the production of a poly(ester amide) triblock copolymer. J. Appl. Polym. Sci. 2003, 90, 3797–3805. [Google Scholar] [CrossRef]
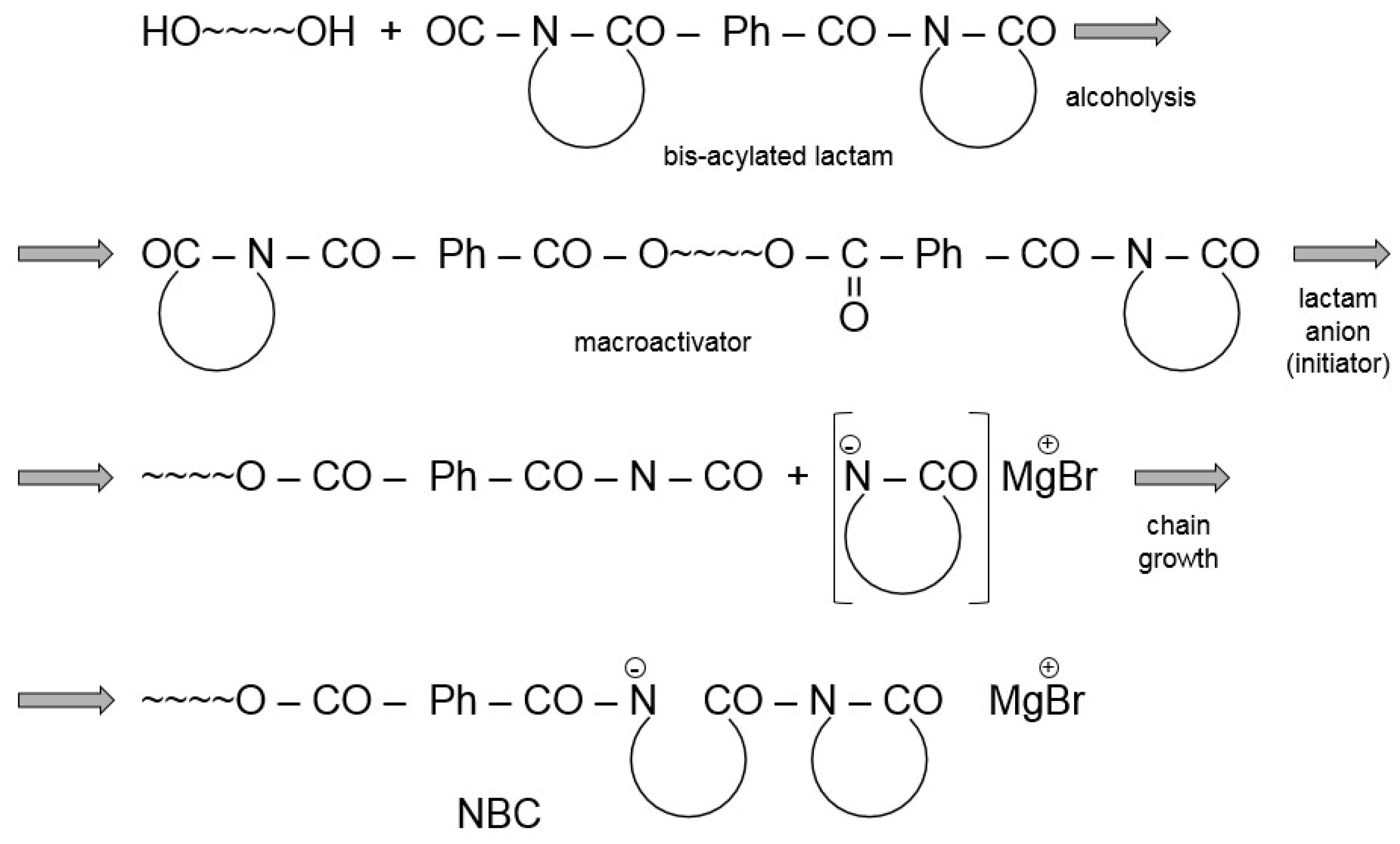
- Sobotík, R.; Šrubař, R.; Roda, J. Polymerization of lactams, 88. Copolymers poly(ϵ-caprolactam)-block-polybutadiene prepared by anionic polymerization, part iii. Model polymerizations initiated with potassium salt of ϵ-caprolactam and accelerated with isocyanates and their derivatives. Macromol. Chem. Phys. 1997, 198, 1147–1163. [Google Scholar] [CrossRef]
- Rached, R.; Hoppe, S.; Jonquieres, A.; Lochon, P.; Pla, F. A new macroinitiator for the synthesis of triblock copolymers PA12-b-pdms-b-PA12. J. Appl. Polym. Sci. 2006, 102, 2818–2831. [Google Scholar] [CrossRef]
- Pae, Y. Structure and properties of polyimide-g-nylon 6 and nylon 6-b-polyimide-b-nylon 6 copolymers. J. Appl. Polym. Sci. 2006, 99, 300–308. [Google Scholar] [CrossRef]
- Hu, G.-H.; Li, H.; Feng, L.-F. Rate of the activated anionic polymerisation of ε-caprolactam onto an isocyanate bearing polypropylene in the melt. Polymer 2005, 46, 4562–4570. [Google Scholar] [CrossRef]
- Hu, G.-H.; Li, H.; Feng, L.-F. Follow-up of the course of the anionic ring-opening polymerization of lactams onto an isocyanate-bearing polymer backbone in the melt. J. Appl. Polym. Sci. 2006, 102, 4394–4403. [Google Scholar] [CrossRef]
- Zhang, C.-L.; Feng, L.-F.; Gu, X.-P.; Hoppe, S.; Hu, G.-H. Kinetics of the anionic polymerization of ε-caprolactam from an isocyanate bearing polystyrene. Polym. Eng. Sci. 2011, 51, 2261–2272. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Xu, W.; Xiong, Y.-Q.; Xu, W.-J. Preparation of PS-g-PA6 copolymers by anionic polymerization of ε-caprolactam using PS precursors with N-carbamated caprolactam pendants as macroactivators. J. Appl. Polym. Sci. 2008, 108, 3177–3184. [Google Scholar] [CrossRef]
- Xu, W.; Liu, Y.-C.; Xiong, Y.-Q.; Miao, Y.-R.; Xu, W.-J. Anionic polymerization and properties of graft copolymers consisting of alternating styrene/maleimide copolymer main chains and polyamide 6 grafts. J. Appl. Polym. Sci. 2008, 108, 1880–1886. [Google Scholar] [CrossRef]
- Jovic, K.; Unold, J.; Naumann, S.; Ullrich, M.; Schmidt, F.G.; Buchmeiser, M.R. In situ copolymerization of lactams for melt spinning. Macromol. Mater. Eng. 2016, 301, 423–428. [Google Scholar] [CrossRef]
- Bouchékif, H.; Tunc, D.; Le Coz, C.; Deffieux, A.; Desbois, P.; Carlotti, S. Controlled synthesis of crosslinked polyamide 6 using a bis-monomer derived from cyclized lysine. Polimer 2014, 55, 5991–5997. [Google Scholar] [CrossRef]
- Tunc, D.; Bouchekif, H.; Améduri, B.; Jérôme, C.; Desbois, P.; Lecomte, P.; Carlotti, S. Synthesis of aliphatic polyamide bearing fluorinated groups from ε-caprolactam and modified cyclic lysine. Eur. Polym. J. 2015, 71, 575–584. [Google Scholar] [CrossRef]
- Volkova, T.V.; Vygodskii, Y.S.; Zabegaeva, O.N.; Zubavichus, Y.V.; Il’ina, M.N.; Krasnov, A.P.; Afonicheva, O.V.; Lozinskaya, E.I.; Garbuzova, I.A.; Shaplov, A.S. Synthesis and characterization of grafted copolymers of aromatic polyimides and ε-caprolactam. J. Appl. Polym. Sci. 2009, 114, 577–586. [Google Scholar] [CrossRef]
- Hou, L.-L.; Liu, H.-Z.; Yang, G.-S. A novel approach to the preparation of thermoplastic polyurethane elastomer and polyamide 6 blends by in situ anionic ring-opening polymerization of ε-caprolactam. Polym. Int. 2006, 55, 643–649. [Google Scholar] [CrossRef]
- Bakkali-Hassani, C.; Tunc, D.; Roos, K.; Planes, M.; Lecomte, P.; Carlotti, S. Simultaneous anionic ring-opening and condensation reactions for the synthesis of aliphatic-N-alkyl aromatic copolyamides. Macromolecules 2017, 50, 175–181. [Google Scholar] [CrossRef]
- Puffr, R.; Stehlíček, J.; Kovářová, J. Block copolymers of hexano-6-lactam with N-methylated aliphatic (co)polyamides. Polymer 2000, 41, 3111–3120. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Z.; Liu, L.; Dai, X.; Zhang, S.; Cao, M. Respective contribution research of soft component and macroinitiator on synthesis and performance of mcpa-pea materials. Polym. Eng. Sci. 2017. [Google Scholar] [CrossRef]
- Lee, B.H.; White, J.L. Formation of a polyetheramide triblock copolymer by reactive extrusion; process and properties. Polym. Eng. Sci. 2002, 42, 1710–1723. [Google Scholar]
- Harkin-Jones, E.; Crawford, R.J. Mechanical properties of rotationally molded nyrim. Polym. Eng. Sci. 1996, 36, 615–625. [Google Scholar] [CrossRef]
- Ning, X.; Ishida, H. Rim-pultrusion of nylon-6 and rubber-toughened nylon-6 composites. Polym. Eng. Sci. 1991, 31, 632–637. [Google Scholar] [CrossRef]
- Grishchuk, S.; Gryshchuk, O.; Weber, M.; Karger-Kocsis, J. Structure and toughness of polyethersulfone (pesu)-modified anhydride-cured tetrafunctional epoxy resin: Effect of pesu molecular mass. J. Appl. Polym. Sci. 2012, 123, 1193–1200. [Google Scholar] [CrossRef]
- Gupta, A.; Singhal, R.; Nagpal, A.K. Crosslinking reaction of epoxy resin (diglycidyl ether of bisphenol a) by anionically polymerized polycaprolactam: I. Mechanism and optimization. J. Appl. Polym. Sci. 2003, 89, 3237–3247. [Google Scholar] [CrossRef]
- Fang, H.; Yang, G. Influence of in situ compatibilization on in situ formation of low-density polyethylene/polyamide 6 blends by reactive extrusion. J. Appl. Polym. Sci. 2010, 116, 3027–3034. [Google Scholar]
- Du, L.; Yang, G. Reactive extrusion for the synthesis of nylon 12 and maleated low-density polyethylene blends via the anionic ring-opening polymerization of lauryllactam. J. Appl. Polym. Sci. 2009, 114, 2662–2672. [Google Scholar] [CrossRef]
- Xu, S.; Ye, L. Preparation and properties of monomer casting nylon-6/peo blend prepared via in situ polymerization. Polym. Eng. Sci. 2015, 55, 589–597. [Google Scholar] [CrossRef]
- Teng, J.; Otaigbe, J.U.; Taylor, E.P. Reactive blending of functionalized polypropylene and polyamide 6: In situ polymerization and in situ compatibilization. Polym. Eng. Sci. 2004, 44, 648–659. [Google Scholar] [CrossRef]
- Pei, A.; Liu, A.; Xie, T.; Yang, G. Blends of immiscible polystyrene/polyamide 6 via successive in-situ polymerizations. Macromol. Chem. Phys. 2006, 207, 1980–1985. [Google Scholar] [CrossRef]
- Wu, B.; Xie, T.; Yang, G. Investigation on particular phase morphology of immiscible polyamide 12 and polystyrene blends prepared via anionic ring-opening polymerization. Polym. Eng. Sci. 2012, 52, 1831–1838. [Google Scholar] [CrossRef]
- Karger-Kocsis, J. Reinforced polymer blends. In Polymer Blends; Paul, D.R., Bucknall, C.B., Eds.; John Wiley & Sons: New York, NY, USA, 2000; Volume 2, pp. 395–428. [Google Scholar]
- Omonov, T.S.; Harrats, C.; Moussaif, N.; Groeninckx, G.; Sadykov, S.G.; Ashurov, N.R. Polyamide 6/ethylene-butylene elastomer blends generated via anionic polymerization of ε-caprolactam: Phase morphology and dynamic mechanical behavior. J. Appl. Polym. Sci. 2004, 94, 2538–2544. [Google Scholar] [CrossRef]
- Yan, D.; Li, G.; Huang, M.; Wang, C. Tough polyamide 6/core–shell blends prepared via in situ anionic polymerization of ε-caprolactam by reactive extrusion. Polym. Eng. Sci. 2013, 53, 2705–2710. [Google Scholar] [CrossRef]
- Utracki, L.A.; Charles, A. Polymer Blends Handbook; Kluwer Academic Pub.: Dordrecht, The Netherlands, 2002; p. 1442. [Google Scholar]
- Mohammadian-Gezaz, S.; Khoshhal, A. Phase morphology and dynamic mechanical properties of nylon 6 based blends prepared via successive in situ ring opening polymerization. J. Macromol. Sci. 2017, 56, 262–278. [Google Scholar] [CrossRef]
- Hou, L.L.; Yang, G.S. Morphology and thermal behavior of mcpa6/san blends prepared by anionic ring-opening polymerization of ε-caprolactam. J. Appl. Polym. Sci. 2006, 100, 1357–1363. [Google Scholar] [CrossRef]
- Wu, T.; Xie, T.; Yang, G. Synthesis and properties of monomer casting polyamide 6/poly(methyl methacrylate) blends. J. Appl. Polym. Sci. 2009, 111, 101–107. [Google Scholar] [CrossRef]
- Wu, T.; Yang, G. Synthesis and characterization of monomer-casting polyamide 6/polymethacrylic ionomer blends. J. Appl. Polym. Sci. 2009, 111, 2970–2979. [Google Scholar] [CrossRef]
- Ji, Y.; Li, W.; Ma, J.; Liang, B. A novel approach to the preparation of nanoblends of poly(2,6-dimethyl-1,4-phenylene oxide)/polyamide 6. Macromol. Rapid Commun. 2005, 26, 116–120. [Google Scholar] [CrossRef]
- Ahmadi, S.; Jahani, Y.; Naderi, G.; Asadollahzadeh, A.H. Supertough (polyamide 6)/(acrylonitrile butadiene rubber) nano alloy through in situ polymerization of caprolactam in the presence of acrylonitrile butadiene rubber nanophase. J. Vinyl Addit. Technol. 2015, 21, 116–121. [Google Scholar] [CrossRef]
- Hou, L.; Liu, H.; Yang, G. Preparation and characterization of thermoplastic polyurethane elastomer and polyamide 6 blends by in situ anionic ring-opening polymerization of ϵ-caprolactam. Polym. Eng. Sci. 2006, 46, 1196–1203. [Google Scholar] [CrossRef]
- Fakirov, S. (Ed.) Transreactions in Condensation Polymers; Wiley-VCH: Weinheim, Germany, 1999; p. 510. [Google Scholar]
- Wang, X.; Yang, G.; Zheng, Q. Structure, morphology and properties of a novel molecular composite by in-situ blending of anionic polyamide 6 with a polyamide copolymer containing rigid segments. Macromol. Mater. Eng. 2007, 292, 197–205. [Google Scholar] [CrossRef]
- Cartier, H.; Hu, G.-H. A novel reactive extrusion process for compatibilizing immiscible polymer blends. Polymer 2001, 42, 8807–8816. [Google Scholar] [CrossRef]
- Okada, A.; Usuki, A. Twenty years of polymer-clay nanocomposites. Macromol. Mater. Eng. 2006, 291, 1449–1476. [Google Scholar] [CrossRef]
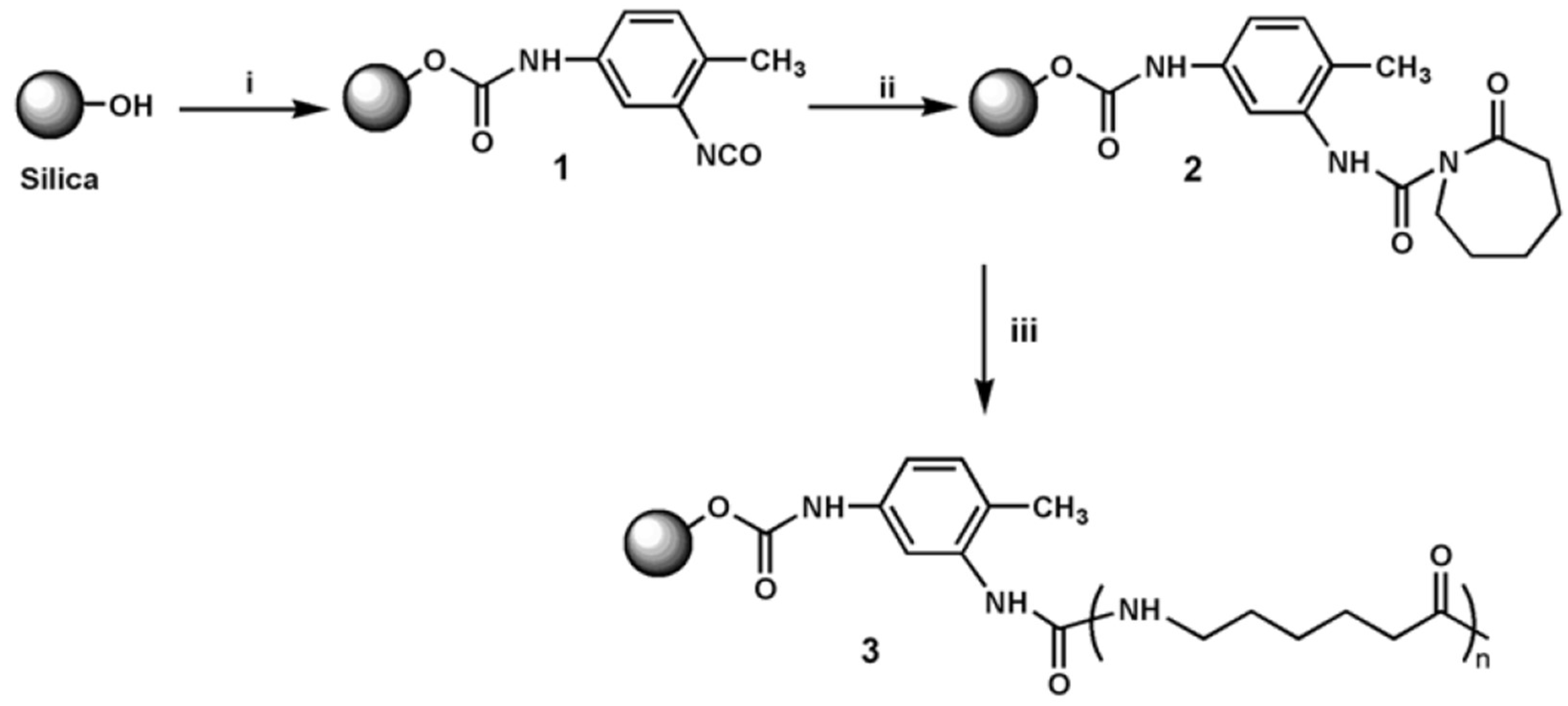
- Yang, M.; Gao, Y.; He, J.P.; Li, H.M. Preparation of polyamide 6/silica nanocomposites from silica surface initiated ring-opening anionic polymerization. eXPRESS Polym. Lett. 2007, 1, 433–442. [Google Scholar] [CrossRef]
- Oliveira, F.; Dencheva, N.; Martins, P.; Lanceros-Méndez, S.; Denchev, Z. Reactive microencapsulation of carbon allotropes in polyamide shell-core structures and their transformation in hybrid composites with tailored electrical properties. eXPRESS Polym. Lett. 2016, 10, 160–175. [Google Scholar] [CrossRef]
- Jia, Y.C.; He, H.; Yu, P.; Chen, J.; Lai, X.L. Synergistically improved thermal conductivity of polyamide-6 with low melting temperature metal and graphite. eXPRESS Polym. Lett. 2016, 10, 679–692. [Google Scholar] [CrossRef]
- Maxian, O.; Pedrazzoli, D.; Manas-Zloczower, I. Conductive polymer foams with carbon nanofillers—Modeling percolation behavior. eXPRESS Polym. Lett. 2017, 11, 406–418. [Google Scholar] [CrossRef]
- Mohamed, M.A.A.; Pedrazzoli, D.; Nady, N.; Kalaitzidou, K. Temperature-dependent rigidity and magnetism of polyamide 6 nanocomposites based on nanocrystalline fe-ni alloy of various geometries. eXPRESS Polym. Lett. 2016, 10, 822–834. [Google Scholar] [CrossRef]
- Faridirad, F.; Ahmadi, S.; Barmar, M. Polyamide/carbon nanoparticles nanocomposites: A review. Polym. Eng. Sci. 2017, 57, 475–494. [Google Scholar] [CrossRef]
- Brêda, C.; Dencheva, N.; Lanceros-Méndez, S.; Denchev, Z. Preparation and properties of metal-containing polyamide hybrid composites via reactive microencapsulation. J. Mater. Sci. 2016, 51, 10534–10554. [Google Scholar] [CrossRef]
- Rusu, G.; Rusu, E. Nylon 6/sio2 nanocomposites synthesized by in situ anionic polymerization. High Perform. Polym. 2006, 18, 355–375. [Google Scholar] [CrossRef]
- Xu, S.; Zhao, X.; Ye, L. Mechanical and crystalline properties of monomer casting nylon-6/sio2 composites prepared via in situ polymerization. Polym. Eng. Sci. 2013, 53, 1809–1822. [Google Scholar] [CrossRef]
- Rusu, G.; Rusu, E. Anionic nylon 6/tio2 composite materials: Effects of tio2 filler on the thermal and mechanical behavior of the composites. Polym. Compos. 2012, 33, 1557–1569. [Google Scholar] [CrossRef]
- Dencheva, N.; Denchev, Z.; Lanceros-Méndez, S.; Ezquerra Sanz, T. One-step in situ synthesis of polyamide microcapsules with inorganic payload and their transformation into responsive thermoplastic composite materials. Macromol. Mater. Eng. 2016, 301, 119–124. [Google Scholar] [CrossRef]
- Chen, J.; Volinsky, A.A.; He, W. Synthesis and characterization of mc nylon/modified yttrium hydroxide nanocomposites. J. Appl. Polym. Sci. 2016, 133, 5. [Google Scholar] [CrossRef]
- Krastev, P.; Mateva, R. In-situ preparation of polyamide-6/polypropylene glycol copolymers with mineral fillers. J. Chem. Technol. Metall. 2014, 49, 535–540. [Google Scholar]
- Ricco, L.; Russo, S.; Monticelli, O.; Bordo, A.; Bellucci, F. E-caprolactam polymerization in presence of polyhedral oligomeric silsesquioxanes (poss). Polymer 2005, 46, 6810–6819. [Google Scholar] [CrossRef]
- Baldi, F.; Bignotti, F.; Ricco, L.; Monticelli, O.; Riccò, T. Mechanical and structural characterization of poss-modified polyamide 6. J. Appl. Polym. Sci. 2006, 100, 3409–3414. [Google Scholar] [CrossRef]
- Dencheva, N.; Gaspar, H.; Filonovich, S.; Lavrova, O.; Busani, T.; Bernardo, G.; Denchev, Z. Fullerene-modified polyamide 6 by in situ anionic polymerization in the presence of pcbm. J. Mater. Sci. 2014, 49, 4751–4764. [Google Scholar] [CrossRef]
- Zuev, V.V.; Ivanova, Y.G. Mechanical and electrical properties of polyamide-6-based nanocomposites reinforced by fulleroid fillers. Polym. Eng. Sci. 2012, 52, 1206–1211. [Google Scholar] [CrossRef]
- Engelmann, G.; Gohs, U.; Ganster, J. Monomer cast polyamide 6 composites and their treatment with high-energy electrons. J. Appl. Polym. Sci. 2012, 123, 1201–1211. [Google Scholar] [CrossRef]
- Li, C.; Xiang, M.; Ye, L. Structure and tribological performance of monomer casting nylon-6/colloidal graphite composites synthesized through in situ polymerization. Polym. Plast. Technol. Eng. 2017, 56, 1345–1357. [Google Scholar] [CrossRef]
- Horský, J.; Kolařík, J.; Fambri, L. Gradient composites of alkaline poly(6-hexanelactam) with graphite: One-step synthesis, structure, and mechanical properties. Macromol. Mater. Eng. 2001, 286, 216–224. [Google Scholar] [CrossRef]
- Qu, L.; Veca, L.M.; Lin, Y.; Kitaygorodskiy, A.; Chen, B.; McCall, A.M.; Connell, J.W.; Sun, Y.-P. Soluble nylon-functionalized carbon nanotubes from anionic ring-opening polymerization from nanotube surface. Macromolecules 2005, 38, 10328–10331. [Google Scholar] [CrossRef]
- Kelar, K.; Jurkowski, B. Properties of anionic polymerized ε-caprolactam in the presence of carbon nanotubes. J. Appl. Polym. Sci. 2007, 104, 3010–3017. [Google Scholar] [CrossRef]
- Yang, M.; Gao, Y.; Li, H.; Adronov, A. Functionalization of multiwalled carbon nanotubes with polyamide 6 by anionic ring-opening polymerization. Carbon 2007, 45, 2327–2333. [Google Scholar] [CrossRef]
- Mhetre, S.K.; Patra, P.K.; Kim, Y.K.; Warner, S.B. In-situ polymerized nylon 6/mwnt nanocomposite fibers. Res. J. Text. Appar. 2007, 11, 35–41. [Google Scholar] [CrossRef]
- Yan, D.; Xie, T.; Yang, G. In situ synthesis of polyamide 6/mwnts nanocomposites by anionic ring opening polymerization. J. Appl. Polym. Sci. 2009, 111, 1278–1285. [Google Scholar] [CrossRef]
- Yan, D.; Yang, G. Synthesis and properties of homogeneously dispersed polyamide 6/mwnts nanocomposites via simultaneous in situ anionic ring-opening polymerization and compatibilization. J. Appl. Polym. Sci. 2009, 112, 3620–3626. [Google Scholar] [CrossRef]
- Penu, C.; Hu, G.-H.; Fonteix, C.; Marchal, P.; Choplin, L. Effects of carbon nanotubes and their state of dispersion on the anionic polymerization of ϵ-caprolactam: 1. Calorimetry. Polym. Eng. Sci. 2010, 50, 2287–2297. [Google Scholar] [CrossRef]
- Penu, C.; Hu, G.-H.; Fonteix, C.; Marchal, P.; Choplin, L.; Feng, L.-F. Effects of carbon nanotubes and their state of dispersion on the anionic polymerization of ϵ-caprolactam: II. Rheology. Polym. Eng. Sci. 2011, 51, 1116–1121. [Google Scholar] [CrossRef]
- Hänsch, S.; Socher, R.; Pospiech, D.; Voit, B.; Harre, K.; Pötschke, P. Filler dispersion and electrical properties of polyamide 12/mwcnt-nanocomposites produced in reactive extrusion via anionic ring-opening polymerization. Compos. Sci. Technol. 2012, 72, 1671–1677. [Google Scholar] [CrossRef]
- Yan, D.; Yang, G. Effect of multiwalled carbon nanotubes on the morphology and electrical properties of polyamide 6/polystyrene blends prepared via successive polymerization. J. Appl. Polym. Sci. 2012, 125, E167–E174. [Google Scholar] [CrossRef]
- Huang, S.; Toh, C.L.; Yang, L.; Phua, S.; Zhou, R.; Dasari, A.; Lu, X. Reinforcing nylon 6 via surface-initiated anionic ring-opening polymerization from stacked-cup carbon nanofibers. Compos. Sci. Technol. 2014, 93, 30–37. [Google Scholar] [CrossRef]
- Rahimi, S.K.; Otaigbe, J.U. Polyamide 6 nanocomposites incorporating cellulose nanocrystals prepared by in situ ring-opening polymerization: Viscoelasticity, creep behavior, and melt rheological properties. Polym. Eng. Sci. 2016, 56, 1045–1060. [Google Scholar] [CrossRef]
- Rahimi, S.K.; Otaigbe, J.U. The effects of the interface on microstructure and rheo-mechanical properties of polyamide 6/cellulose nanocrystal nanocomposites prepared by in-situ ring-opening polymerization and subsequent melt extrusion. Polymer 2017, 127, 269–285. [Google Scholar] [CrossRef]
- Liu, A.; Xie, T.; Yang, G. Synthesis of exfoliated monomer casting polyamide 6/Na+-montmorillonite nanocomposites by anionic ring opening polymerization. Macromol. Chem. Phys. 2006, 207, 701–707. [Google Scholar] [CrossRef]
- Liu, A.; Xie, T.; Yang, G. Comparison of polyamide-6 nanocomposites based on pristine and organic montmorillonite obtained via anionic ring-opening polymerization. Macromol. Chem. Phys. 2006, 207, 1174–1181. [Google Scholar] [CrossRef]
- Wu, T.; Liu, A.; Xie, T.; Yang, G. Evaluation of polymethacrylic ionomer as compatibilizers for MCPA6/clay composites. J. Appl. Polym. Sci. 2008, 110, 2727–2732. [Google Scholar] [CrossRef]
- Rothe, B.; Elas, A.; Michaeli, W. In situ polymerisation of polyamide-6 nanocompounds from caprolactam and layered silicates. Macromol. Mater. Eng. 2009, 294, 54–58. [Google Scholar] [CrossRef]
- Rothe, B.; Kluenker, E.; Michaeli, W. Masterbatch production of polyamide 6-clay compounds via continuous in situ polymerization from caprolactam and layered silicates. J. Appl. Polym. Sci. 2012, 123, 571–579. [Google Scholar] [CrossRef]
- Cabrera Álvarez, E.N.; Ramos de Valle, L.F.; Rodríguez González, F.J.; Soriano-Corral, F.; Díaz De León, R.E. Influence of laurolactam content on the clay intercalation of polyamide 6,12/clay nanocomposites synthesized by open ring anionic polymerization. J. Nanomater. 2012, 2012, 7. [Google Scholar] [CrossRef]
- Dencheva, N.; Denchev, Z. Clay distribution and crystalline structure evolution in polyamide 6/montmorillonite composites prepared by activated anionic polymerization. J. Appl. Polym. Sci. 2013, 130, 1228–1238. [Google Scholar] [CrossRef]
- Vyas, A.; Iroh, J.O. Thermal behavior and structure of clay/nylon-6 nanocomposite synthesized by in situ solution polymerization. J. Therm. Anal. Calorim. 2014, 117, 39–52. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Y.; Zhao, X.; Zhao, D.; Yang, G. A commercial production route to prepare polymer-based nanocomposites by unmodified multilayer graphene. J. Appl. Polym. Sci. 2015, 132. [Google Scholar] [CrossRef]
- Minář, J.; Brožek, J. E-caprolactone as a medium for improving dispersability of graphene oxide in polyamide based composites. Eur. Polym. J. 2017, 91, 212–220. [Google Scholar]
- Schmidhuber, S.; Fries, E.; Zimmermann, P. It couldn’t be more hybrid. Thermoplastic-matrix rtm on the roof frame of the roading roadter. Kunstst. Int. 2017, 1–2, 36–38. [Google Scholar]
- Sealy, C. Molding the future: Engel takes composite approach to composites. Reinf. Plast. 2016, 60, 138–141. [Google Scholar] [CrossRef]
- Harte, A.M.; Mc Namara, J.F. Overinjection of thermoplastic composites: I. Processing and testing of components. J. Mater. Proc. Technol. 2007, 182, 12–20. [Google Scholar] [CrossRef]
- Harte, A.M.; Mc Namara, J.F. Overinjection of thermoplastic composites: Ii. Numerical simulation. J. Mater. Proc. Technol. 2007, 182, 21–27. [Google Scholar] [CrossRef]
- Litt, M.H.; Brinkmann, A.W. Nylon 6/graphite fiber composites by in situ polymerization. J. Elastoplast. 1973, 5, 153–160. [Google Scholar] [CrossRef]
- Gabbert, J.D.; Garner, A.Y.; Hedrick, R.M. Reinforced nylon 6 block copolymers. Polym. Compos. 1983, 4, 196–199. [Google Scholar] [CrossRef]
- Horský, J.; Kolařík, J.; Fambri, L. Composites of alkaline poly(6-caprolactam) and short glass fibers: One-step synthesis, structure and mechanical properties. Die Angew. Makromol. Chem. 1999, 264, 39–47. [Google Scholar] [CrossRef]
- Holmes, M. Expanding the market for long fiber technology. Reinf. Plast. 2017. [Google Scholar] [CrossRef]
- Luisier, A.; Bourban, P.-E.; Manson, J.-A.E. Reaction injection pultrusion of PA12 composites: Process and modelling. Composites 2003, 34, 583–595. [Google Scholar] [CrossRef]
- Ringenbach, S.; Richeton, J.; Coulton, J. Hyundai’s breakthrough front bumper crash beam. JEC Compos. Mag. 2015, 98, 39–41. [Google Scholar]
- Szaplonczay, P.; Karger-Kocsis, J.; Czigány, T.; Zsigmond, B. Process and Equipment for Producing Composite Core with Thermoplastic Matrix for Recyclable and Thermally Stable Electrical Transmission Line Conductor. U.S. Patent WO 2009/130525, 23 April 2009. [Google Scholar]
- Van Rijswijk, K.; Teuwen, J.J.E.; Bersee, H.E.N.; Beukers, A. Textile fiber-reinforced anionic polyamide-6 composites. Part i: The vacuum infusion process. Composites 2009, 40, 1–10. [Google Scholar] [CrossRef]
- Van Rijswijk, K.; Bersee, H.E.N.; Jager, W.F.; Picken, S.J. Optimization of anionic polyamide-6 for vacuum infusion of thermoplastic composites: Choice of activator and initiator. Composites 2006, 37, 949–956. [Google Scholar] [CrossRef]
- Van Rijswijk, K.; Bersee, H.E.N.; Beukers, A.; Picken, S.J.; van Geenen, A.A. Optimization of anionic polyamide-6 for vacuum infusion of thermoplastic composites: Influence of polymerization temperature on matrix properties. Polym. Test. 2006, 25, 392–404. [Google Scholar] [CrossRef]
- Van Rijswijk, K.; Lindstedt, S.; Bersee, H.E.N.; Gleich, K.F.; Titzschkau, K.; Mc Dade, E.J. Reactively processed polyamide-6 structural composites for automotive applications. In Proceedings of the 6th Annual SPE Automotive Composites Conference, Troy, MI, USA, 12–14 September 2006; pp. 435–442. [Google Scholar]
- Van Rijswijk, K.; van Geenen, A.A.; Bersee, H.E.N. Textile fiber-reinforeced anionic polyamide-6 composites. Part ii: Investigation on interfacial bond formation by short beam shear test. Composites 2009, 40, 1033–1043. [Google Scholar] [CrossRef]
- Zingraff, L.; Michaud, V.; Bourban, P.-E.; Manson, J.-A.E. Resin transfer moulding of anionically polymerized polyamide 12. Composites 2005, 36, 1675–1686. [Google Scholar] [CrossRef]
- Prabhakaran, R.T.D. Are reactive thermoplastic polymers suitable for future wind turbine composite materials blades? Mech. Adv. Mater. Struct. 2014, 21, 213–221. [Google Scholar] [CrossRef]
- Yan, C.; Li, H.; Zhang, X.; Zhu, Y.; Fan, X.; Yu, L. Preparation and properties of continuous glass fiber reinforeced anionic polyamide-6 thermoplastic composites. Mater. Des. 2013, 46, 688–695. [Google Scholar] [CrossRef]
- Pillay, S.; Vaidya, U.K.; Janowski, G.M. Liquid molding of carbon fabric-reinforced nylon matrix composite laminates. J. Thermoplast. Compos. Mater. 2005, 18, 509–527. [Google Scholar] [CrossRef]
- Kan, Z.; Yang, M.-B.; Yang, W.; Liu, Z.-Y.; Xie, B.-H. Investigation on the reactive processing of textile-ramie fiber reinforeced anionic polyamide-6 composites. Compos. Sci. Technol. 2015, 110, 188–195. [Google Scholar] [CrossRef]
- Barfknecht, P.W.; Martin, J.; Pillay, B.; Vaidya, U.K.; Gray, G.M. Single-stream processing technique for in situ polymerization of glass fiber/polyamide-6 laminates. J. Thermoplast. Compos. Mater. 2016, 30, 1639–1653. [Google Scholar] [CrossRef]
- Bitterlich, M.; Ehleben, M.; Wollny, A.; Desbois, P.; Renkl, J.; Schmidhuber, S. Tailored to reactive polyamide 6. Kunstoffe Int. 2014, 3, 47–51. [Google Scholar]
- Wakeman, M.D.; Zingraff, L.; Bourban, P.-E.; Månson, J.-A.E.; Blanchard, P. Stamp forming of carbon fibre/PA12 composites—A comparison of a reactive impregnation process and a commingled yarn system. Compos. Sci. Technol. 2006, 66, 19–35. [Google Scholar] [CrossRef]
- Reith, L.; Weissinger, M.; Mueller, N.; Sperneder, G.; Schoefer, G. Method for the Production of Plastic Parts. U.S. Patent Appl. 2018/0029312, 1 February 2018. [Google Scholar]
- Cischino, E.; Di Paolo, F.; Mangino, E.; Pullini, D.; Elizetxea, C.; Maestro, C.; Alcalde, E.; Christiansen, J.D. An Advanced Technological Lightweighted Solution for a Body in White, Proceedings of the 6th Transport Research Arena, Warsaw, Poland, 18–21 April 2016; Transportation Research Procedia: Warsaw, Poland, 2016; pp. 1021–1030. [Google Scholar]
- Karger-Kocsis, J.; Yuan, Q.; Czigány, T. Assignment of acoustic emission of the failure sequence and damage zone growth in glass fiber strand mat-reinforced structural nylon rim composites. Polym. Bull. 1992, 28, 717–723. [Google Scholar] [CrossRef]
- Karger-Kocsis, J. Instrumented impact testing of a glass swirl mat-reinforced reaction injection-molded polyamide block copolymer (NBC). J. Appl. Polym. Sci. 1992, 45, 1595–1609. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Czigany, T. Fracture behaviour of glass-fibre mat-reinforced structural nylon rim composites studied by microscopic and acoustic emission techniques. J. Mater. Sci. 1993, 28, 2438–2448. [Google Scholar] [CrossRef]
- Rosso, P.; Friedrich, K.; Wollny, A.; Mülhaupt, R. A novel polyamide 12 polymerization system and its use for a lcm-process to produce cfrp. J. Thermoplast. Compos. Mater. 2005, 18, 77–90. [Google Scholar] [CrossRef]
- Connor, M.T.; Eder, R.; Schmid, E.; Wild, U. Ems Polymerisation Moulding (EPM): A Novel Solution to Thermoplastic Composite Manufacturing. In Proceedings of the Progress Through Innovation and Cost Effectiveness. JEC-1998, Paris, France, 22–23 April 1998; SAMPE Europe: Paris, France, 1998; pp. 385–395. [Google Scholar]
- Máirtín, P.Ó.; McDonnell, P.; Connor, M.T.; Eder, R.; Ó Brádaigh, C.M. Process investigation of a liquid pa-12/carbon fibre moulding system. Composites 2001, 32, 915–923. [Google Scholar] [CrossRef]
- Karger-Kocsis, J.; Bárány, T. Single-polymer composites (SPCs): Status and future trends. Compos. Sci. Technol. 2014, 92, 77–94. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, A.; Yang, G. Polyamide single polymer composites prepared via in situ anionic polymerization of ε-caprolactam. Composites 2010, 41, 1006–1011. [Google Scholar] [CrossRef]
- Gong, Y.; Yang, G. All-polyamide composites prepared by resin transfer molding. J. Mater. Sci. 2010, 45, 5237–5243. [Google Scholar] [CrossRef]
- Dencheva, N.; Denchev, Z.; Pouzada, A.S.; Sampaio, A.S.; Rocha, A.M. Structure-properties relationship in single polymer composites based on polyamide 6 prepared by in-mold anionic polymerization. J. Mater. Sci. 2013, 48, 7260–7273. [Google Scholar] [CrossRef]
- Dencheva, N.; Sampaio, A.S.; Oliveira, F.M.; Pouzada, A.S.; Brito, A.M. Preparation and properties of polyamide-6-based thermoplastic laminate composites by a novel in-mold polymerization technique. J. Appl. Polym. Sci. 2014, 131, 1097–1107. [Google Scholar] [CrossRef]
- Karger-Kocsis, J. Interphase with lamellar interlocking and amorphous adherent—A model to explain effects of transcrystallinity. Adv. Compos. Lett. 2000, 9, 225–227. [Google Scholar]
- Kishi, H.; Nakao, N.; Kuwashiro, S.; Matsuda, S. Carbon fiber reinforced thermoplastic composites from acrylic polymer matrices: Interfacial adhesion and physical properties. Express Polym. Lett. 2017, 11, 334–342. [Google Scholar] [CrossRef]
- Dencheva, N.; Vale, D.M.; Denchev, Z. Dually reinforced all-polyamide laminate composites via microencapsulation strategy. Polym. Eng. Sci. 2017, 57, 806–820. [Google Scholar] [CrossRef]
- Lauke, B. Fracture toughness modelling of polymers filled with inhomogeneously distributed rigid spherical particles. Express Polym. Lett. 2017, 11, 545–554. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rotational Molding | Classical | Reactive |
|---|---|---|
| Temperature | T ~ 240 °C | T ~ 150 °C |
| Cycle time | t >40 min | t = 15–20 min |
| Speed ration (S1/S2) | 5/4 | 5/4 |
| Melting point Tm (°C) | 224.3 | 224 |
| Degree of crystallinity (%) | 28 | 49 |
| Degree of conversion (%) | 98.9% | 98.9% |
| Intrinsic viscosity (dL/g) | 1.07 | 7 |
| Molecular weight (g/mol) | 30,778 | 182,594 |
| Tensile Properties | ||
| Young’s modulus (MPa) | 750 | 1560 |
| Yield stress (MPa) | 62 | 80 |
| Elongation at break (%) | 32 | 64 |
| Nanofiller (Type, Amount) | Monomer | Initiator/Activator (Type, Amount) | Preparation | Testing | Results, Comments | Refs. |
|---|---|---|---|---|---|---|
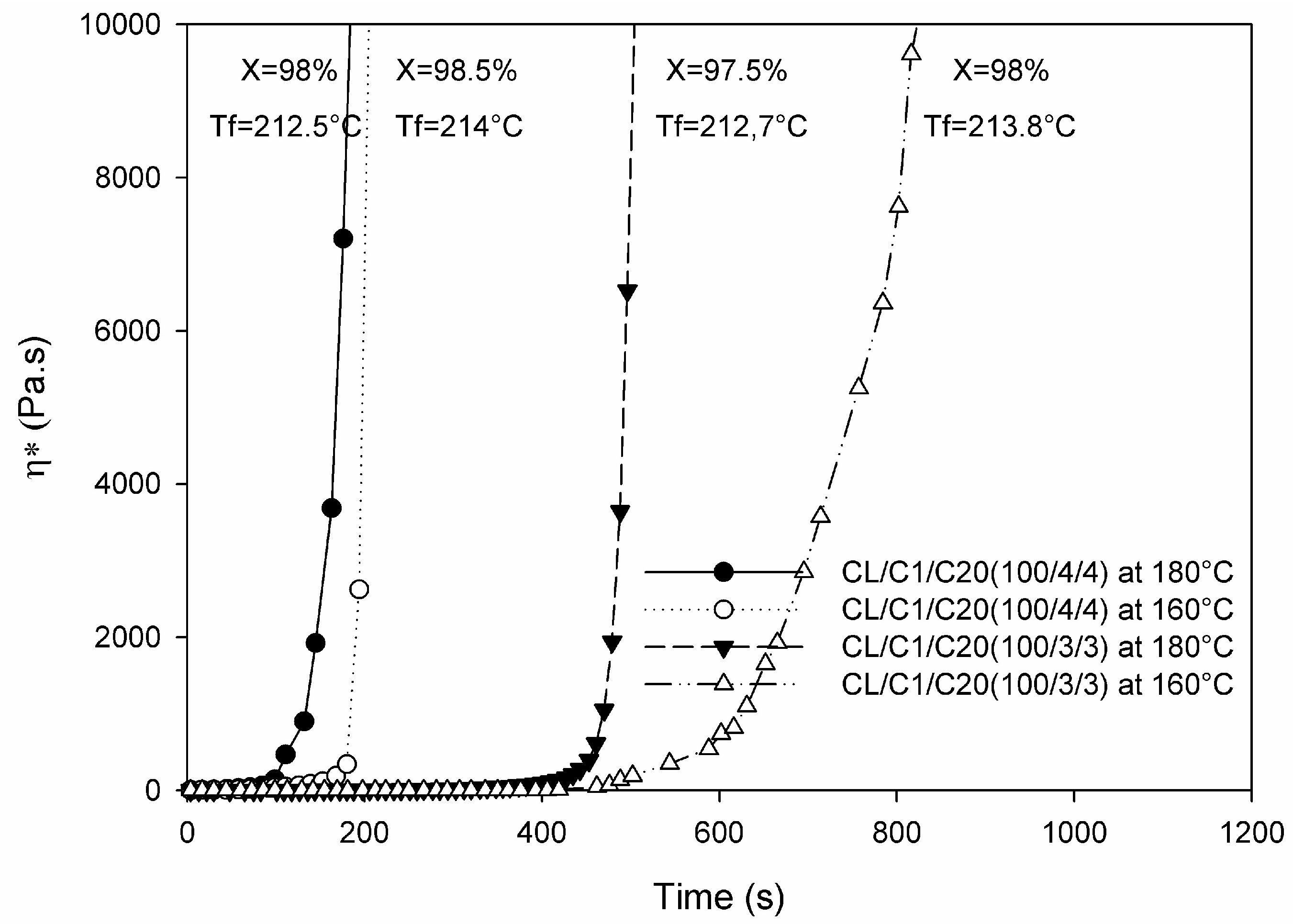
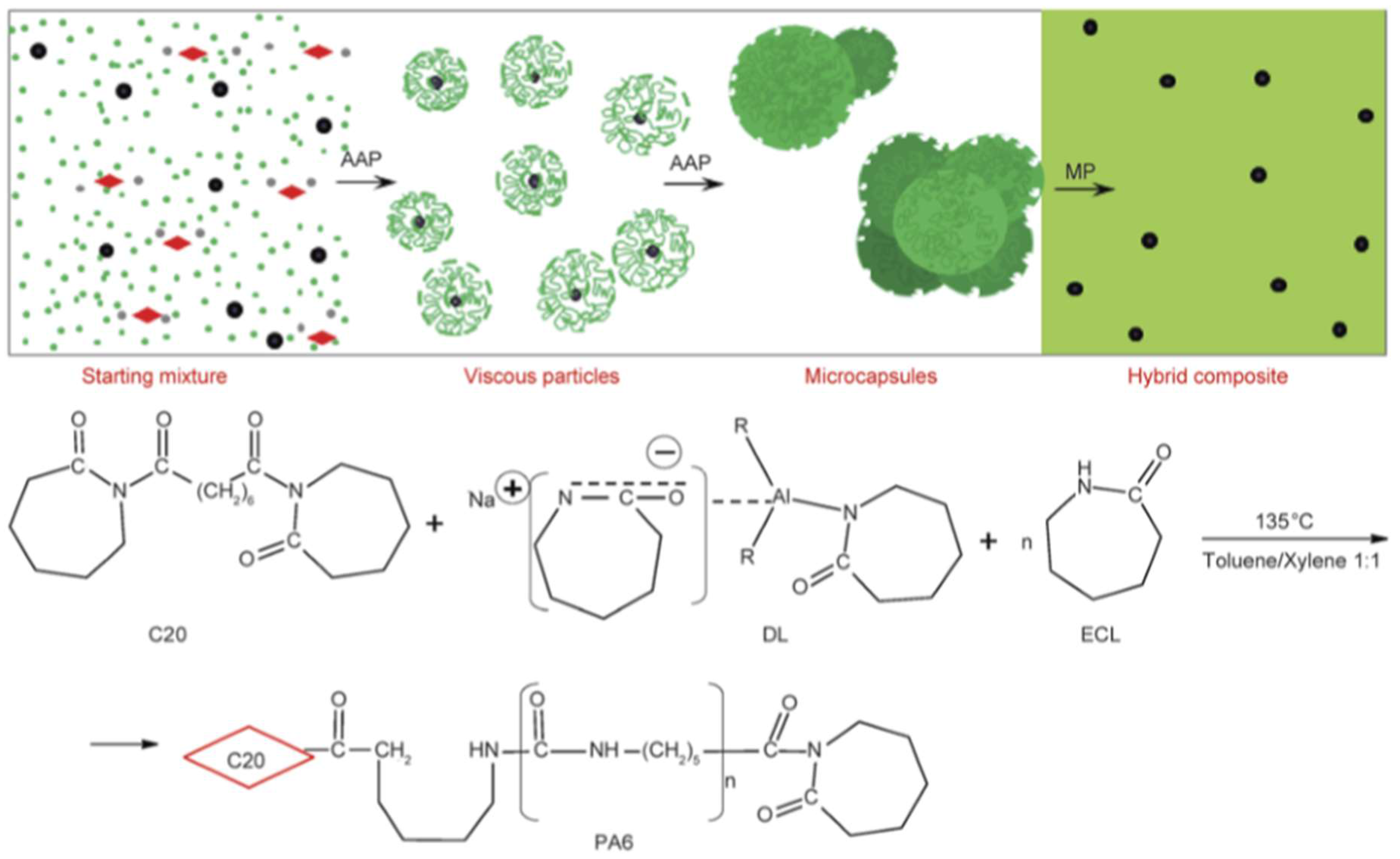
| Cu-, Zn-, Fe- particles (micron-scale, up to 8 wt %) Al- particles (micron-scale, up to 30 wt %) | CL | Dilactamate®/C20 (3 mol %/1.5 mol %) | In solvent (toluene/xylene = 1:1) at 135 °C, followed by filtering, drying and compression molding at T = 230 °C | Optical microscopy, viscosimetry (Mv), DSC, TGA, synchrotron WAXS, electric conductivity, tensile tests | PA-6 microcapsules (see Figure 7) produced and their formation mechanism proposed. PA-6 nanocomposites exhibited enhanced E-modulus and tensile strength. γ-polymorph formed upon nanofiller loading. No electric percolation observed. Potential for energy storage deduced. | [121] |
| SiO2 (7 nm) with and without silane surface modification (2–10 wt %) | CL | Dilactamate®/N,N′-[methylene-di(4,4′-phenylene)bis-carbamoyl]bis-ε-caprolactam (0.8 mol %/0.4 mol %) | CL with initiator +CL with activator mixed separately at T = 100 °C. Two melts mixed and polymerized at 160 °C for 40 min via rotational molding | Viscosimetry (Mv), water uptake, DSC, TGA, WAXS, TEM, FTIR, impact and flexural properties | Silane treatment of silica improved the polymer yield (>95%), reduced the water absorption, enhanced the flexural modulus and strength. Notched Izod impact strength (IS) (peaked at 4 wt %) was also improved by contrast to the unmodified silica. | [122] |
| Porous SiO2 (20 nm) functionalized with TDI (<14 wt %) | CL | Na/SiO2 with carbamoyl and group (see Figure 6) (molar ratio = 6:1) | CL + initiator + activator mixed at 80 °C under N2 and sonicated for 30 min. Polymerization at 170 °C for 6 h will varies feed ratios | Viscosimetry (Mv), FTIR, DSC, TGA, TEM | Feed ration CL/(initiator + activator) affected Mv reaching ~12 kDa. TGA proved the “grafting from” approach, i.e., CL polymerization according to the scheme in Figure 6. At higher SiO2 content prominent agglomeration found (Figure 8) | [115] |
| SiO2 (5 μm) acicular—aspect ratio ~15 with amino coupling agent | CL | NaOH/isocyanate (TDI) | Particles introduced in CL melt at 130 °C. NaOH added upon stirring for 30 min followed by dosing TDI. Cast polymerization at 170 °C for 20 min | FTIR, DMA, DSC, WAXS, SEM, mechanical properties | Particles well dispersed. Tensile and notched Charpy IS increased, reaching a maximum at 3–5 wt % silica, then decreased. Nucleation and crystallization affected by the silica presence. Silica needles pulled out thereby enhancing the toughness | [123] |
| TiO2 (<10 μm) with and without surface treatment with aminosilane ≤ 8 wt % | CL | Dilactamate®/N,N′-[methylene-di(4,4′-phenylene)bis-carbamoyl]bis-ε-caprolactam (0.8 mol %/0.4 mol %) | Polymerization via rotational molding at T = 160 °C for 30 min | DSC, TGA, tensile properties, notched Izod | Tensile and flexural moduli increased with increasing filler content without any effect of surface treatment. The latter surface treatment improved the strength. The toughness and tensile elongation were reduced with increasing TiO2 content whereby marginal effect of silane coupling was observed | [124] |
| Metals, Metal oxides, Carbon black (CB), Graphite, CNT, CNF Organoclays | CL | Dilactamate®/C20 (3 mol %/1.5 mol %) | Microcapsules’ production in solvent (see Figure 7). Capsules filtered, dried and specimens produced by compression molding at T = 230 °C at 5 MPa pressure | Optical microscopy, SEM, mechanical properties, electric and magnetic behavior | Conversion up to 85% and filler content (“pay-load”) up to 30 wt %. Mechanical and dielectrical properties tailored upon amount, type and combination of the additives. | [125] |
| Yttrium hydroxide with and without surface treatment, diameter: ~400 nm, length: few microns (<0.8 wt %) | CL | NaOH/TDI | Cast polymerization at T = 180 °C for 1 h | SEM, WAXS, tensile and impact testing | Good dispersion of the filler. Tensile strength and water absorption reduced, whereas impact strength increased, peaking at ~0.3 wt % | [126] |
| Boron carbid (B4C) 15–62 μm, Graphite ~10 wt % | CL + isophorone diisocyanate functionalized polypropylene-glycol (PPG) macroactivator (NBC-type) | NaCL/macroactivator | Bulk polymerization in ampule and in mold casting. Mixing with filler, in situ macroactivator preparation at 120 °C under N2. Initiator added at 140 °C. Polymerization at 180 °C. | Degree of conversion (DOC), 1H-NMR, FTIR, Charpy impact | Copolymer formation between CL and PPG verified. At high macroactivator content polymerization rate and yield are influenced by the filler (B4C and graphite). Charpy IS strongly improved but its change with the filler content differed between B4C and graphite | [127] |
| POSS with –NH2 functionality (≤16 wt %) | CL | NaH (NaCL)/cyclohexyl-carbamoylcaprolactam or POSS-CL (reaction product of POSS-NH2 with carbonylbiscaprolactam, activator content varied between 0.6 and 1.8 mol %) | Different polymerization techniques: hydrolytic, quasi-adiabatic AROP, isothermal AROP, anionic suspension polymerization | DOC, viscosimetry (Mv), DSC, WAX, SEM | In AROP techniques DOC was higher than 93%, Mv varied between 13 and 176 kDa as a function of AROP technique and activator type/amount. AROP performed better than the hydrolytic route. The tensile behavior of the POSS-containing nanocomposites featured improved ductility at cost of stiffness and strength | [128,129] |
| CB, MWCNT, CNF, Graphite (≤10 wt %) | CL | Dilactamate®/C20 (3 mol %/1.5 mol %) | Microcapsules production in solvent—see Figure 7. Capsules filtered and dried prior to compression molding | Optical microscopy, viscosimetry (Mv) DSC, TGA, synchrotron WAXS electrical, dielectrical behavior | All fillers enhanced the stiffness and reduced the deformation at break with increasing content. Tensile strength improvement was found out for MWCNT. The conductivity, permeability strongly changed as fraction of the type, amount and combination of these fillers | [116] |
| C60 (fullerene) [6,6]phenyl-C61-butyric acid methyl ester (≤3 wt %) | CL | Dilactamate®/C20 | Modified fullerene dispersed in molten CL at 110 °C under N2 blanket. Then initiator/activator introduced, homogenized and polymerized at T = 170 °C for 30 min | DOC, viscosimetry (Mv) DSC, TGA, DMA, FTIR, SEM, WAXS, electric conductivity | Complex interaction between the π-electrons of fullerene and CL revealed that strongly effected the polymerization. Formation mechanism for the linear/crosslinked chain formation proposed. The volume resistivity above 0.1 wt % fullerene content was reduced by 2–4 order of magnitude | [130] |
| C60, C60/C70 mixture, fullerene soot (0.5–2 μm) | CL | Na/toluene-2,6-diisocyanate | Bulk polymerization at T = 140–160 °C for 12 h | DSC, electrical resistivity, tensile and compression properties, tribology | Small enhancement in stiffness and strength with increasing fullerene content. Volume resistivity decreased with 6 order of magnitudes at a fullerene content of 0.10 wt %. The coefficient of friction was halved in presence of fullerenes. | [131] |
| CB nanoscale SiO2 micronscale SiC submicronscale SCF ≤ 15 wt % | CL | C10/C20 also in presence of a curing agent for electron beam irradiation | Filler introduced in the activator-containing CL at 120 °C. AROP performed at 160 °C for 30 min | Viscosimetry (Mv) DSC, TGA, heat distortion temperature (HDT), DOC, SEM, TEM, mechanical testing | Stiffness, strength and HDT improved in the range of 10%–30% at 2 wt % filler content. 15 wt % short carbon fibers (SCF) enhanced the tensile strength from 78 to 93 MPa and doubled the E-modulus. Crystallinity slightly reduced. Effect of the dose of electron beam irradiation was moderate for nanoscaled CB. | [132] |
| Graphite (colloidal) with and without titanate coupling agent 4 μm ≤ 8 wt % | CL | NaOH/TDI (0.5 mol %/0.5 mol %) | Filler dispersed in molten CL at 130 °C before adding the initiator and activator under vacuum. Cast polymerization at 175 °C for 30 min | Viscosimetry (Mv), FTIR, DSC, WAXS, DMA, mechanical properties, friction/wear | MW reduced from 85 to 55 kDa with the graphite content. Graphite worked as heterogeneous nucleant during crystallization. The tensile strength did not change until 4 wt % Graphite before drastic reduction. Notched Charpy IS improved only at 0.5–1 wt %. PA-6 with 4 wt % graphite exhibited more than 10-fold increase in wear resistance. | [133] |
| Graphite, 5 μm (5 wt %) | CL | Dilactamate®/PUs (2/1; 1.8/0.8) | CL molten under N2 and mixed with the PU (macroactivator), followed by introduction of the graphite powder and the initiator. Casting at 170 °C for 1 h | Optical microscopy, DSC, DMA, tensile tests, flexural creep, tribology | Composites with gradient structure produced. Polyether-urethane as macroactivator yielded high MW with crosslinking. Graphite filling reduced MW, the spherulite diameter, the tensile strength, elongation at break and the coefficient of friction (by 50%) | [134] |
| SWCNT functionalized with CL | CL | Na/CL-functionalized SWNT | Polymerization at T = 140 °C for 24 h. | SEM, 1H-NMR, Raman spectroscopy, TGA, AFM, UV spectroscopy | “Grafting from” approach. i.e., covalent bonding of CL to CNT followed by the AROP of CL, proved | [135] |
| MWCNT (<0.3 wt %) | CL | Dilactamate®/TDI (0.3 mol %/0.15 mol %) | CL mixed with MWCNT at 100 °C, then initiator introduced at 135 °C followed by the activator and mixing. Cast polymerization at 175 °C for 3.5–4.5 min. | DOC, DSC, TGA, DMA, mechanical properties | DOC >96%. All nanocomposites showed increased tensile modulus and strength compared to neat PA-6. The elongation at break did not change whereas the Charpy IS decreased with increasing MWCNT content | [136] |
| MWCNT with –OH functionality | CL | Na(NaCL)/CL-functionalized MWCNT (MWCNT-OH reacted first with TDI and then with CL), see Figure 6 | CL + Na + CL-functionalized MWCNT mixed/sonicated at 70 °C for 30 min. Polymerization at 170 °C for 6 h | FTIR, TGA, UV-Vis, TEM | “Grafting from” approach in two steps (CL-functional MWCNT activator formation and acyl-CL initiated AROP of CL) confirmed | [137] |
| MWCNT (purified) <1 wt % | CL | NaH (NaCL)/N-acetyl-caprolactam | CL + polyoxyethylene + MWCNT + acetyl-caprolactam mixed/sonicated, then NaH added and polymerized at 120 °C for 6 min. Fibers produced at different stretching ratios. | Viscosimetry (Mv), SEM, DSC, tensile tests | MWCNT dispersed by ultrasonication. Tensile E-modulus and tensile strength increased by ~40% in case of 1 wt % MWCNT containing nanocomposite with a stretching ratio of 4. | [138] |
| MWCNT with –OH functionality (≤0.2 wt %) | CL | NaCL/TDI | MWCNT-OH dispersed in molten CL through a water-assisted method. Water removed at 170 °C. Then NaCL and TDI introduced, polymerization at 160 °C for 10 min. | Optical microscopy, DSC, TEM, TGA | Fine dispersion of MWCNT-OH acting as heterogeneous nucleating agent. DOC ~96% | [139] |
| MWCNT with –OH functionality (≤1.5 wt %) | CL | NaCL(C10)/MWCNT-NCO + TDI (prepared by reacting MWCNT-OH with TDI) | To CL solution in DMF MWCNT-NCO was added and ultrasonicated at RT. DMF removed in vacuo and heated to 170 °C. After adding TDI, NaCL was added and cast polymerization performed at 160 °C for 10 min. | FTIR, SEM, DSC, TGA, tensile properties | PA-6 chains covalently attached to the sidewalls of MWCNT which were uniformly dispersed. MWCNT worked as nucleating agent and also improved the thermal stability. Tensile modulus and strength were markedly improved at cost of the elongation at break. | [140] |
| MWCNT | CL | C10/C20 (0.3 wt %/0.3 wt %) | Small samples produced for DSC and rheology tests at T = 180–220 °C | DOC, DSC, design of experiments, GPC, rheology | MWCNT had inhibiting effect on the AROP of CL. DOC was simulated. The MW was not affected by MWCNT. It was suggested that MWCNT may react with the initiator. | [141,142] |
| MWCNT (≤5 wt %) | LL | NaH/N,N′-ethylene bis(stearamide) (molar ratio = 1/0.5) | Polymerization in microcompounder: premixing at 170 °C for 5 min and polymerization at 270 °C for 4 min under N2 | TGA, GPC, optical microscopy, TEM, electrical conductivity | DOC at ~99%. Mv values between 10 and 41 kDa along with polydispersity in the range of 1.5–2.2. MWCNT delayed the polymerization. Volume resistivity dropped 8 order of magnitudes at 5 wt % MWCNT compared to the neat PA-6. Similar results obtained by classical melt mixing of PA-12 with MWCNT. | [143] |
| MWCNT (1 wt %) | CL + styrene (successive polymerization, styrene first) PA-6/PS blend ratio: 80/20 | NaCL/TDI | First PS/CL/MWCNT mixture obtained after the polymerization of styrene. To this mixture NaCL and TDI were added at 150 °C and residual styrene removed. Cast polymerization of CL at 180 °C for 20 min. | SEM, TEM, dielectric spectroscopy | PS became the dispersed phase and MWCNTs were selectively located in the interphase between PA-6 matrix and PS. | [144] |
| CNF (stacked-cup) ≤ 0.8 wt % | CL | Na (NaCL)/caprolactam-functionalized CNF + caprolactam-capped diisocyanate | CNF was acid treated and functionalized with HMDI in DMF, then capped with CL. CL melted at 80 °C and CL-functionalized CNF + CL-capped diisocyanate added. Polymerization at 150 °C for 30 min. | Viscosimetry (Mv), TEM, FTIR, TGA, SEM, PSC, WAXS, mechanical and impact tests | Stiffness and strength significantly enhanced along with slight improvement in toughness. CNF promoted the formation of the γ-phase. Mv data scattered between 54 and 59 kDa. | [145] |
| Cellulose nanocrystal (CNC) (≤2 wt %) | CL | NaH (NaCL)/phenyl isocyanate (1.5 mol %/1.2 mol %) | CNC dispersed in molten CL under sonication. Initiator added in N2 atmosphere. Activator, prepared separately by reacting CL with the isocyanate, added and polymerization at 150 °C for 30 min. | DOC, TGA, DMA, AFM, SEM, creep melt rheology | CNC was efficient reinforcement: improved the creep resistance, enhanced the DMA properties. The zero shear viscosity was prominently higher in CNC presence compared to the neat PA-6, suggesting the onset of a percolated structure that was prone for breaking upon shear. | [146] |
| CNC with and without aminosilane surface modification (≤3 wt %) | CL | EtMgBr (CLMgBr)/C20 | CL + CNC +initiator was mixed with CL + activator and polymerized at 150 °C. Samples produced by extrusion. For comparison purpose classical melt blending served. | SEM, TEM, TGA, FTIR, solid state NMR, rheology (nano) mechanical tests | Based on solid state NMR CNC-grafted PA-6 was proposed (involving transamidation, urea bond formation). Tensile stiffness and strength strongly improved at cost of elongation at break. Melt elasticity and strength enhanced by CNC reinforcement. | [147] |
| MMT, pristine (NaMMT) and organophil (intercalant: dioctadecyl dimethyl ammonium chloride) versions (OMMT) (≤2 wt %) | CL | C10/TDI | NaMMT dispersed in aqueous CL under ultrasonication. Afterward water removed in vacuo at 170 °C, then initiator added followed by TDI. Polymerization at 160 °C for 10 min. OMMT introduced directly or in acetone—assisted dispersion. | GPC, X-ray diffraction (XRD), TEM, TGA, DSC | DOC was higher than 94% except OMMT (86%). Mn and Mw values were at about 20 and 50 kDa respectively. NaMMT was exfoliated based or XRD results below 1.5 wt % content. Above this intercalation took place. The thermal stability was prominently improved by NaMMT. NaMMT acted as heterogeneous nucleant and promoted also the γ-phase formation. OMMT appeared in intercalated structure and did not improve the PA-6 matrix properties. | [148,149] |
| NaMMT (pristine clay) (3 wt %) | CL | NaCL/TDI in presence PMMA-Na+ ionomer as compatibilizer | NaMMT + CL + PMMA-Na+ ionomer mixed in aqueous solution, then water evaporated. Initiator and activator added and cast polymerized at 180 °C for 10 min. | XRD, DSC, TEM, shear viscosity | NaMMT was intercalated in absence of the compatibilizer or in its low amount. Exfoliated structure received in the blend PA-6/clay/ionomer = 97/3/4.5. Well dispersed clay layers reduced the crystallinity and favored the formation of the γ-polymorph. | [150] |
| Clay (MMT) with and without organophile modification (≤4 wt %) | CL | Initiator/activator undefined | Preparation via reactive extrusion. (CL + initiator) and (CL + activator) were separately introduced into an extruder. Extruder temperatures: polymerization and processing zones 180 °C and 220 °C, respectively. Clay added differently. | TEM, optical microscopy, tensile properties | Continuous production of PA-6/clay nanocomposites is feasible. Clay particles are intercalated/partly exfoliated. The E-modulus of PA-6 is increased by 20% and 30% by the incorporation of 2 and 4 wt % clay, respectively. | [151,152] |
| NaMMT (clay) (2 wt %) | CL, LL, CL + LL | NaCL or CLMgBr/N-acetyl caprolactam (0.5 mol %/0.5 mol %) | AROP of lactams performed at 180 °C for 30 min in N2 atmosphere | DOC, GPC, XRD, DSC, SEM, TEM | NaCL produced random, whereas CLMgBr tended to result in block copolymers. The intercalation was reduced with increasing LL content. In the block-type copolymer the intercalation of clay remained the same with increasing LL content. LL content reduced the DOC and MW of the final copolymer. Crystallinity strongly reduced by LL content. | [153] |
| OMMT (≤10 wt %) | CL | Dilactamate®/C20 (1.5 mol %/0.75 mol %) | CL melt mixed with OMMT under N2 at 110 °C. Then initiator and activator added. Polymerization in a mold placed in a hot press (165 °C, 10 MPa) | DOC, Synchrotron WAXS, FTIR, TEM | Conversion > 97%. Up to 1 wt % OMMT was exfoliated, above this intercalated. Micronscale OMMT agglomerates also revealed. The matrix in the nanocomposites was α-phase. After melting/recrystallization the γ-form appeared. | [154] |
| OMMT (≤10 wt %) | CL | NaH/N-acetyl caprolactam | Polymerization in solution using NMP at 160 °C for 30–45 min | DSC, SEM, WAXS, viscosimetry (Mv) | MW dropped with increasing OMMT content. Crystallinity increased up to 1 wt %. OMMT then decreased. At higher OMMT content PA-6 crystallized in γ-form. OMMT intercalation was supported by the polymerization in solvent. | [155] |
| Graphene (≤0.5 wt %) | CL | NaOH/TDI | Graphene added to molten CL and ultrasonicated. NaOH introduced and water removed in vacuum at 180 °C followed by dosing TDI. Cast polymerization at 160 °C for 15 min | GPC, TEM, SEM, XPS, Raman, DSC, TGA, mechanical properties | MW (both Mn and Mw) slightly reduced with increasing graphene content. Nanocomponents displayed higher thermooxidative stability than PA-6. Flexural modulus, strength and impact strength drastically enhanced while the formation of γ-polymorph promoted. | [156] |
| Graphene oxide (GO) (≤1 wt %) | CL + ε-caprolactone (ratio: 90/10 and 80/20) | CLMgBr/ε-caprolactone (activator) | GO dispersed in molted CL at 80 °C in Ar atmosphere. Mixture heated to 110 °C and initiator added, followed by ε-caprolactone. Cast polymerization at 150 °C for 1 h. | XPS, TGA, TEM, viscosimetry (Mv) DSC, mechanical tests | Mv decreased with GO content. The formed poly(ester amid) was random type. GO acted as nucleating and reinforcing additive. E-modulus increased while impact strength decreased with increasing GO content. | [157] |
| Reinforcement | Monomer/Solvent or Copolymer (Amount) | Initiator/Activator (Amount) | Technology | Process Parameters | Testing | Results, Comments | Refs. |
|---|---|---|---|---|---|---|---|
| GF | CL/- | Sodium dihydridobis(2-methoxyethoxo)aluminate/PIC (0.3/0.3 mol %) | Casting | Tpolym = 135 °C; T0 = 133–134 °C; Tmax = 205 °C; | ρ, XC (X-ray diffraction), Vf, SEM, DMA, mechanical tests, DSC |
| [164] |
| Sodium tetra(6-caprolactamo) aluminate/PIC (0.3/0.3 mol %) | |||||||
| CL/- | NaCL/HMDI (0.75/0.75 mol %) | Pultrusion | Vf = 72 %; Tproc = 140–160 °C; Vpul = 40.6 cm/min; Treact = 52 s. | FTIR, DMS, viscosity, IS, SEM |
| [91] | |
| LL/Dimethylpropylene urea | NaCL (0.75 wt %; 1 wt %; 3 wt %)/ cycloaliphatic monocarbodiimide (0.75/0.75 wt %) (1/1 wt %) (3/3 wt %) | Pultrusion | Tproc = 230–290 °C Vpul = 0.8–3.4 m/min dpolym = 3.15 m; 2.10 m; 1.05 m. Fpul = f (Tproc) = 300–1450 N | DOC, XC |
| [25,166] | |
| NBC | Not mentioned/acyllactam end groups & carbonyl groups of the polyesteramide prepolymer | Casting | Tpolym = 130 °C | Mechanical properties, thermal expansion, water absorption. | GF in NBC gives increased resistance to expansion from moisture absorption and thermal changes. Temperature resistance of stiffness and resistance to heat sag improved. Losses in IS may be partially restored by moisture absorption and/or changes in resin matrix formulation. | [163] | |
| NBC | Acyllactam end groups of the prepolymer */not mentioned *Polyesteramide prepolymer terminated by acyllactam | Rotation molding | Tmold = 110–190 °C | XC = f (Tmold); IS = f (Tmold, tcycle, filler); E = f (Tmold, filler); Shrinkage = f (Tmold); Water uptake = f (Tmold) |
| [90] | |
| CF | CL/- | NaCL/tert-butyl acetate (1/2 mol %) | Casting | treact = 30–40 min; Tpolym = 195–220 °C | Mechanical properties |
| [162] |
| NaCL/ε-caprolactone (1/2 mol %) | |||||||
| NaCL/benzyl benzoate (1/2 mol %) | |||||||
| NaCL/benzyl acetate (1/2 mol %) | |||||||
| NaCL/phenyl acetate (2/1 mol %) | |||||||
| CBT, AROP of lactams and their copolymers | Not disclosed | Pultrusion | 3 heating zones in the die: T1 = 170 °C, T2 = 180 °C, T3 = 190 °C. Toven = 240 °C | E; σ; εR | The field of the invention relates to the conductor for electrical transmission lines having composite load bearing core produced by pultrusion using a thermoplastic polymer matrix, by in situ polymerization of the cyclic monomers and/or oligomers, optionally in the presence of polymers prone to melt phase transreactions, with reinforcement consisting of high modulus and strength fibers. | [168] |
| Reinforcement | Monomer | Initiator/Activator (Amount) | Technology | Production Parameters | Testing | Results, Comments | Refs. |
|---|---|---|---|---|---|---|---|
| GF 8-harness satin weave, 300 gsm, E-glass | CL | C1/C20 1.2 mol %/1.2 mol % | VARTM | Pp = 250 mbar tcycle = 60 min Tm = 110 °C Tmold = 160 °C | DOC, XC, ILSS, ultrasonic analysis, microscopy, VC, mechanical tests | The highest XC = 41% and DOC = 96% were achieved at Tmold = 160 °C with 6% void content (VC) and ILSS of 62 MPa. Vf = 50%. The highest ILSS ≈ 68 MPa and the lowest VC = 2% were achieved at Tmold = 180 °C with a DOC = 93% and the XC = 32% respectively. Vf = 50%. In both cases a special aminosilane sizing was used. Melt degassing in a buffer vessel. | [169,173] |
| PP = 250 mbar tcycle = 60 min Tm = 110 °C Tmold = 180 °C | |||||||
| PP = 250 mbar tcycle = 60 min Tm = 110 °C Tmold = 170 °C | Mechanical characteristics were measured in dry as molded, and 23 °C/50% RH conditions: Compressive strength, modulus and strain; Tensile strength, modulus and strain; Shear strength, modulus; | [172] | |||||
| GF—plain woven S-glass, 400 gsm | CL | C10/C20 1–3/0.5–1.5 mol % molar ratio 2:1 | VARTM | C20: 0.5–1.5 mol % tpolym = 60 min Tmold = 180 °C | Mv, XC, mechanical tests, morphology, ILSS | Mv: 10–12 kDa ILSS: 33–43 MPa Tensile strength: 328–434 MPa Flexural strength: 320–407 MPa; XC: 37%–43% | [176] |
| C20 = const tpolym = 60 min Tmold = 150–190 °C | Mv: 10–12 kDa ILSS: 38–44 MPa Tensile strength: 363–434 MPa Flexural strength: 333–396 MPa; XC: 45%–40% | ||||||
| C20 = const tpolym = 5–120 min Tmold = 160 °C | Mv—almost unchanged ILSS: 38–44 MPa Tensile strength: 382–437 MPa; Flexural strength: 364–395 MPa; XC: 41%–44% | ||||||
| GF-plain weave, 588 gsm, E-glass | CL | C1/4,4′-methylenediphenyl diisocyanate 5/0.9 wt % | VARTM | tcycle = 60 min Tm = 120 °C Tmold = 160 °C | Microscopy, 1H-NMR, FTIR, TGA, DOC | A single-stream processing technique was introduced. An organosilane activator was deposited on the GF surface (N-[5-(trimethoxysilyl)-2-aza-1-oxopentyl]caprolactam), and different isocyanate-based activators used. DOC: inlet and outlet: close to 100%, middle: below 25%. | [179] |
| GF—continuous strand mat (swirl mat), 450 gsm | NBC | - | SRIM | - | Acoustic emission, mechanical tests, IS, microscopy | Fracture toughness (KC) improved with increasing of Vf of GF. Increasing of the crosshead speed resulted in increased Kc that is untypical. IS went through a maximum as a function of temperature. Failure sequence analysis was performed using AE and optical microscopy simultaneously. Fracture mechanics data depended on specimen size and type: the reasonable ligament width and length to span ratio were defined as >12 and >1.7 respectively. | [184,185,186] |
| CF—4 harness satin weave, 200 gsm | CL | C10/C20 3/1.5 mol % | VARTM | Pp = 98 kPa Tm = 100 °C Tmold1 = 100 °C Tmold2 = 150 °C | TGA, DSC, DOC | The mold and the melt temperatures were 100 °C. After a complete impregnation of a preform the temperature was raised up to 150 °C. Average DOC = 98.01%. Average XC = 40%. Reinforcement’s weight fraction, Wf = 64% (uniform). | [177] |
| Pp = 98 kPa Tm = 150 °C Tmold = 150 °C | The mold and melt temperatures were 150 °C. Infusion was incomplete (75% impregnation) due to fast polymerization of the melt. Average DOC = 97.98%. Average XC = 36% (more consistent). Wf = 52%. | ||||||
| CF—2/2 twill fabric | LL | NaH/N,N′-ethylenebisstearmide | T-RTM | Tm = 160 °C Pclamp = 10 bar Tmold = 270 °C tcycle = 10 min | SEM, TGA, density, XC, mechanical tests, DMTA | Vf = 30% Flexural strength = 311 MPa; Flexural modulus = 21.2 GPa; XC = 29% Residual monomer content = 0.9% System working reliably. | [187] |
| Grilonit LA 2.5 wt % | Vf = 54%; Flexural strength = 321.2 MPa; Flexural modulus = 37.8 GPa; XC = 52%. | ||||||
| CF—woven 2/2 twill, 240 gsm | LL | Grilonit LA 1.5–5 wt % | T-RTM | PP = 1 bar (over) Tmold = 180–250 °C LA = 2 wt % | Mechanical tests, ILSS | tpolym = 5 min at 250 °C. Processing window: 20 min at 200 °C and 5 min at 250 °C. Tensile strength and stiffness of epoxy based and PA12 based laminates were compared; Testing in ILSS demonstrated plastic deformation instead of failure. | [188] |
| Tm = 180 °C Tmold = 240 °C tinj = 10 sec tpolym = 8.5 min PP = 0.4 bar (over) | Performance of in situ produced PA-12 plate with Vf = 54% and commingled CF/PA-12 at Vf = 56% compared. Composites with in situ PA-12 matrix showed good tensile properties under different conditionings while the compressive performance was lower than that of the CF/PA-12 from commingled yarns. ILSS could not be assessed due to plastic deformation of specimens. | [189] | |||||
| CF—satin weave, 440 gsm | LL | Liquid activating system: NaCL/Carbodiimide 1.5 % | T-RTM | - | Infiltration, diffusion, shrinkage, VC | Matrix shrinkage and residual N2 are specified as potential sources of VC growth. At the equilibrium 1.86 kg of N2 is dissolved in 1 m3 of lactam at 170 °C. Due to pumping of mixture into the mold at 170–190 °C the released gas can generate porosity. To minimize the VC the N2 content in the melt should be minimized and an optimal capillary number set for infusion. Bleeding was used to reduce the VC in parts with degassed matrix. The average VC was reduced from 15% to 1% | [174] |
| CF—5-harness satin weave, 440 gsm | LL | Liquid activator 2–4 % | T-RTM | Tm = 180 °C Tmax = 255 °C Tcool = 140 °C PP1 = 1.5 bar PP2 = 55 bar Pinj = 0.2 bar (over) Flow rate = 200 cm3/min tcycle = 25 min | Mechanical tests, VC | Two types of composites are compared for thermoforming application: commingled CF/PA-12 and in situ polymerized CF/PA-12 Tensile properties were defined before and after heat stamping. P1—injection/P2—compression ratio varied. Reconsolidation of the composites after preheating remained incomplete. Recycling and overinjection molding strategies presented. VC below 1%. | [181] |
| NF—ramie, warp/weft yarn 21S × 21S, 52 × 36 | CL | C1/C20 1.2/0.6 mol % | VARTM | Tmold = 150 °C PP1 = 100 mbar | DOC, XC, mechanical tests, viscosimetry | DOC = 94.4% XC = 48.0% Mv = 101 kDa Wf = 40%. | [178] |
| C10/C20 1.2/0.6 mol % | - | FTIR, atomic absorption spectroscopy | Drastic inhibition and discoloration observed with NaOH and C10 initiators in reactive processing due to the byproducts generated by the “peeling reaction” of cellulose in alkaline environment under heat. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ageyeva, T.; Sibikin, I.; Karger-Kocsis, J. Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends. Polymers 2018, 10, 357. https://doi.org/10.3390/polym10040357
Ageyeva T, Sibikin I, Karger-Kocsis J. Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends. Polymers. 2018; 10(4):357. https://doi.org/10.3390/polym10040357
Chicago/Turabian StyleAgeyeva, Tatyana, Ilya Sibikin, and József Karger-Kocsis. 2018. "Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends" Polymers 10, no. 4: 357. https://doi.org/10.3390/polym10040357
APA StyleAgeyeva, T., Sibikin, I., & Karger-Kocsis, J. (2018). Polymers and Related Composites via Anionic Ring-Opening Polymerization of Lactams: Recent Developments and Future Trends. Polymers, 10(4), 357. https://doi.org/10.3390/polym10040357