Synthesis and Luminescence Properties of New Metal-Organic Frameworks Based on Zinc(II) Ions and 2,5-Thiophendicarboxylate Ligands





Abstract
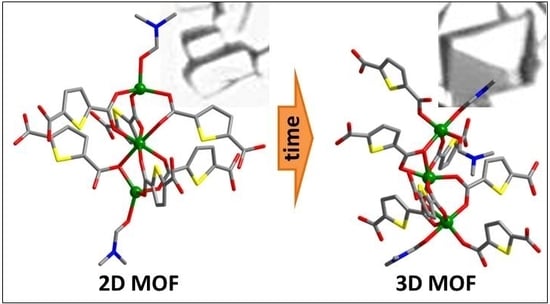
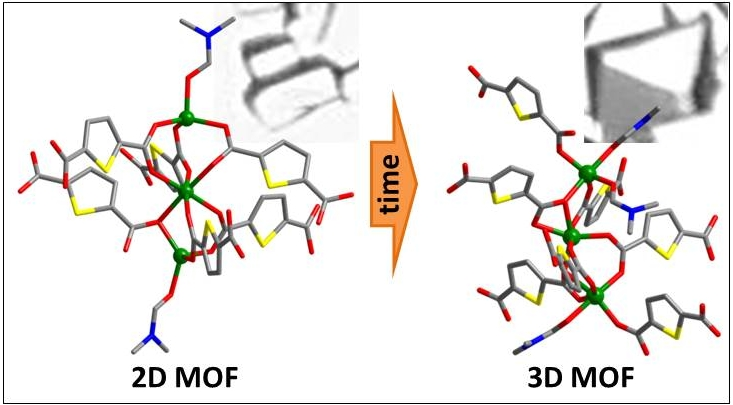
1. Introduction
2. Materials and Methods
2.1. Reagents and Instruments
2.2. Single-Crystal X-ray Diffraction
2.3. Synthetic Procedures and Analyses
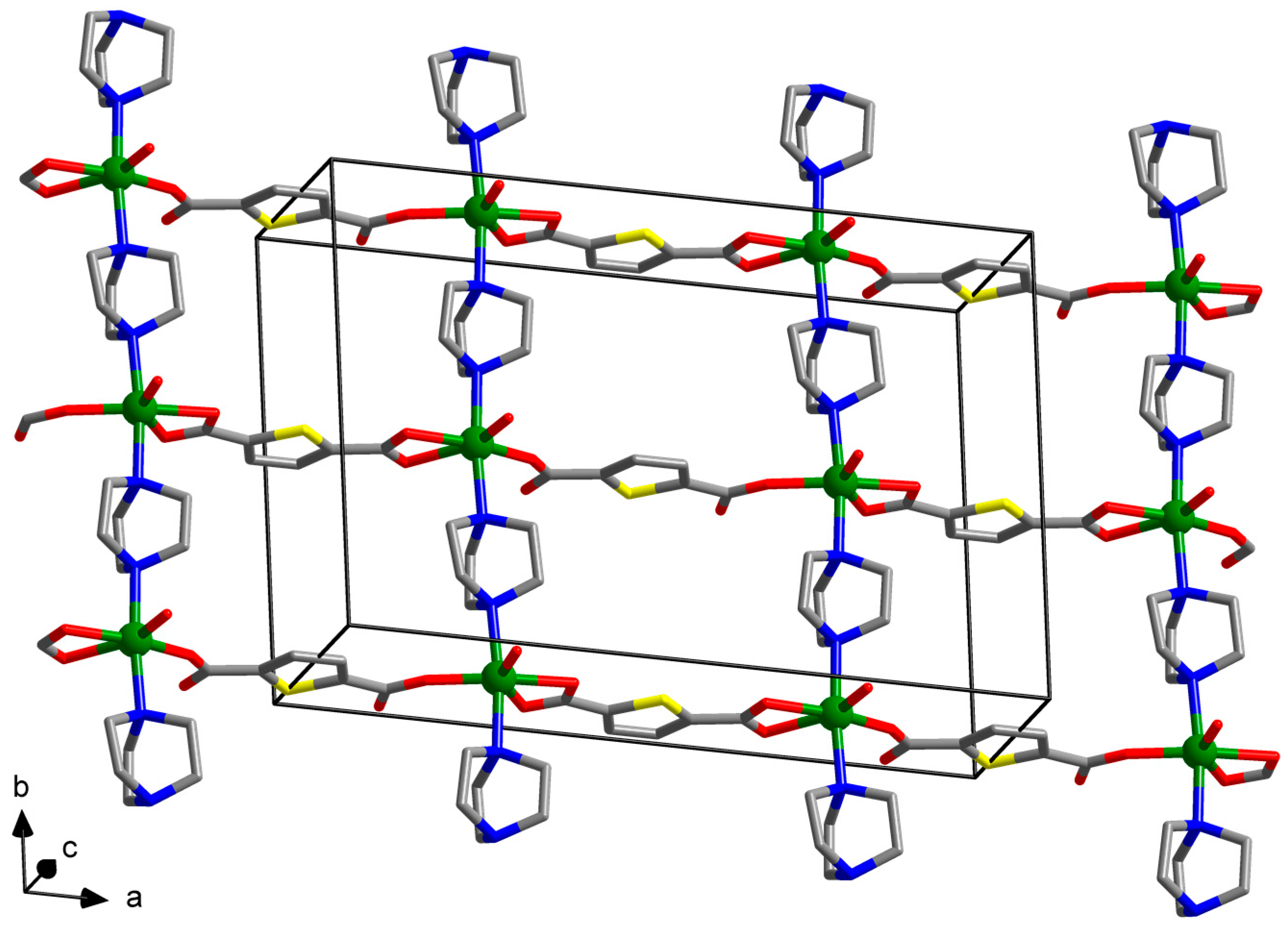
2.3.1. Synthesis of [Zn(tdc)(dabco)(H2O)]∙DMF (1)
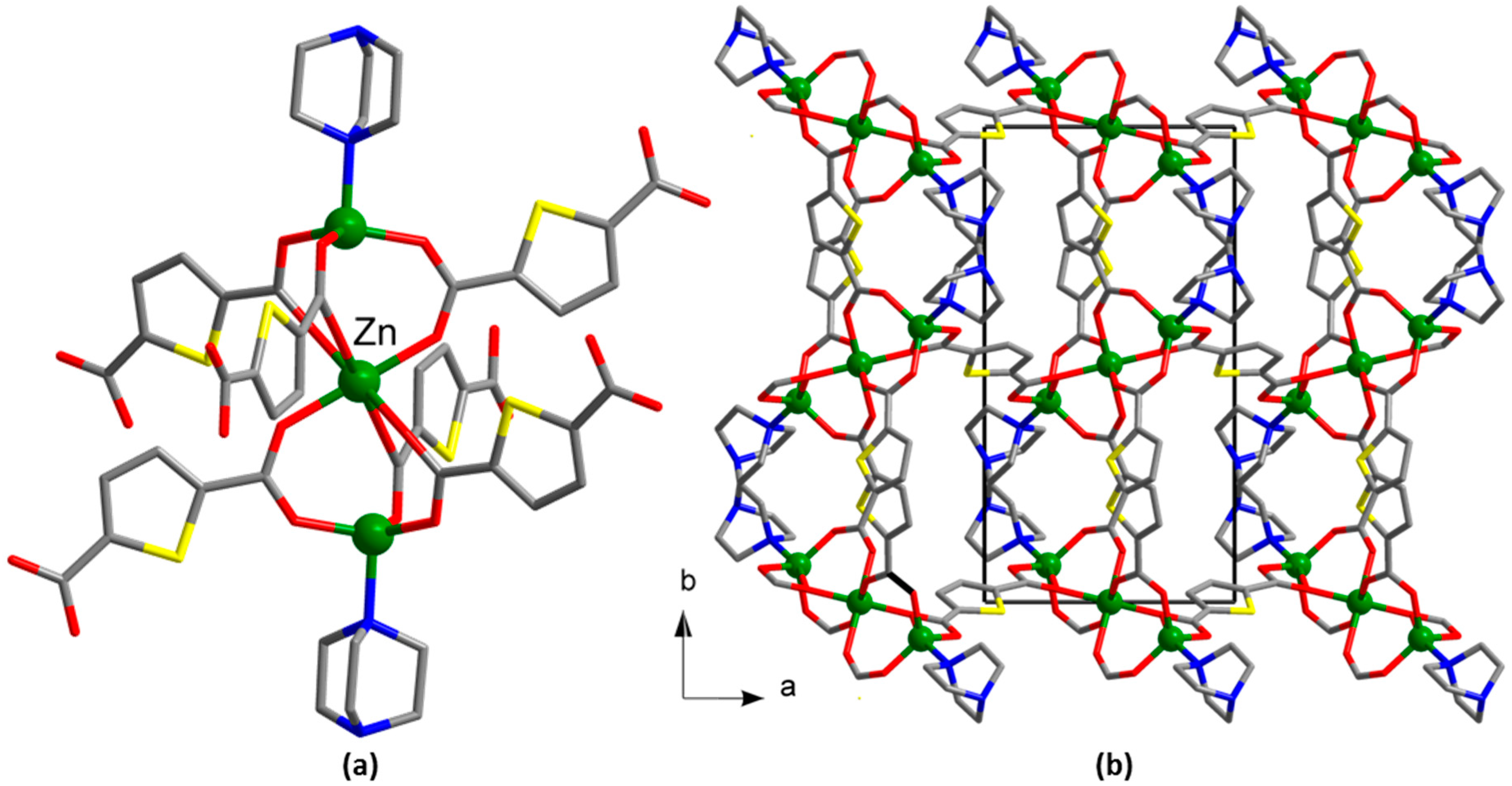
2.3.2. Synthesis of [Zn3(tdc)3(dabco)2] (2)
2.3.3. Synthesis of [Zn5(tdc)4(Htdc)2(dabco)2]∙4DMF∙14H2O (3)
2.3.4. Synthesis of [Na2Zn(tdc)2(DMF)2] (4)
2.3.5. Synthesis of [Zn3(tdc)3(DMF)2]⋅0.8DMF⋅1.1H2O (5)
2.3.6. Synthesis of [Zn3(tdc)3(DMF)3]∙0.8DMF⋅1.3H2O (6)
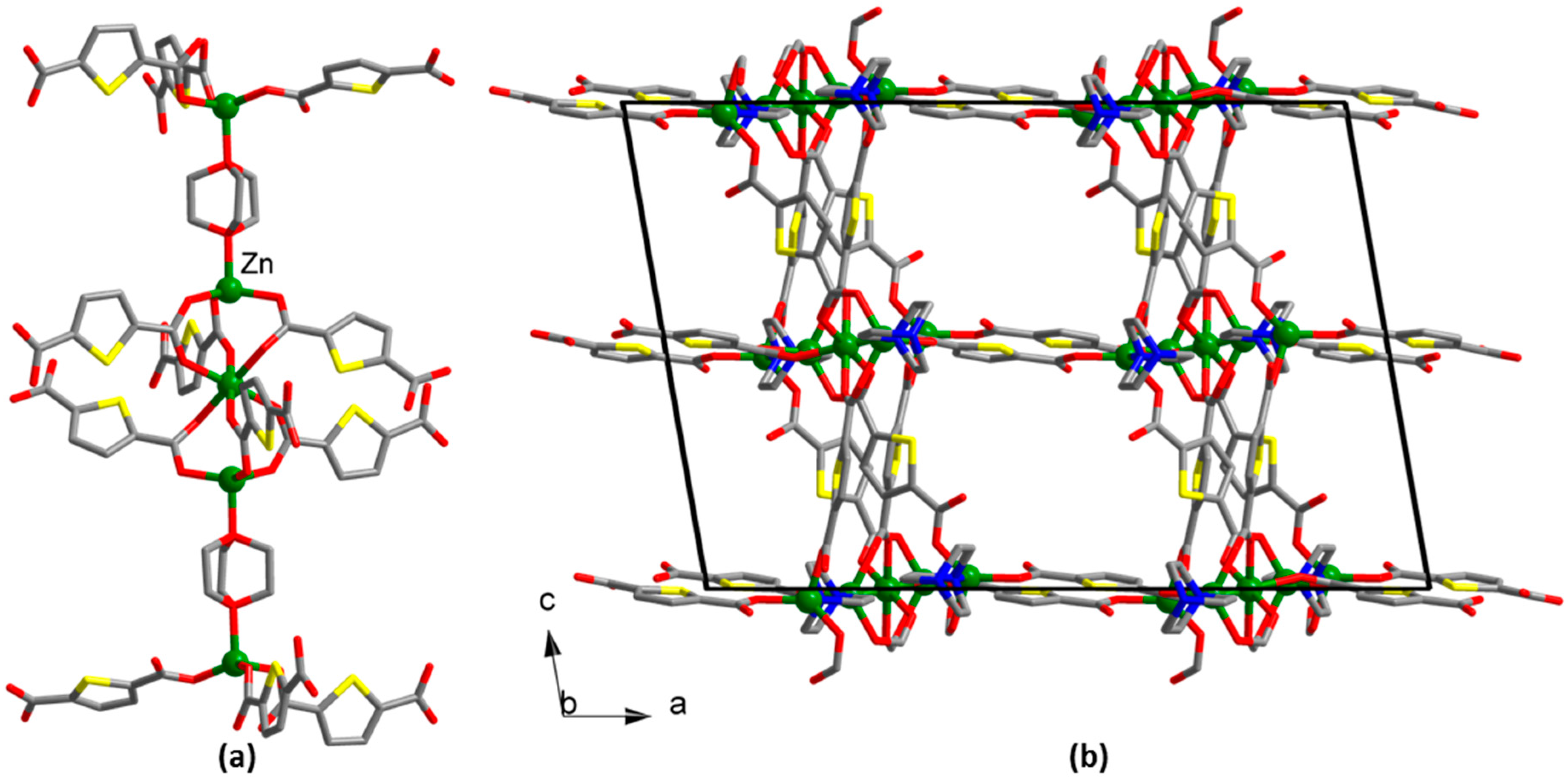
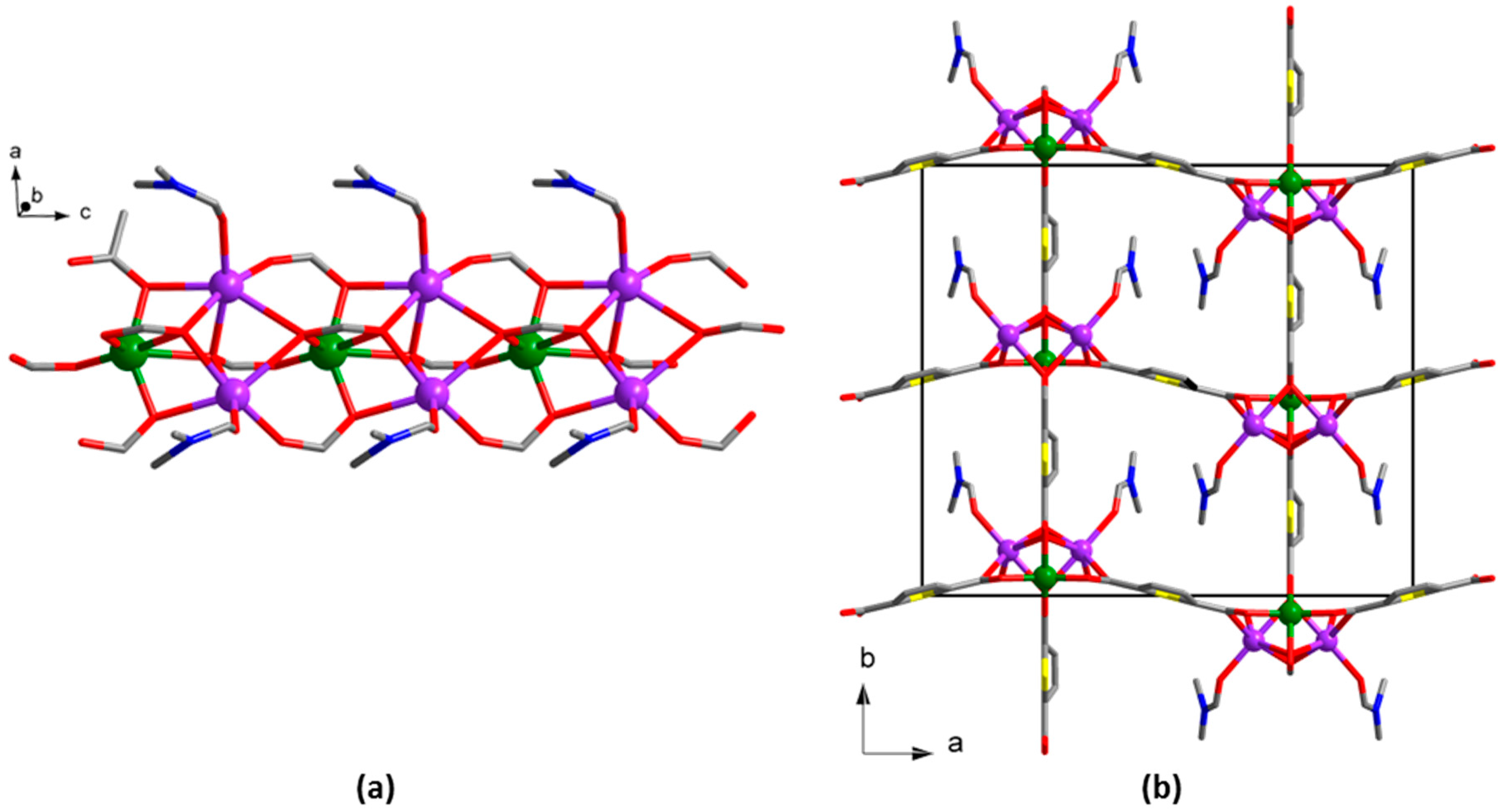
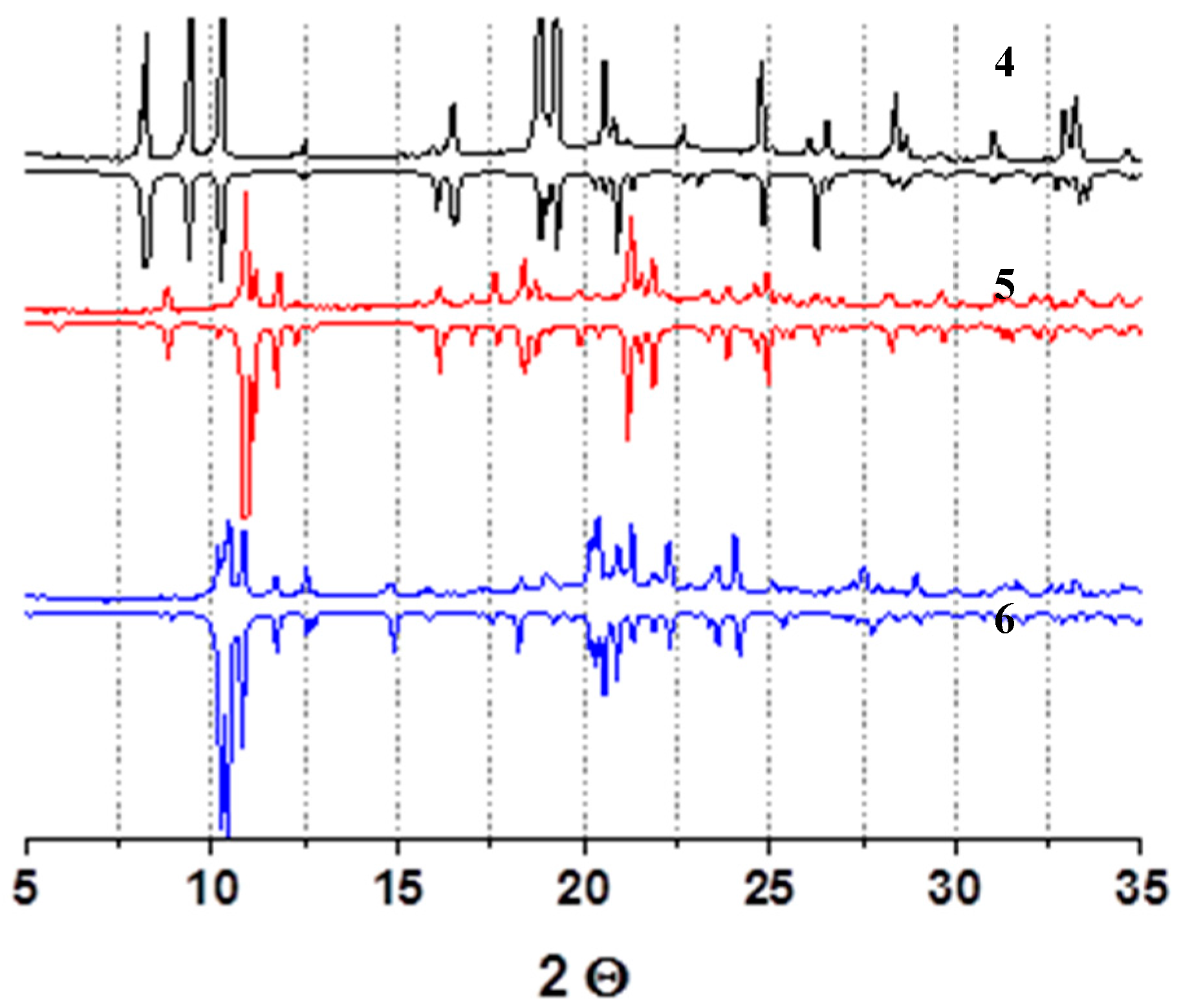
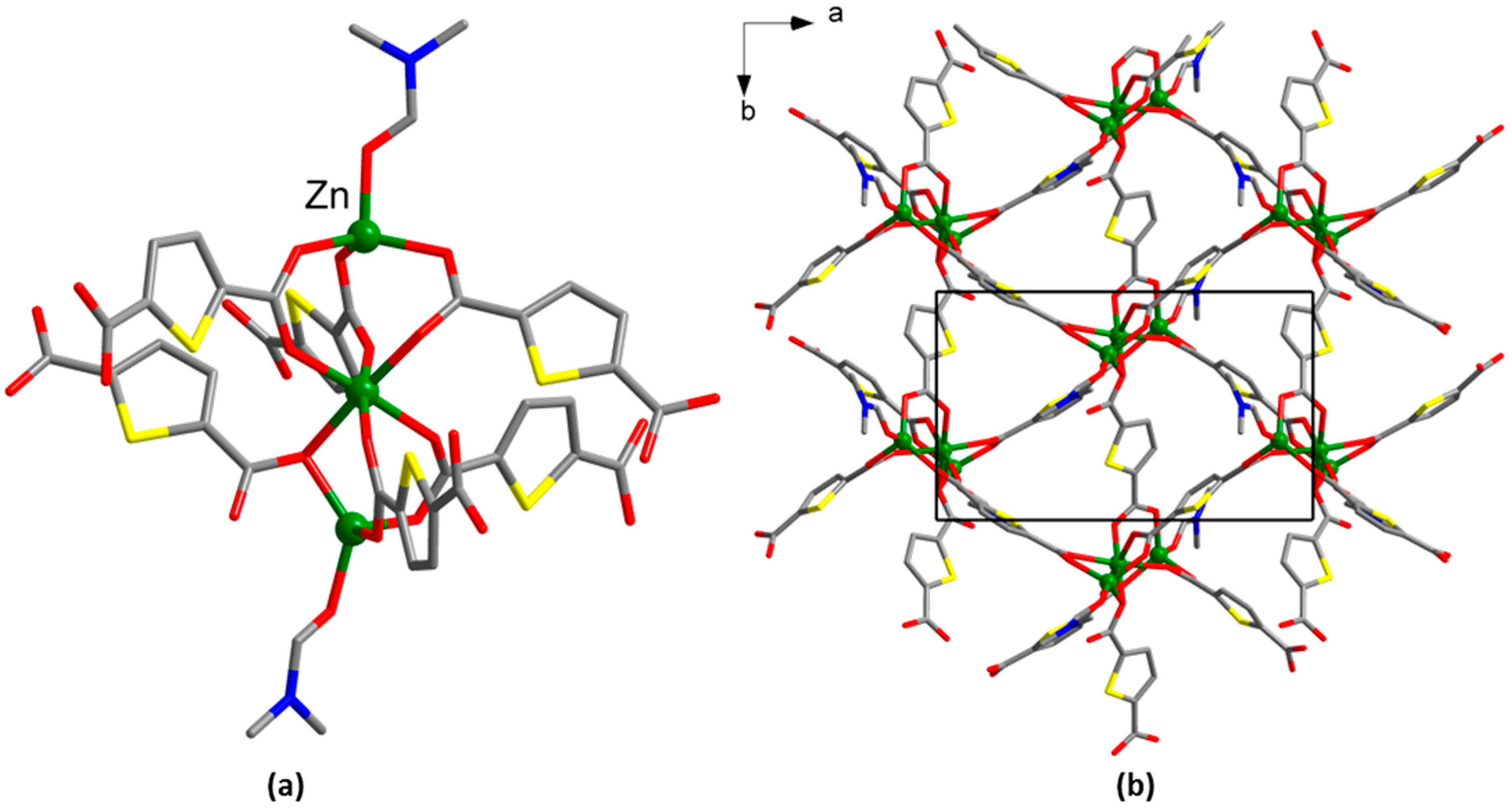
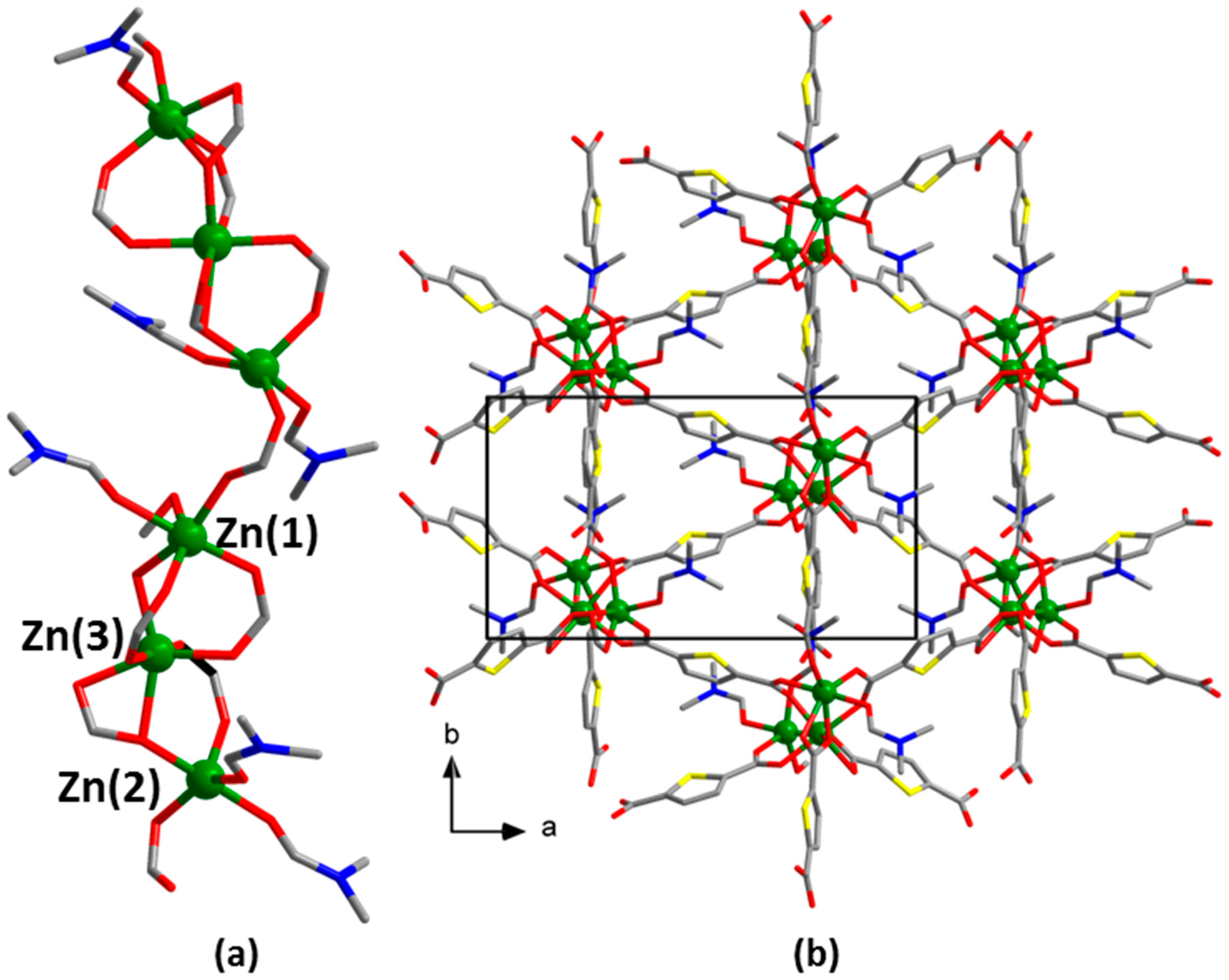
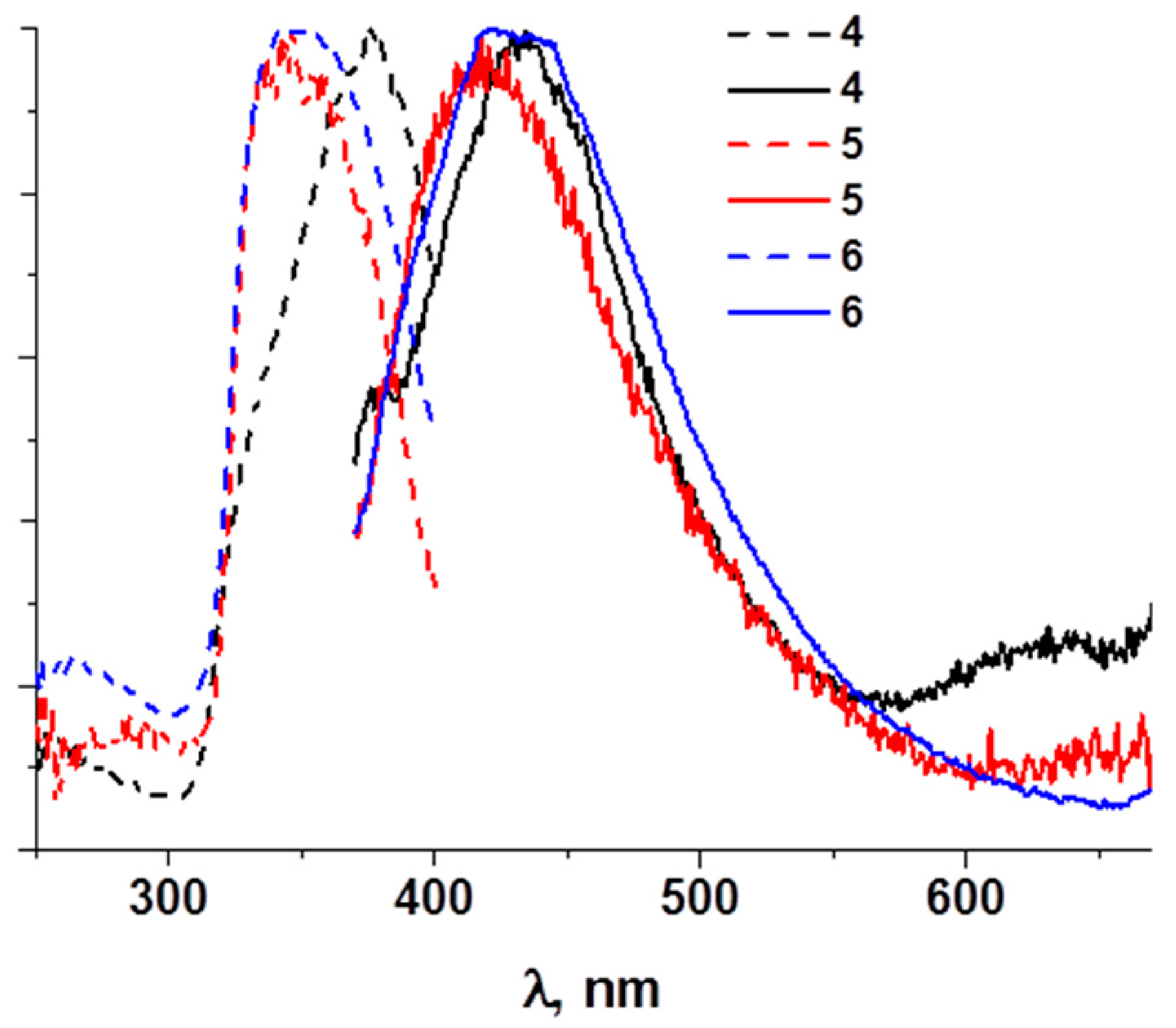
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houka, R.J.T. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wu, Y.-P.; Zhou, Z.-H.; Qin, Z.-S.; Ye, X.; Tian, F.-Y.; Li, D.-S. Construction of a series of lanthanide metal–organic frameworks (Ln-MOFs) based on a new symmetrical penta-aromatic carboxylate strut: Structure, luminescent and magnetic properties. Inorg. Chim. Acta 2016, 453, 757–763. [Google Scholar] [CrossRef]
- Takashima, Y.; Martínez, V.M.; Furukawa, S.; Kondo, M.; Shimomura, S.; Uehara, H.; Nakahama, M.; Sugimoto, K.; Kitagawa, S. Molecular decoding using luminescence from an entangled porous framework. Nat. Commun. 2011, 2, 168. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Deibert, B.J.; Li, J. Luminescent metal–organic frameworks for chemical sensing and explosive detection. Chem. Soc. Rev. 2014, 43, 5815–5840. [Google Scholar] [CrossRef] [PubMed]
- Müller-Buschbaum, K.; Beuerle, F.; Feldmann, C. MOF based luminescence tuning and chemical/physical sensing. Micropor. Mesopor. Mater. 2015, 216, 171–199. [Google Scholar] [CrossRef]
- Duan, J.; Jin, W.; Kitagawa, S. Water-resistant porous coordination polymers for gas separation. Coord. Chem. Rev. 2017, 332, 48–74. [Google Scholar] [CrossRef]
- Agarwal, R.A.; Gupta, N.K. CO2 sorption behavior of imidazole, benzimidazole and benzoic acid based coordination polymers. Coord. Chem. Rev. 2017, 332, 100–121. [Google Scholar] [CrossRef]
- Zhai, Q.-G.; Bu, X.; Zhao, X.; Li, D.-Zh.; Feng, P. Pore space partition in metal–organic frameworks. Acc. Chem. Rev. 2017, 50, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Aquirre-Díaz, L.M.; Reinares-Fisac, D.; Iglesias, M.; Gutiérrez-Puebla, E.; Gándara, F.; Snejko, N.; Ángeles Monge, M. Group 13th metal-organic frameworks and their role in heterogeneous catalysis. Coord. Chem. Rev. 2017, 335, 1–27. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Applications of metal–organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef] [PubMed]
- Chughtai, A.H.; Ahmad, N.; Younus, H.A.; Laypkov, A.; Verpoort, F. Metal–organic frameworks: Versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, S.; Lismont, M.; Escudero, A.; Rungtaweevoranit, B.; Parak, W.J. Positioning metal-organic framework nanoparticles within the context of drug delivery—A comparison with mesoporous silica nanoparticles and dendrimers. Biomaterials 2017, 123, 172–183. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Geib, S.J.; Rosi, N.L. Cation-triggered drug release from a porous zinc−adeninate metal−organic framework. J. Am. Chem. Soc. 2009, 131, 8376–8377. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, S.; Zimpel, A.; Bein, T.; Braig, S.; Stoiber, K.; Vollmar, A.; Müller, D.; Haastert-Talini, K.; Schaeske, J.; Stiesch, M.; et al. Validating metal-organic framework nanoparticles for their nanosafety in diverse biomedical applications. Adv. Healthc. Mater. 2017, 6, 1600818. [Google Scholar] [CrossRef] [PubMed]
- Osborn Popp, T.M.; Yaghi, O.M. Sequence-Dependent Materials. Acc. Chem. Res. 2017, 50, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Doonan, C.J.; Furukawa, H.; Ferreira, R.B.; Towne, J.; Knobler, C.B.; Wang, B.; Yaghi, O.M. Multiple functional groups of varying ratios in metal-organic frameworks. Science. 2010, 327, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Müller, U.; Yaghi, O.M. “Heterogeneity within order” in metal–organic frameworks. Angew. Chem. Int. Ed. 2015, 54, 3417–3430. [Google Scholar] [CrossRef] [PubMed]
- Kleist, W.; Jutz, F.; Maciejewski, M.; Baiker, A. Mixed-Llinker metal-organic frameworks as catalysts for the synthesis of propylene carbonate from propylene oxide and CO2. Eur. J. Inorg. Chem. 2009, 2009, 3552–3561. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of metal-organic frameworks (MOFs): Routes to various MOF topologies, morphologies, and composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro 1.171.38.41, Rigaku Oxford Diffraction: The Woodlands, TX, USA, 2015.
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C 2015, C71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Henke, S.; Kieslich, G.; Schwedler, J.; Yang, M.; Fraser, D.A.X.; O’Hare, D. Time-resolved in situ X-ray diffraction relates metal-dependent metal-organic framework formation. Angew. Chem. Int. Ed. 2016, 55, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Marti-Rujas, J.; Kawano, M. Kinetic products in coordination networks: Ab initio X-ray powder diffraction analysis. Acc. Chem. Res. 2013, 46, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Tranchemontagne, D.J.; Mendoza-Cortés, J.L.; O’Keeffe, M.; Yaghi, O.M. Secondary building units, nets and bonding in the chemistry of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1257–1283. [Google Scholar] [CrossRef] [PubMed]
- Dincă, M.; Long, J.R. Strong H2 binding and selective gas adsorption within the microporous coordination solid Mg3(O2C-C10H6-CO2)3. J. Am. Chem. Soc. 2005, 127, 9376–9377. [Google Scholar] [CrossRef] [PubMed]
- Sapchenko, S.; Dybtsev, D.; Fedin, V. Cage amines in the metal-organic frameworks chemistry. Pure Appl. Chem. 2017, 89, 1049–1064. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| CCDC deposition number | 1586559 | 1586560 | 1586561 | 1586562 | 1586563 | 1586564 |
| Empirical formula | C15H23N3O6SZn | C30H30N4O12S3Zn3 | C60H94N8O42S6Zn5 | C18H18N2Na2O10S2Zn | C26H23N3O14S3Zn3 | C27H27N3O15S3Zn3 |
| M, g/mol | 438.79 | 930.87 | 2128.16 | 597.81 | 893.76 | 925.80 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Cmm2 | P21/c | C2/c | Ama2 | P21/n | P21/c |
| a, Å | 20.7976(8) | 10.3232(4) | 26.8663(7) | 21.4918(6) | 16.8119(5) | 17.7558(4) |
| b, Å | 14.0097(6) | 17.9750(4) | 19.2844(4) | 18.8280(5) | 9.6409(2) | 9.7309(2) |
| c, Å | 6.0284(2) | 10.5364(4) | 18.3894(4) | 5.76625(15) | 21.1725(6) | 21.0282(5) |
| β, deg. | 90 | 113.613(4) | 99.832(2) | 90 | 108.925(3) | 103.575(2) |
| V, Å3 | 1756.48(12) | 1791.43(12) | 9387.6(4) | 2333.30(11) | 3246.17(16) | 3531.75(14) |
| Z | 4 | 2 | 4 | 4 | 4 | 4 |
| D(calc.), g/cm3 | 1.659 | 1.726 | 1.506 | 1.702 | 1.829 | 1.741 |
| μ, mm−1 | 1.556 | 2.234 | 1.481 | 1.327 | 2.465 | 2.271 |
| F(000) | 912 | 944 | 4388 | 1216 | 1800 | 1872 |
| Crystal size, mm | 0.48 × 0.09 × 0.03 | 0.29 × 0.08 × 0.03 | 0.28 × 0.26 × 0.14 | 0.45 × 0.15 × 0.05 | 0.17 × 0.15 × 0.05 | 0.25 × 0.21 × 0.09 |
| θ range for data collection, deg. | 3.28–28.83 | 3.74–29.66 | 3.41–29.56 | 3.57–29.10 | 3.32–29.56 | 3.42–29.01 |
| Index ranges | −28 ≤ h ≤ 20, −19 ≤ k ≤ 17, −7 ≤ l ≤ 7 | −10 ≤ h ≤ 14, −24 ≤ k ≤ 17, −13 ≤ l ≤ 13 | −25 ≤ h ≤ 34, −26 ≤ k ≤ 19, −25 ≤ l ≤ 22 | −28 ≤ h ≤ 24, −16 ≤ k ≤ 23, −6 ≤ l ≤ 7 | −22 ≤ h ≤ 16, −13 ≤ k ≤ 12, −21 ≤ l ≤ 29 | −23 ≤ h ≤ 23, −10 ≤ k ≤ 12, −28 ≤ l ≤ 23 |
| Reflections collected / independent | 3486 / 1797 | 9004 / 4233 | 26598 / 11162 | 4891 / 2302 | 17138 / 7667 | 17555 / 7800 |
| Rint | 0.0185 | 0.0193 | 0.0260 | 0.0178 | 0.0242 | 0.0238 |
| Reflections with I > 2σ(I)] | 1713 | 3491 | 9856 | 2291 | 6469 | 6807 |
| Goodness-of-fit on F2 | 1.031 | 1.054 | 1.089 | 1.276 | 1.038 | 1.031 |
| Final R indices [I > 2σ(I)] | R1 = 0.0233, wR2 = 0.0576 | R1 = 0.0450, wR2 = 0.1045 | R1 = 0.0391, wR2 = 0.0977 | R1 = 0.0582, wR2 = 0.1593 | R1 = 0.0271, wR2 = 0.0584 | R1 = 0.0271, wR2 = 0.0599 |
| R indices (all data) | R1 = 0.0251, wR2 = 0.0584 | R1 = 0.0578, wR2 = 0.1117 | R1 = 0.0454, wR2 = 0.1008 | R1 = 0.0584, wR2 = 0.1594 | R1 = 0.0371, wR2 = 0.0620 | R1 = 0.0343, wR2 = 0.0630 |
| Largest diff. peak / hole, e/Å3 | 0.336 / −0.286 | 1.161 / −0.732 | 0.820 / −0.638 | 0.874 / −1.437 | 0.544 / −0.376 | 0.525 / −0.402 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lysova, A.; Samsonenko, D.; Dybtsev, D.; Fedin, V. Synthesis and Luminescence Properties of New Metal-Organic Frameworks Based on Zinc(II) Ions and 2,5-Thiophendicarboxylate Ligands. Crystals 2018, 8, 7. https://doi.org/10.3390/cryst8010007
Lysova A, Samsonenko D, Dybtsev D, Fedin V. Synthesis and Luminescence Properties of New Metal-Organic Frameworks Based on Zinc(II) Ions and 2,5-Thiophendicarboxylate Ligands. Crystals. 2018; 8(1):7. https://doi.org/10.3390/cryst8010007
Chicago/Turabian StyleLysova, Anna, Denis Samsonenko, Danil Dybtsev, and Vladimir Fedin. 2018. "Synthesis and Luminescence Properties of New Metal-Organic Frameworks Based on Zinc(II) Ions and 2,5-Thiophendicarboxylate Ligands" Crystals 8, no. 1: 7. https://doi.org/10.3390/cryst8010007
APA StyleLysova, A., Samsonenko, D., Dybtsev, D., & Fedin, V. (2018). Synthesis and Luminescence Properties of New Metal-Organic Frameworks Based on Zinc(II) Ions and 2,5-Thiophendicarboxylate Ligands. Crystals, 8(1), 7. https://doi.org/10.3390/cryst8010007