1. Introduction
Anomalous amplification of weak interactions through synchronized behavior of components is ubiquitous in Nature on many scales, from elementary particles to the global scale. In contrast, the synchronous behavior, which should seriously affect material properties, of molecules has not been paid much attention by the chemistry community. Earlier, we proposed the possibility that molecules could be synchronized under very rapid environmental changes (e.g., pH or solvent polarity) in flowing micro-solutions [
1,
2]. Although those results were very primitive, we established simple models for chemists to consider the synchronized behaviors of molecules.
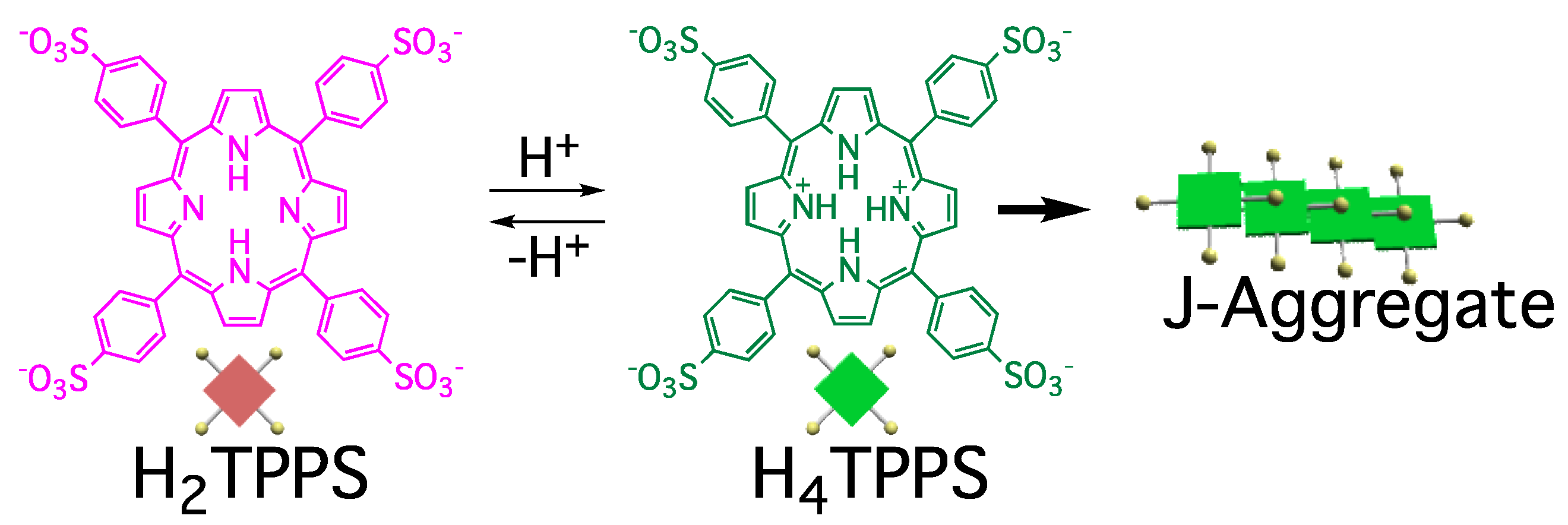
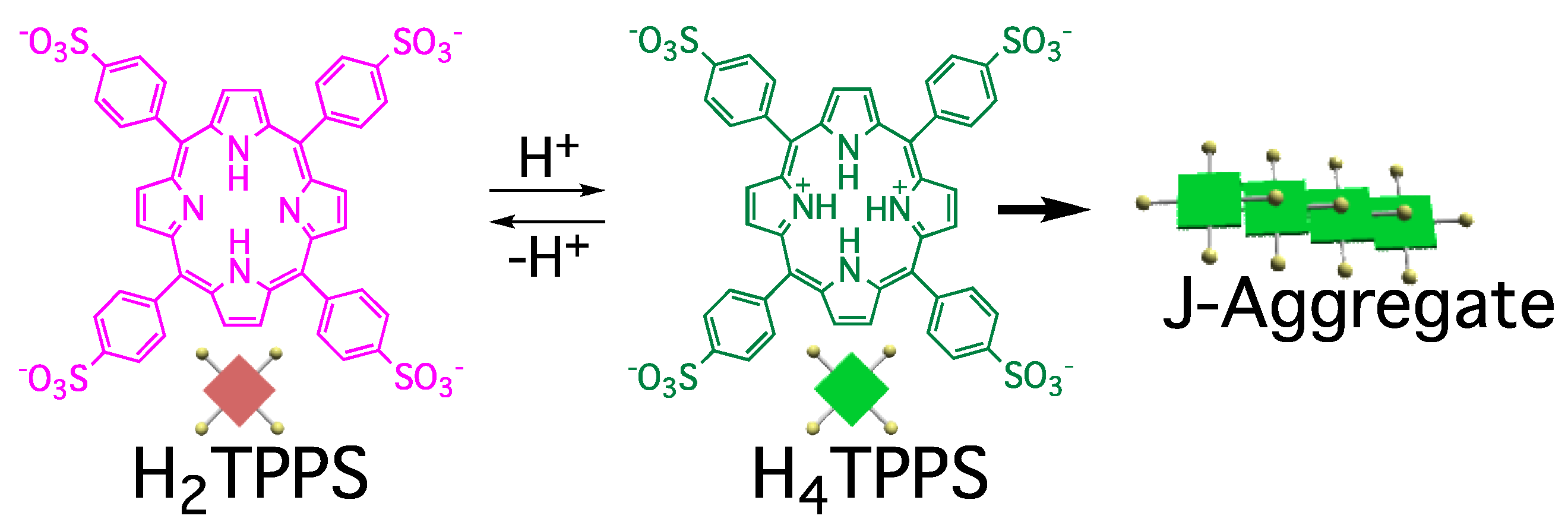
We have employed tetrakis (4-sulfonatophenyl) porphyrin (TPPS, with H
2TPPS and H
4TPPS representing its free and protonated forms, respectively) as a model compound for such studies (
Figure 1) [
3,
4,
5,
6,
7,
8]. J-aggregates formation is triggered by protonation of the central pyrrole groups. The driving force for self-assembly of the resultant H
4TPPS is hydrogen bonding and other electrostatic intermolecular interactions. In a bulk solution, TPPS self-assembles into J-aggregates under acidic conditions, where protonation/deprotonation processes between H
2TPPS and H
4TPPS are under equilibrium. The efficiency of J-aggregate formation is governed predominantly by the concentration of H
4TPPS; therefore, J-aggregate formation is accelerated at lower values of pH. In contrast to this general behavior in solution, we have found that the efficiency of J-aggregate formation is correlated to the time required for protonation of H
4TPPS [
1]. Very rapid proton diffusion in a microflow channel [
9,
10,
11,
12,
13,
14] results in increasing temporal H
4TPPS concentrations, even at a constant value of pH [
15,
16].
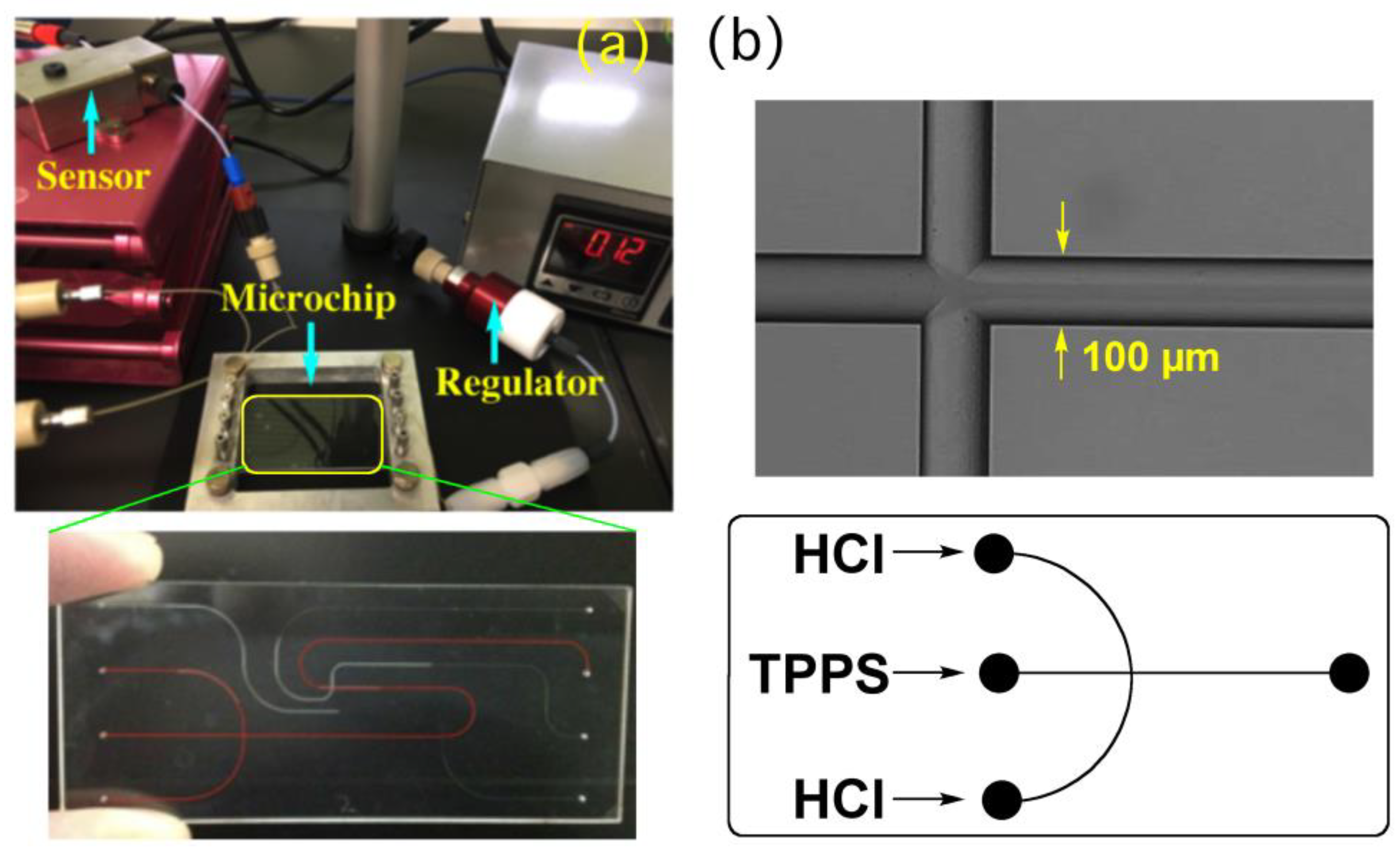
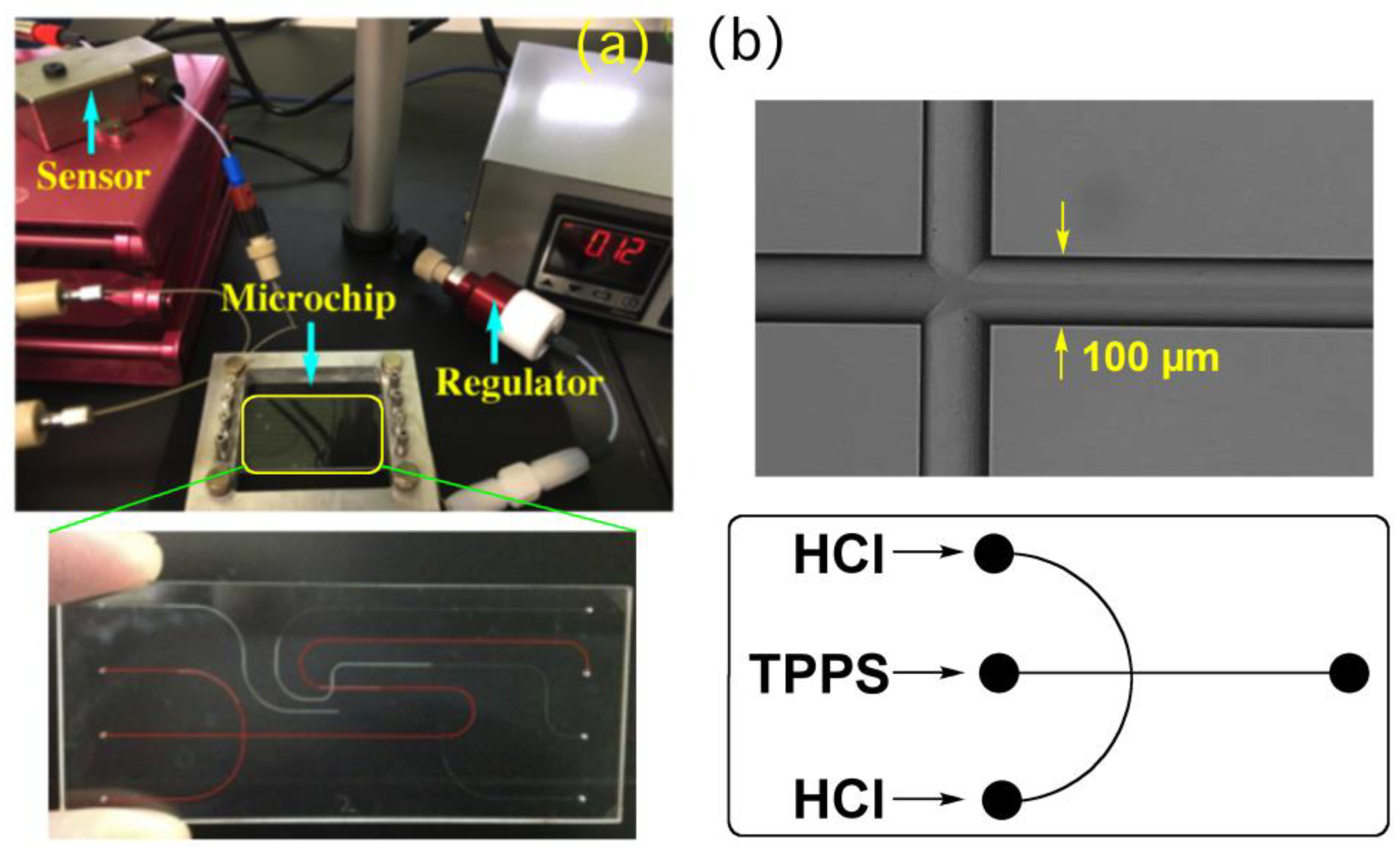
In the present study, we have employed focusing hydrodynamic flow as self-assemble fields [
17,
18,
19], where the central flow is the main channel along which self-assembly proceeds with time and aqueous HCl solutions are injected from the side flow (
Figure 2). The time required for complete diffusion, the residence time in the channel, and final pH values are precisely regulated by changing the flow rate ratios, which enable precise control over the self-assembly dynamics.
Flowing TPPS molecules in a micro-channel are always under the influence of back-pressure. We have demonstrated recently that J-aggregate formation can also be accelerated by back-pressure in a micro-channel [
2]. Accordingly, precise evaluation of the effect of simultaneous protonation of H
2TPPS on its self-assembly should carefully subtract the effect of back-pressure. In this present study, we found that pressure effects have little influence on the overall self-assembly behavior, strongly supporting our finding that very rapid protonation and adjusting time phases of protonation/deprotonation oscillation play essential roles leading to overall acceleration of the self-assembly process.
3. Results and Discussion
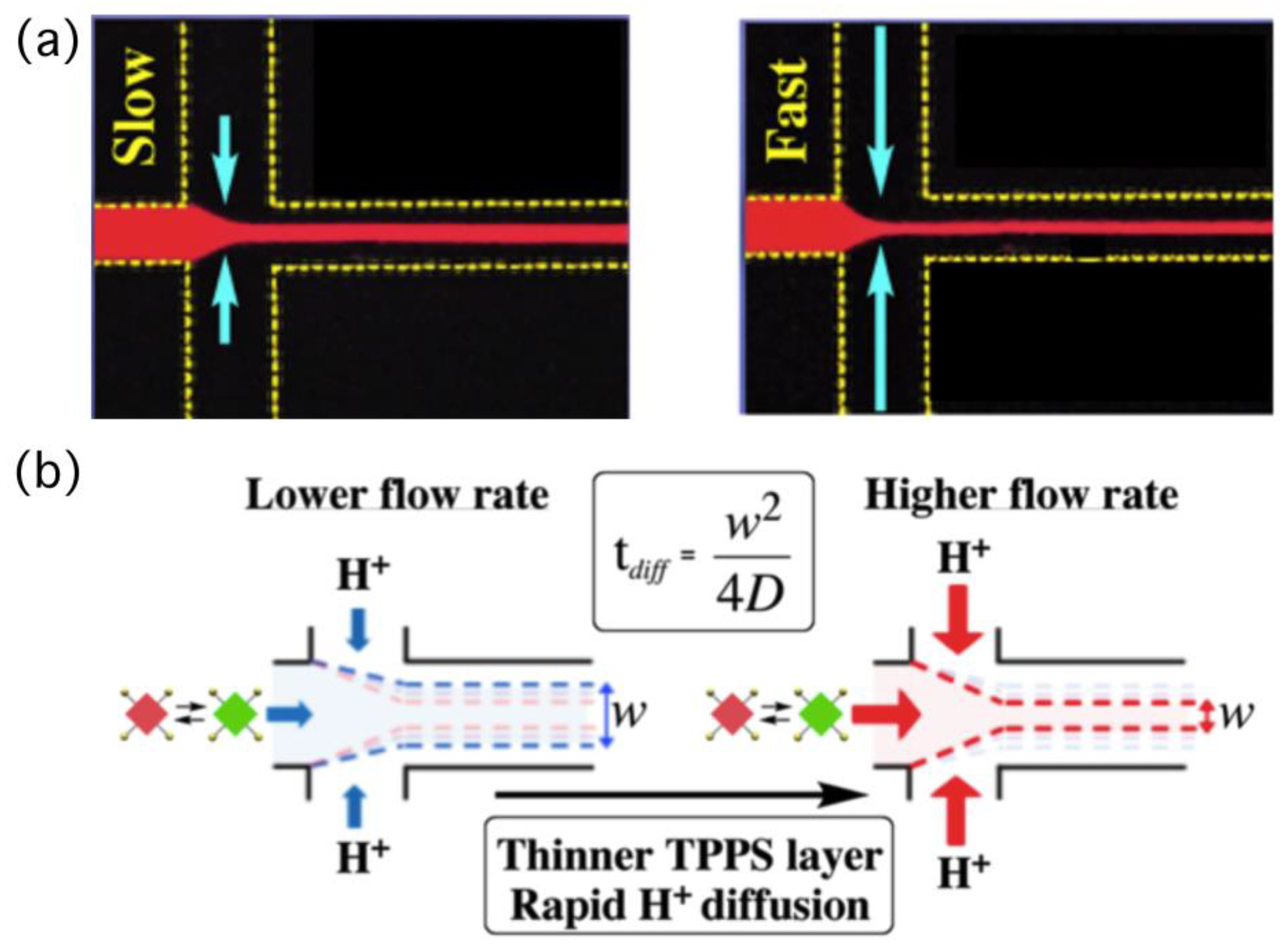
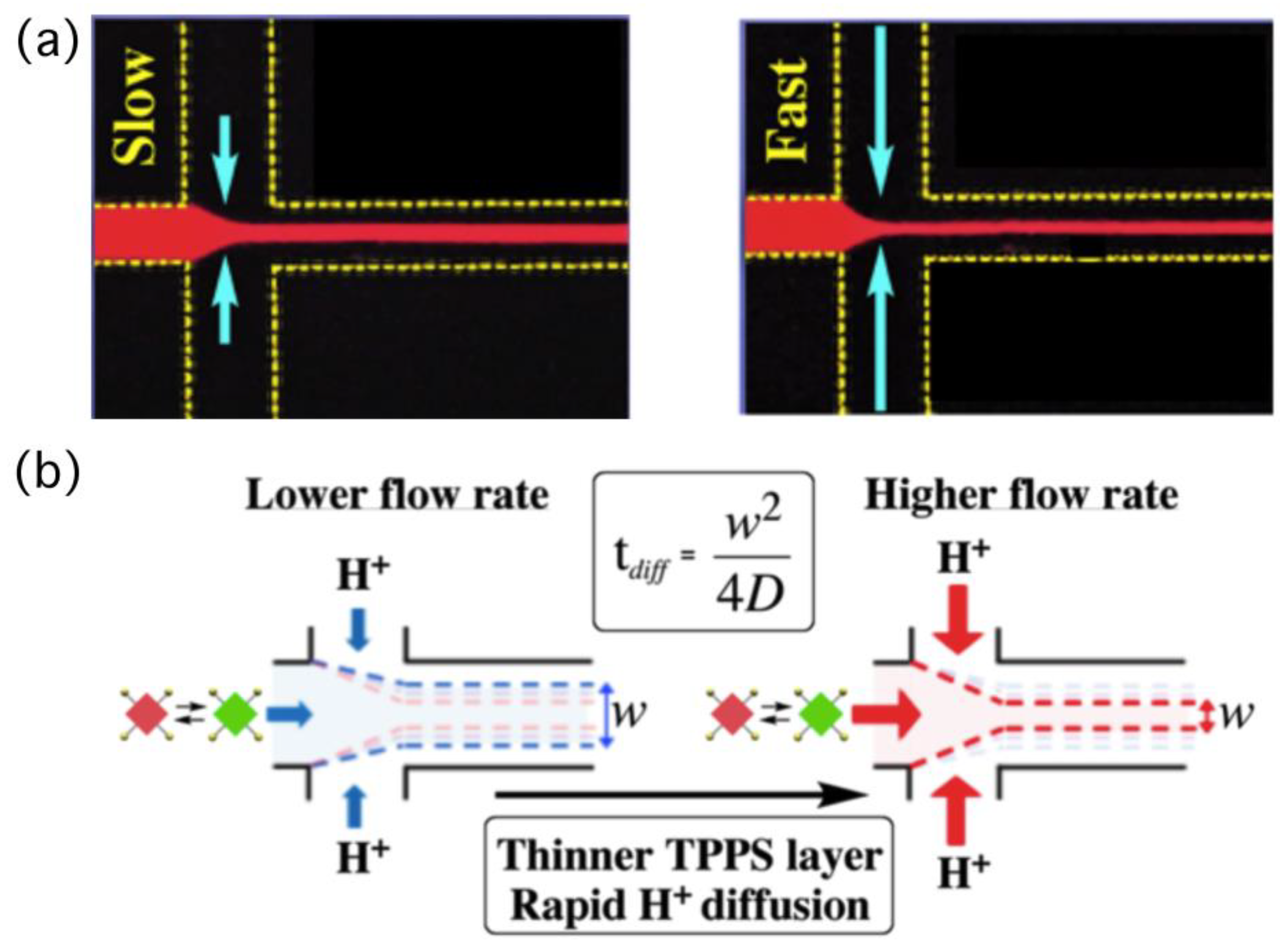
Figure 3a presents a fluorescence microscopy image of a flowing solution of TPPS in a microflow channel. In this study, we injected aqueous hydrochloric acid as lateral solutions, resulting in protons always diffusing into the central thin-layered TPPS solution. In this particular system, the time required for proton diffusion was directly correlated to the width of the central flow, which we could control through the flow rate—a faster flow led to a narrower width (
Figure 3b and
Figure 4a). Controlling the time for proton diffusion was essential because the effective self-assembly of TPPS molecules would occur only during periods of rapid proton diffusion. To evaluate the effect of the time on self-assembly quantitatively, the mole fraction of aggregated dye (α
agg) was defined as below:
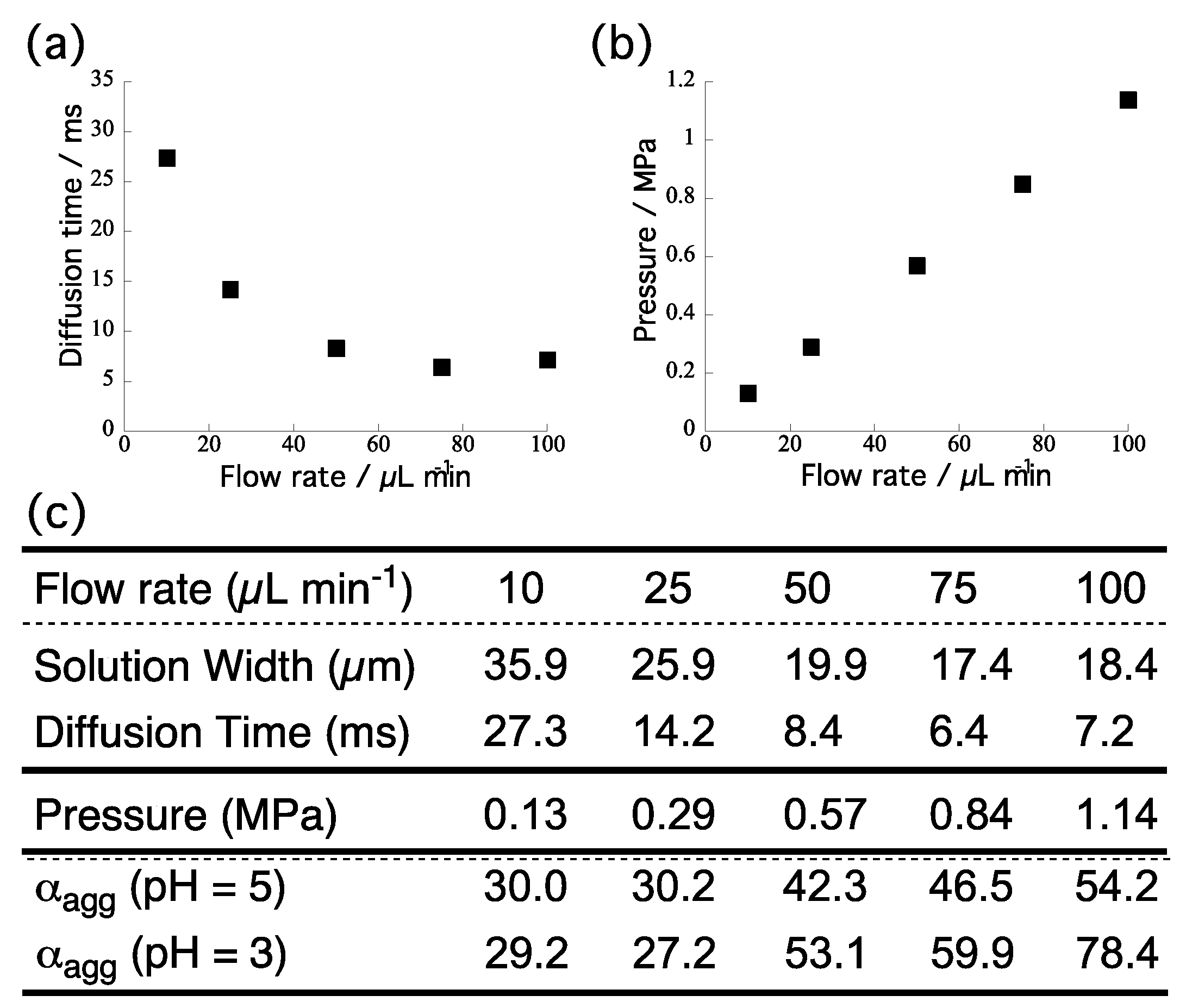
Figure 4c displays the α
agg with respect to the width of the central TPPS layer, as well as the time required for proton diffusion. The time required for proton diffusion in the channel was evaluated according to the reference (see the
Supplementary Materials). We observe a direct correlation between the
αagg and the time required for proton diffusion.
Based on these results, next, we thoroughly examined how the rapid diffusion contributed to the total increase in the
αagg upon increasing the flow rate. To measure the back-pressure applied inside the channel, we connected a differential pressure gage to the inlet and outlet of the channel. According to the Hagen-Poisuille equation, the pressure applied inside the channel would be proportional to the flow rate (
Figure 4b).
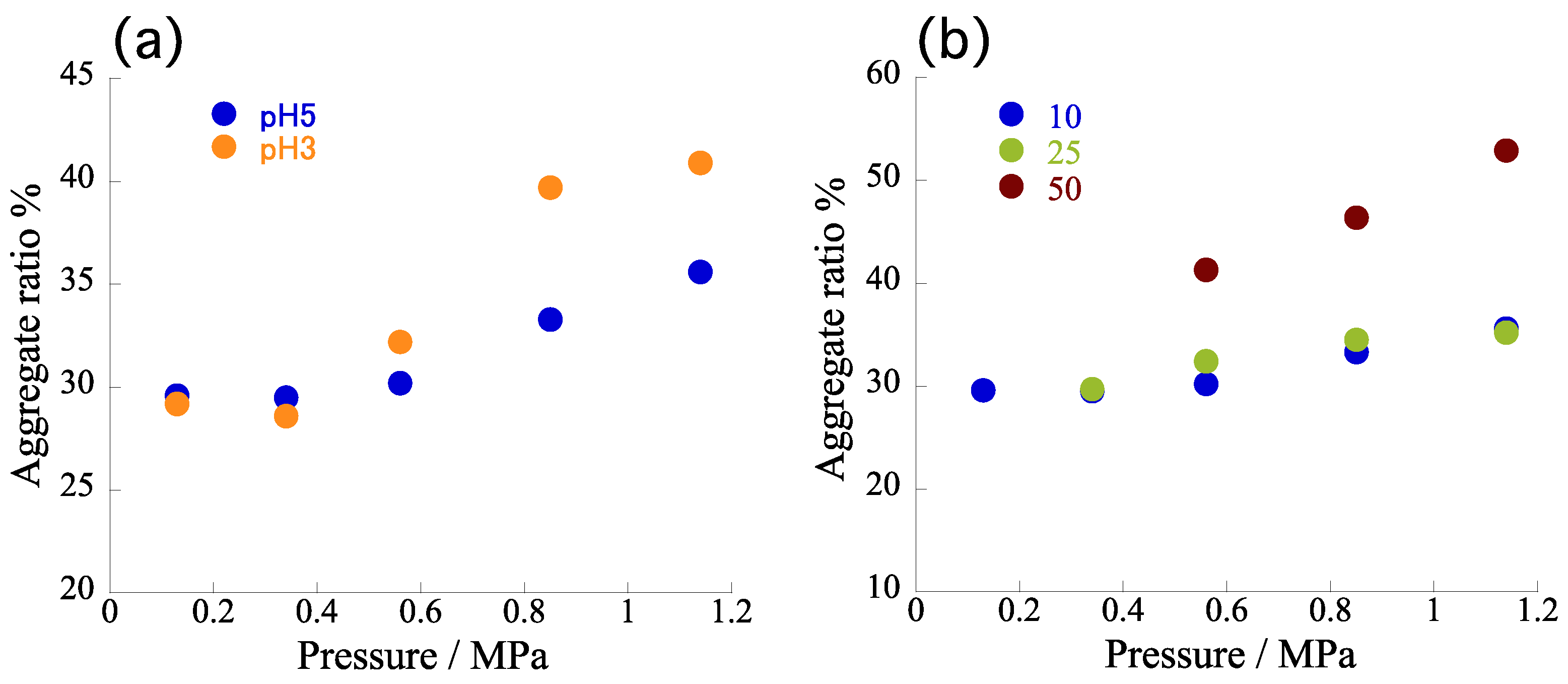
Figure 4c summarizes the evaluated pressures at each flow rate (i.e., 10, 25, 50, 75, 100 μL/min). We observed a proportional correlation between the flow rate and back-pressure, in good accordance with the theoretical prediction.
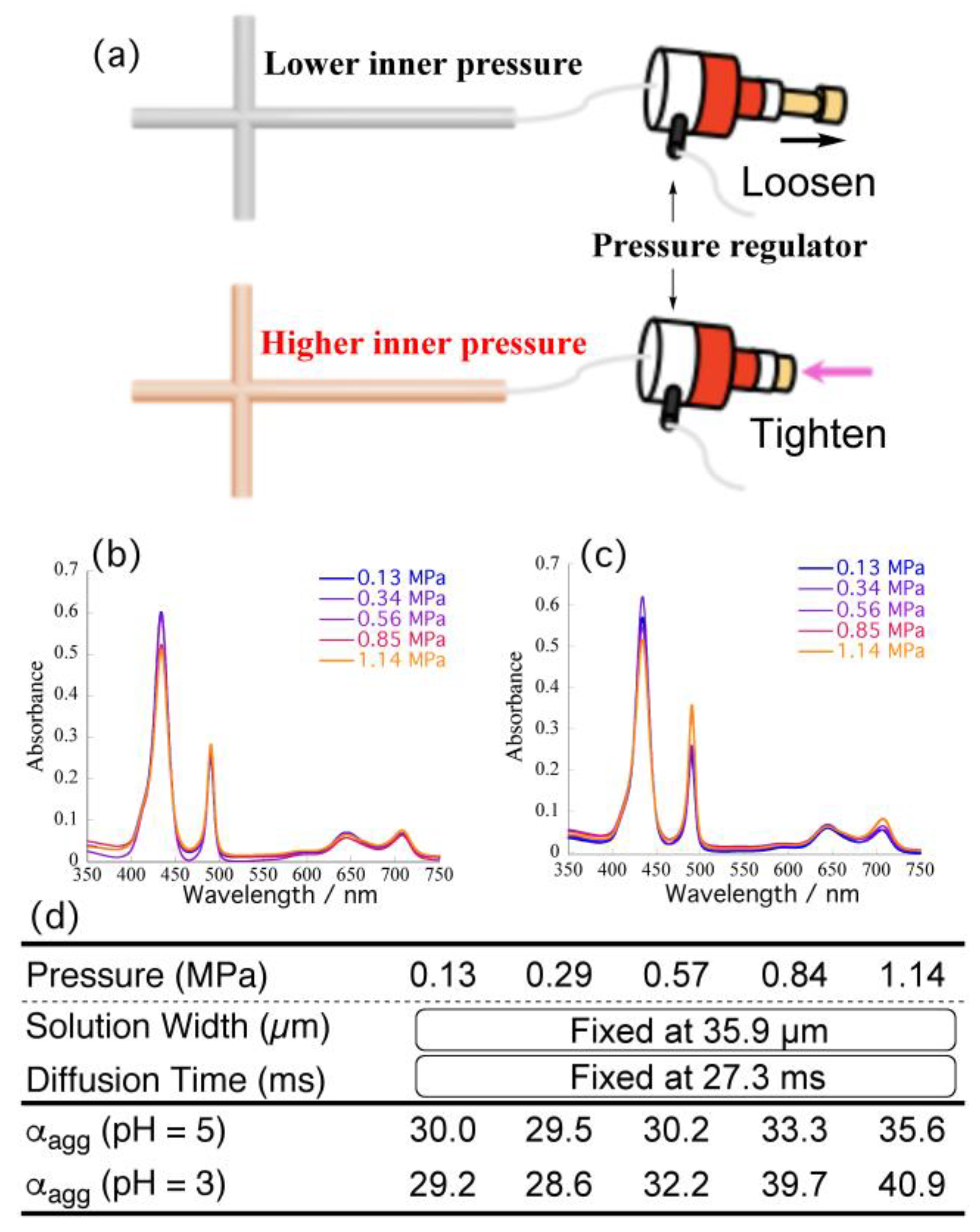
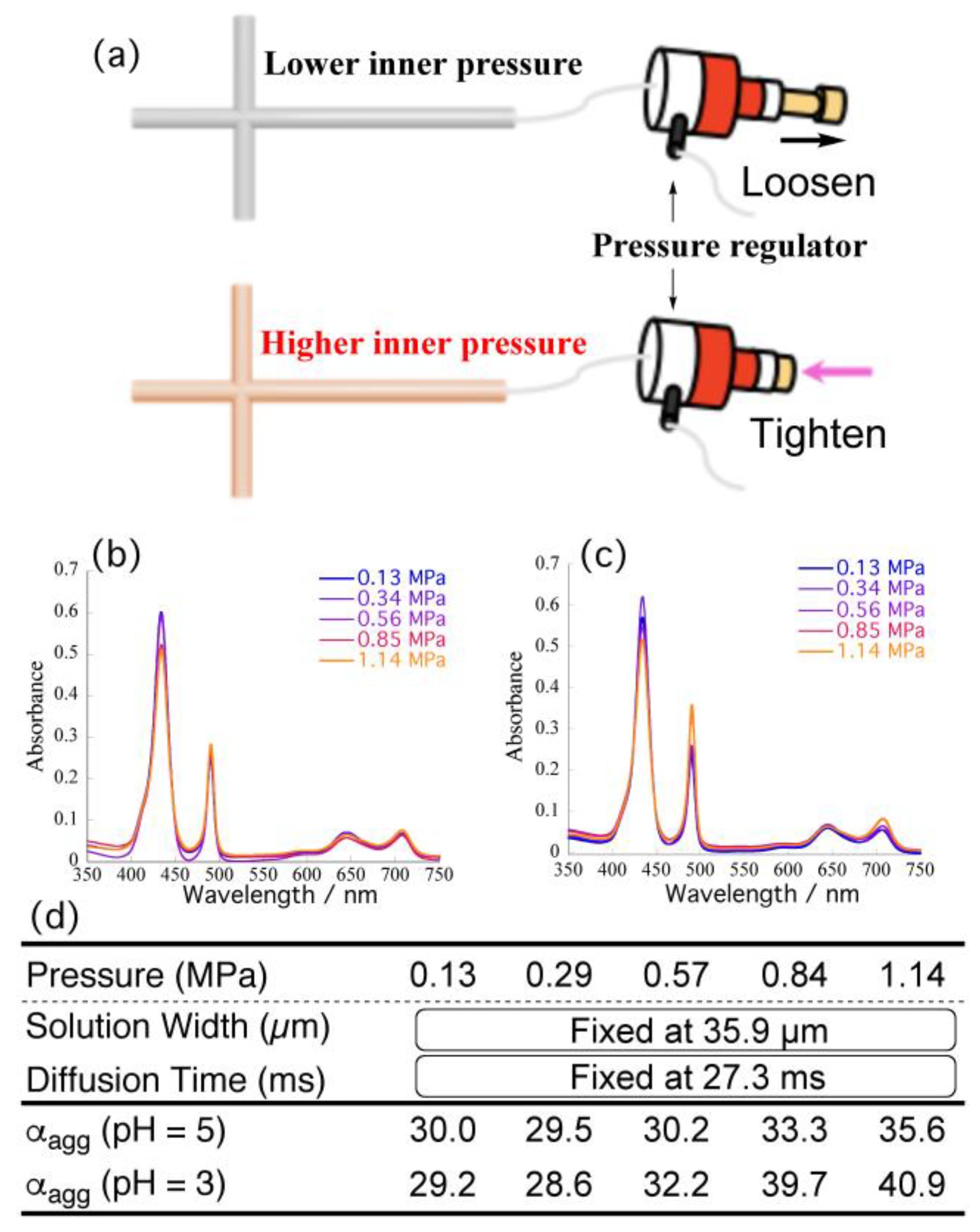
To subtract the effects of pressure on the self-assembly behavior of TPPS, we fixed the flow rate at 10 μL/min (width: 35.9 μm; diffusion time: 27.3 ms) and increased the pressure from 0.13 to 1.14 MPa by means of a back-pressure regulator connected at the end of outlet capillary (
Figure 5a). Each pressure was regulated precisely to equal the back-pressure observed under each flow rate in
Figure 4c. The width of the central layer (time required for proton diffusion) was always constant, regardless of pressure. Therefore, please note that this approach should reveal the effects of the pressure on the total
αagg.
Figure 5b,c indicate that increasing the pressure caused the intensity of the absorption signal at 434 nm (corresponding to H
4TPPS) to decrease, whereas that at 490 nm (assignable to the J-aggregate) increased.
Figure 5d summarizes the resultant
αagg obtained at each pressure. Plots of the
αagg with respect to pressure (
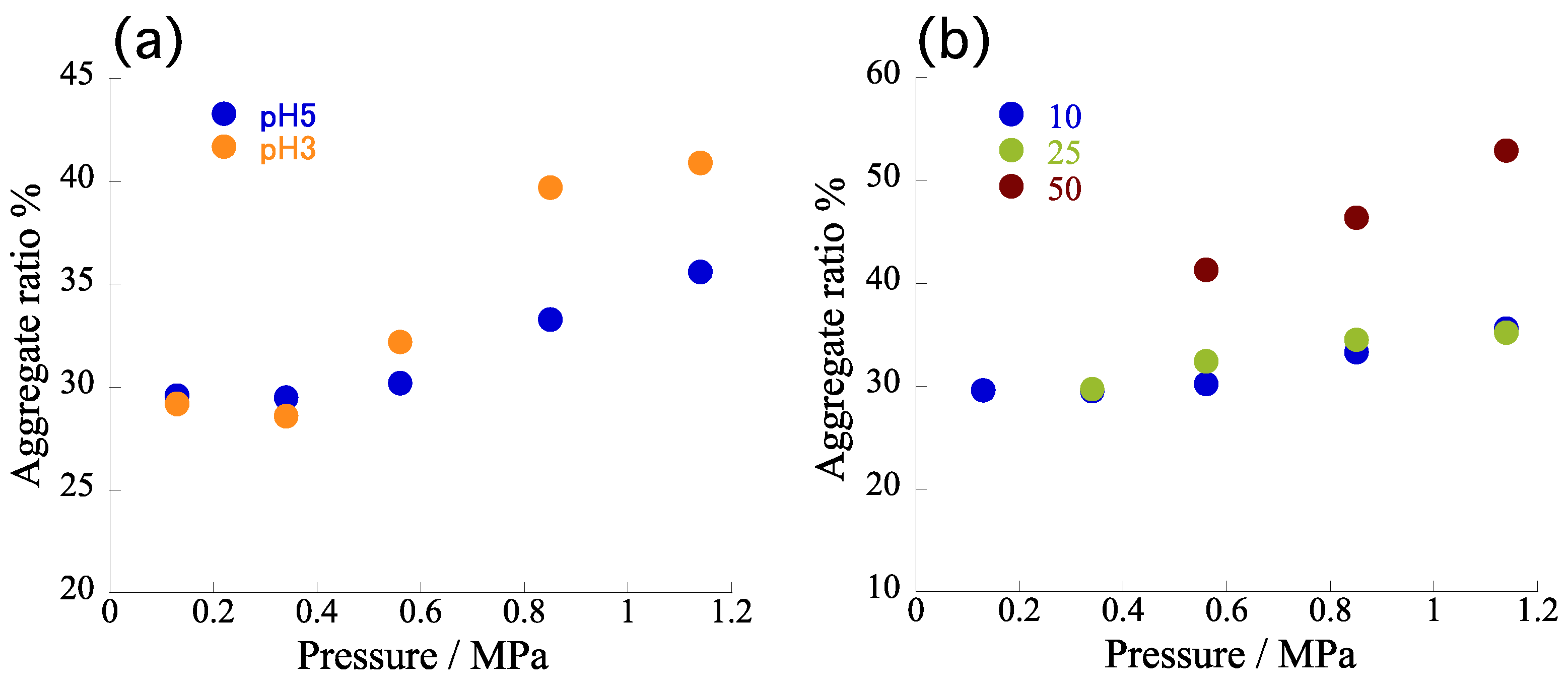
Figure S1; red dots) revealed that the α
agg tended to increase slightly upon increasing the inner pressure (e.g., from 30.0 to 35.6% at pH 5). Considering the overall acceleration rates (i.e., from 30.0 to 54.2 % at pH 5), however, the observed increase rates were very minor. Therefore, we conclude that the pressure in the channel had little effect on the self-assembly, at least in the tested pressure range, suggesting that the accelerated self-assembly in the microflow was attributable mainly to the shorter diffusion time and simultaneous protonation. Given the limited effect of the back-pressure at pH 5, we performed the same experiments at pH 3. Fixing the flow rate at 10 μL/min (i.e., a diffusion time of 27.3 ms), we varied the back-pressure up to 1.1 MPa.
Figure 5d summarizes the measured
αagg, in comparison with those obtained under pH 5.
Figure 6a reveals that the pressure effect was more predominant than it was at pH 5, as expected because the concentration of H
4TPPS was increased at pH 3. We observed that the
αagg also tended to increase upon increasing the pressure, consistent with the behavior observed at pH 5. Nevertheless, it should be noted that the observed pressure effect remained very weak in terms of the total microflow effect.
To further certify the correlation between pressure effects and rapid proton diffusion, we performed the same experiments at higher flow rates of 25 and 50 μL/min (
Figure S2). With much shorter diffusion times of 14.2 and 8.4 ms, respectively, we expected the pressure effect to be enhanced further because the temporal concentration of H
4TPPS would increase as a result of much faster proton diffusion. As expected, a flow rate of 25 μL/min caused the
αagg to increase more sensitively upon increasing the pressure than it did at 10 μL/min. This tendency was even clearer at 50 μL/min; that is, the slope in the plot (
Figure 6b and
Figure S3) became steeper at higher flow rates. Notably, a shorter diffusion time had the same effect as a lower pH on the self-assembly of TPPS. This finding strongly supports our view that very rapid proton diffusion led to an apparent decrease in pH in the area around H
2TPPS. In other words, protons could be concentrated on H
4TPPS through rapid protonation, resulting, ultimately, in a fixed state as a J-aggregate. Here, note again that the α
agg tended to increase upon increasing the inner pressure (e.g., from 29.7 to 35.2% under 25 μL/min at pH 5). With the overall acceleration rates (e.g., from 30.0 to 54.2% at pH 5) taken into consideration, however, the observed increase rates were still partial. The pressure effect on the
αagg became predominant only under the much faster proton diffusion conditions (i.e., at 50 μL/min). The pressure effect appeared depending on the concentration of H
4TPPS.
This series of experiments has brought the pressure effect to the fore; it is an additional effect to consider, one that depends on the pH and/or the flow rates (i.e., the diffusion time of the protons). Lowering the pH and shortening the diffusion time (increasing the flow rates) would both lead to an increase in temporal H4TPPS concentration. We conclude that the pressure effect was a secondary effect that appeared only under such a concentrated state of H4TPPS.
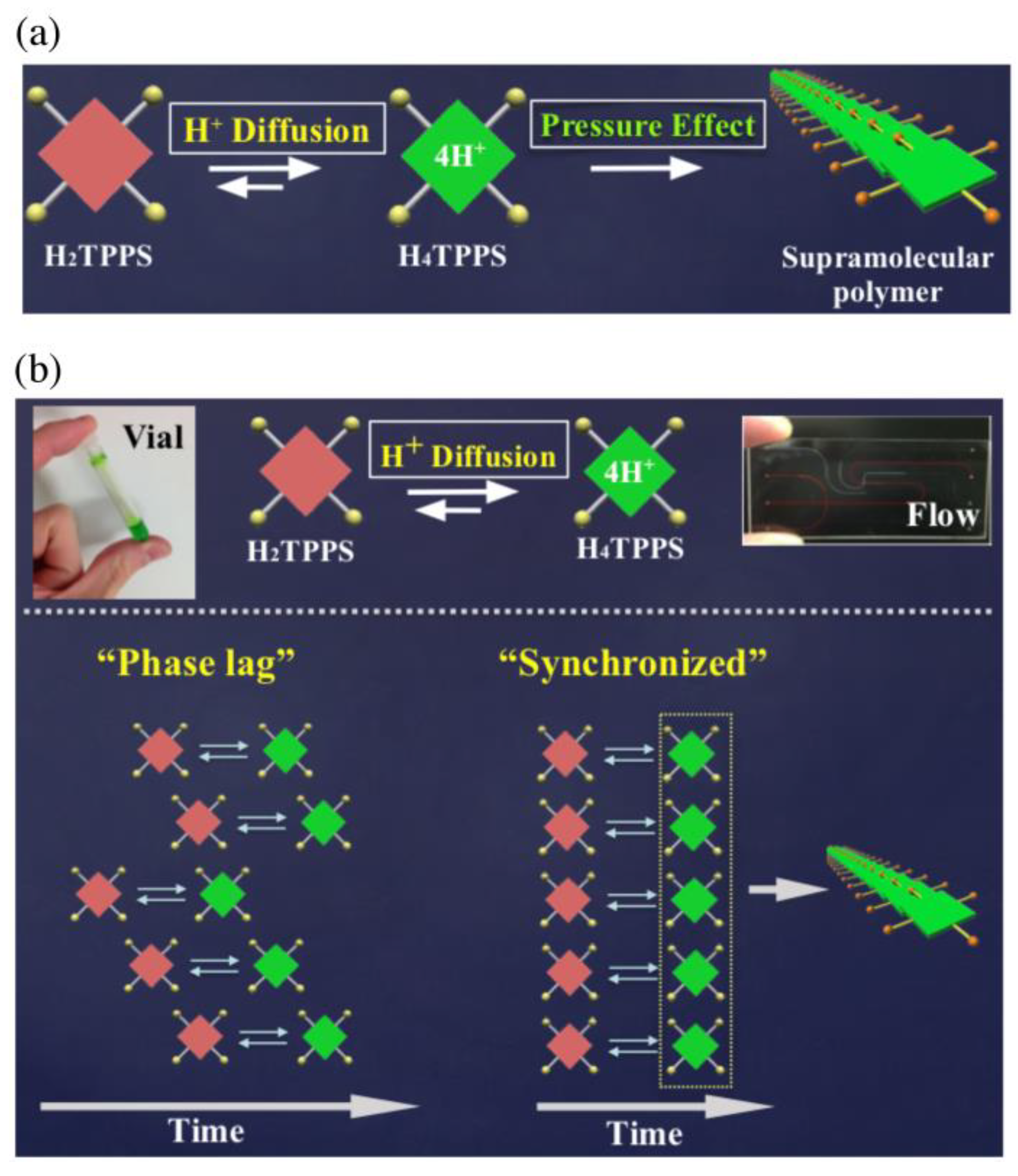
Figure 7a summarizes the mechanism of the self-assembly process. TPPS obeys the cooperative polymerization mechanism, characterized by the existence of a nucleation process as a rate-determining step. Rapid diffusion of protons generated a temporally concentrated state of H
4TPPS. The activation energy for the nucleation step would decrease under such a concentrated state. Thus, shortening the diffusion time, which would cause the equilibrium shift toward H
4TPPS, would be essential for accelerating the self-assembly of TPPS (first step of
Figure 7a). Increasing the pressure would accelerate the intermolecular interactions of H
4TPPS (second step of
Figure 7a), which should occur especially at a higher H
4TPPS concentration, thereby leading to effective nucleation. As a feature point of self-assembly in microflow, same assembly processes occur in a parallel fashion along the channel. To clarify the self-assembly of a set of activated species along time progress, we propose a time-phase correlation scheme, among several equilibrium reactions, which would highlight the synchronous behavior of molecules toward self-assembly (
Figure 7b). Here, the synchronous behavior of molecules can be defined as the state that numerous numbers of molecules are under the same chemical environments in a restricted space and temporally behave as a sort of cluster, which would realize only through kinetic pathway under non-equilibrium. Synchronous behavior would allow more than thousands of molecules to acquire self-assembly abilities at the same timing, thus leading to acceleration of intermolecular interactions.
Finally, we have highlighted the effect of rapid proton diffusion on the self-assembly of TPPS. In this present system, laminar flow provided an environment for rapid and homogeneous proton diffusion. A micromixer would lead to further acceleration of proton diffusion into the TPPS solution, where a turbulent flow generated a fragmental solution on the micrometer scale. Following the laminar flow system, we injected the same TPPS solution from one leg into the channel of micromixer and mixed with aqueous hydrochloric acid injected from another leg. Notably, as summarized in
Figure S4, the pressure measured inside the channel was almost equal to atmospheric pressure, even at higher flow rates; therefore, pressure effects would be negligible in comparison with that in a flow focus-type microchannel. Nevertheless, the
αagg remained high on average, even at lower flow rates. Accordingly, we attribute the observed accelerated effects on the protonation and self-assembly of TPPS to the very rapid diffusion of protons [
19].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}