Solvent Dependent Disorder in M2(BzOip)2(H2O)·Solvate (M = Co or Zn)

 , , and
, , and
Abstract
:1. Introduction
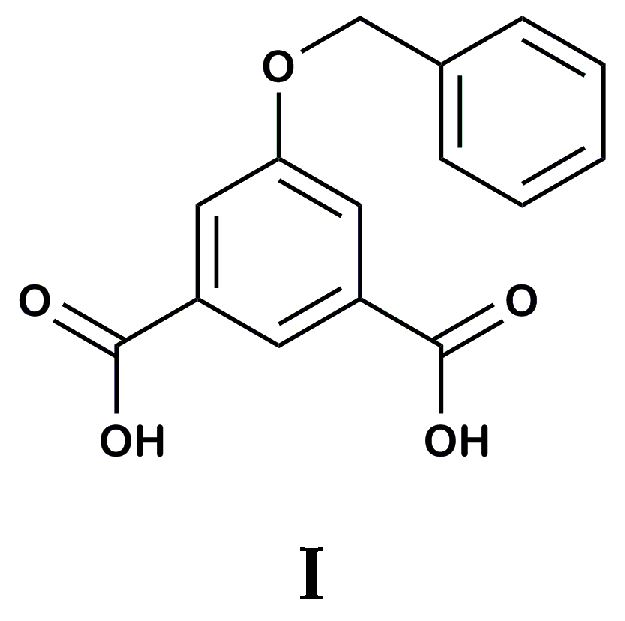
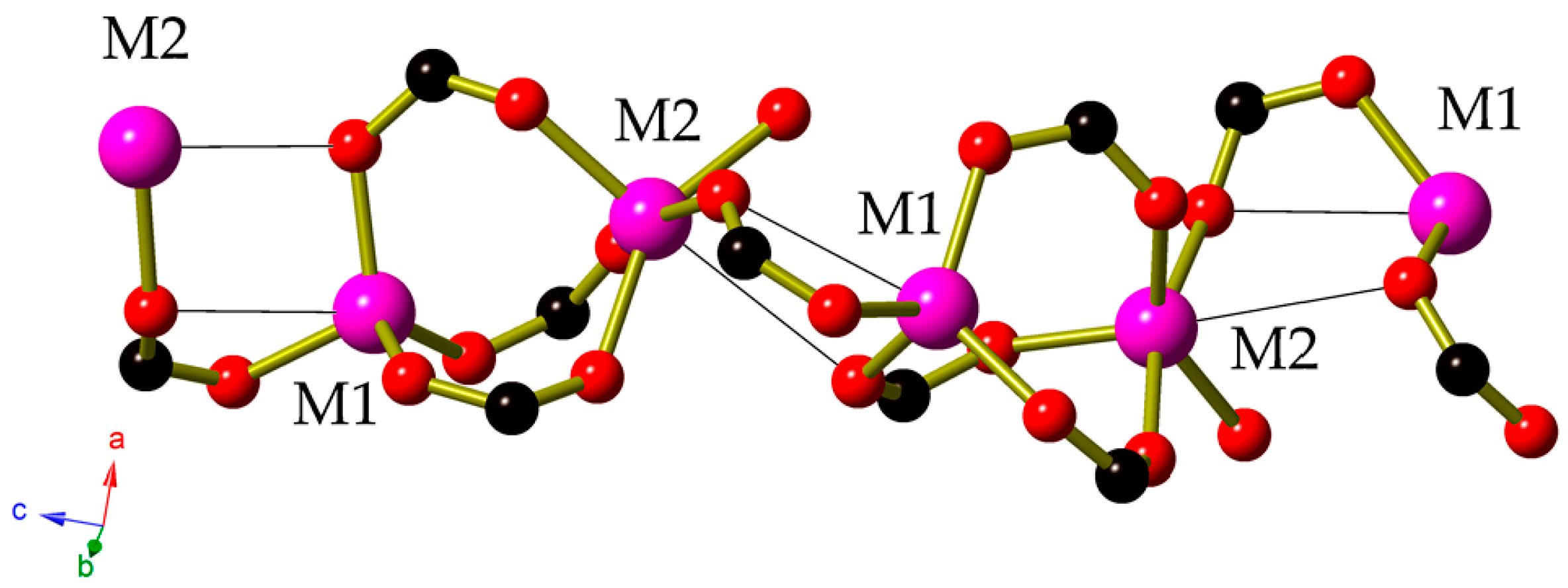
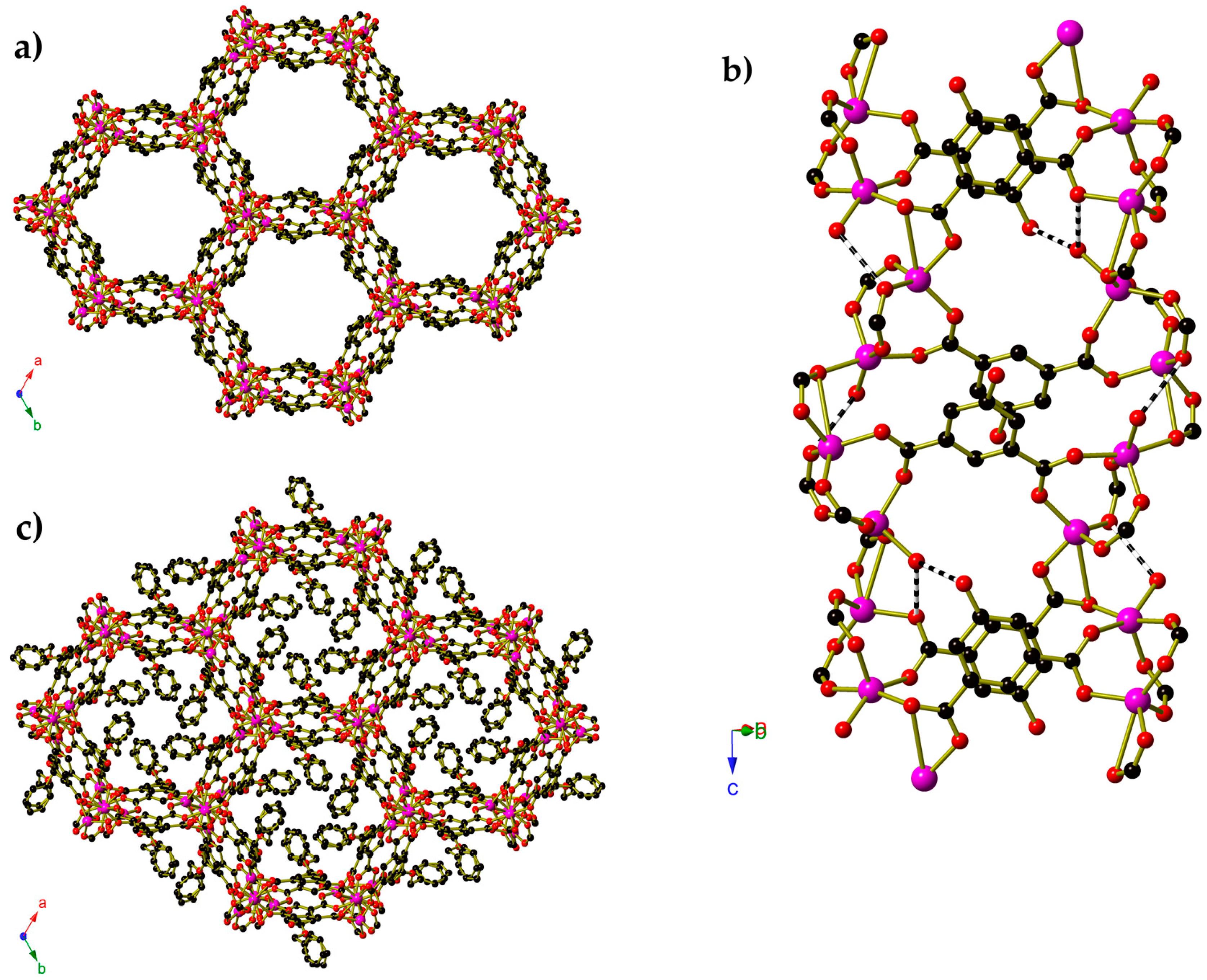
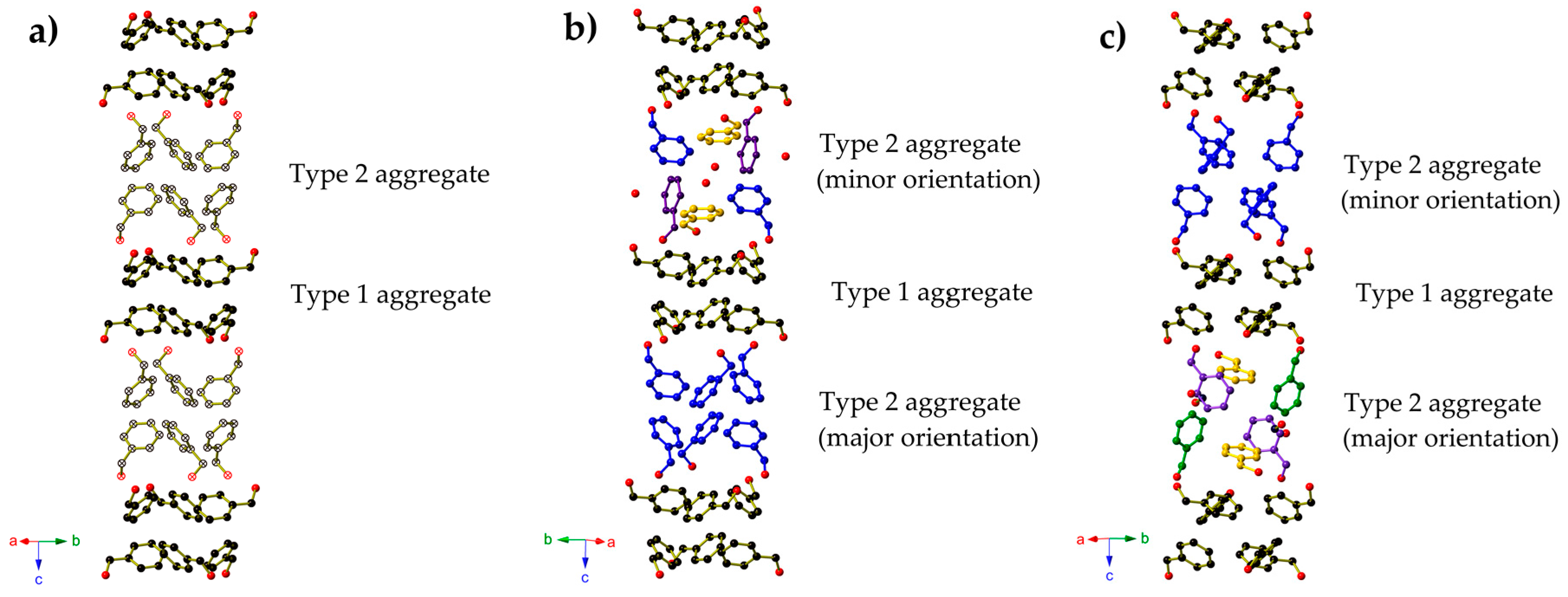
2. Results
3. Discussion
4. Materials and Methods
4.1. Dimethyl 5-Hydroxy Isophthalate
4.2. Dimethyl 5-Benzyloxy Isophthalate
4.3. 5-Benzyloxy Isophthalic Acid
4.4. M2(BzOip)2(H2O)·Solvate (M = Co or Zn)
4.5. Crystallographic Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Serre, C.; Millange, F.; Thouvenot, C.; Nogues, M.; Marsolier, G.; Louer, D.; Ferey, G. Very Large Breathing Effect in the First Nanoporous Chromium(III)-Based Solids: MIL-53 or CrIII(OH)·{O2C−C6H4−CO2}·{HO2C−C6H4−CO2H}x·H2Oy. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surble, S.; Margiolaki, I. A Chromium Terephthalate-Based Solid with Unusually Large Pore Volumes and Surface Area. Science 2005, 309, 2040–2042. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O.M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Dietzel, P.D.C.; Blom, R.; Fjellvåg, H. Base-Induced Formation of Two Magnesium Metal-Organic Framework Compounds with a Bifunctional Tetratopic Ligand. Eur. J. Inorg. Chem. 2008, 2008, 3624–3632. [Google Scholar] [CrossRef]
- Gao, Q.; Jiang, F.-L.; Wu, M.-Y.; Huang, Y.-G.; Wei, W.; Zhang, Q.-F.; Hong, M.-C. Crystal Structures, Topological Analyses, and Magnetic Properties of Manganese-Dihydroxyterephthalate Complexes. Aust. J. Chem. 2010, 63, 286–292. [Google Scholar] [CrossRef]
- Bloch, E.D.; Murray, L.J.; Queen, W.L.; Chavan, S.; Maximoff, S.N.; Bigi, J.P.; Krishna, R.; Peterson, V.K.; Grandjean, F.; Long, G.J.; et al. Selective Binding of O2 over N2 in a Redox–Active Metal–Organic Framework with Open Iron(II) Coordination Sites. J. Am. Chem. Soc. 2011, 133, 14814–14822. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, P.D.C.; Morita, Y.; Blom, R.; Fjellvåg, H. An In Situ High-Temperature Single-Crystal Investigation of a Dehydrated Metal–Organic Framework Compound and Field-Induced Magnetization of One-Dimensional Metal–Oxygen Chains. Angew. Chem. Int. Ed. 2005, 44, 6354–6358. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, P.D.C.; Panella, B.; Hirscher, M.; Blom, R.; Fjellvåg, H. Hydrogen adsorption in a nickel based coordination polymer with open metal sites in the cylindrical cavities of the desolvated framework. Chem. Commun. 2006, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Sanz, R.; Martínez, F.; Orcajo, G.; Wojtas, L.; Briones, D. Synthesis of a honeycomb-like Cu-based metal–organic framework and its carbon dioxide adsorption behavior. Dalton Trans. 2013, 42, 2392–2398. [Google Scholar] [CrossRef] [PubMed]
- Rosi, N.L.; Kim, J.; Eddaoudi, M.; Chen, B.; O’Keeffe, M.; Yaghi, O.M. Rod Packings and Metal−Organic Frameworks Constructed from Rod-Shaped Secondary Building Units. J. Am. Chem. Soc. 2005, 127, 1504–1518. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Rosi, N.; Vodak, D.; Wachter, J.; O’Keeffe, M.; Yaghi, O.M. Systematic Design of Pore Size and Functionality in Isoreticular MOFs and Their Application in Methane Storage. Science 2002, 295, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.G.; Luz, I.; Llabres i Xamena, F.X.; Corma, A.; Garcia, H. Water Stable Zr–Benzenedicarboxylate Metal–Organic Frameworks as Photocatalysts for Hydrogen Generation. Chem. Eur. J. 2010, 16, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Ahnfeldt, T.; Gunzelmann, D.; Loiseau, T.; Hirsemann, D.; Senker, J.; Ferey, G.; Stock, N. Synthesis and Modification of a Functionalized 3D Open-Framework Structure with MIL-53 Topology. Inorg. Chem. 2009, 48, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Couck, S.; Grzywa, M.; Denayer, J.F.M.; Volkmer, D.; Van Der Voort, P. Vanadium Analogues of Nonfunctionalized and Amino-Functionalized MOFs with MIL-101 Topology–Synthesis, Characterization, and Gas Sorption Properties. Eur. J. Inorg. Chem. 2012, 2012, 2481–2486. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Ferey, G.; Serre, C.; Mellot-Draznieks, C.; Millange, F.; Surble, S.; Dutour, J.; Margiolaki, I. A Hybrid Solid with Giant Pores Prepared by a Combination of Targeted Chemistry, Simulation, and Powder Diffraction. Angew. Chem. Int. Ed. 2004, 43, 6296–6301. [Google Scholar] [CrossRef] [PubMed]
- Horcajada, P.; Surble, S.; Serre, C.; Hong, D.-Y.; Seo, Y.-K.; Chang, J.-S.; Greneche, J.-M.; Margiolaki, I.; Ferey, G. Synthesis and catalytic properties of MIL-100(Fe), an iron(III) carboxylate with large pores. Chem. Commun. 2007, 2820–2822. [Google Scholar] [CrossRef] [PubMed]
- McCormick, L.J.; Morris, S.A.; Slawin, A.M.Z.; Teat, S.J.; Morris, R.E. Coordination Polymers of 5-Alkoxy Isophthalic Acids. Cryst. Growth Des. 2016, 16, 5771–5780. [Google Scholar] [CrossRef]
- Yang, H.-B.; Northrop, B.H.; Zheng, Y.-R.; Ghosh, K.; Stang, P.J. Facile Self-Assembly of Neutral Dendritic Metallocycles via Oxygen-to-Platinum Coordination. J. Org. Chem. 2009, 74, 7067–7074. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-X.; Zhang, X.; Cheng, J.-K.; Yao, Y.-G. A novel 1D→2D interdigitated framework directed by hydrogen bonds. J. Mol. Struct. 2011, 991, 31–34. [Google Scholar] [CrossRef]
- Gole, B.; Bar, A.K.; Mukherjee, P.S. Modification of Extended Open Frameworks with Fluorescent Tags for Sensing Explosives: Competition between Size Selectivity and Electron Deficiency. Chem. Eur. J. 2014, 20, 2276–2291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-W.; Zhao, G.-Y. Synthesis, Crystal Structure, and Photoluminescent Property of a New 1D→2D Interdigitated Framework. Synth. React. Inorg. Met. Org. Nano-Met. Chem. 2015, 45, 524–526. [Google Scholar] [CrossRef]
- Su, Y.; Li, X.; Li, X.; Pan, H.; Wang, R. Effects of hydroxy substituents on Cu(II) coordination polymers based on 5-hydroxyisophthalate derivatives and 1,4-bis(2-methylimidazol-1-yl)benzene. CrystEngComm 2015, 17, 4883–4894. [Google Scholar] [CrossRef]
- Gole, B.; Bar, A.K.; Mukherjee, P.S. Multicomponent Assembly of Fluorescent-Tag Functionalized Ligands in Metal–Organic Frameworks for Sensing Explosives. Chem. Eur. J. 2014, 20, 13321–13336. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, J.; Li, M.-K.; Fei, Z. Synthesis, structures and photocatalytic properties of two new Co(II) coordination polymers based on 5-(benzyloxy)isophthalate ligand. J. Mol. Struct. 2014, 1059, 294–298. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, J.-X.; Yao, Y.-G. Tail of the Organic Ligand Templated Metal-Organic Framework. J. Inorg. Organomet. Polym. Mater. 2012, 22, 1189–1193. [Google Scholar] [CrossRef]
- Perry, J.J.; McManus, G.J.; Zaworotko, M.J. Sextuplet phenyl embrace in a metal–organic Kagomé lattice. Chem. Commun. 2004, 2534–2535. [Google Scholar] [CrossRef] [PubMed]
- Dance, I.; Scudder, M. The Sextuple Phenyl Embrace, a Ubiquitous Concerted Supramolecular Motif. J. Chem. Soc. Chem. Commun. 1995, 1039–1040. [Google Scholar] [CrossRef]
- Dance, I.; Scudder, M. Supramolecular Motifs: Concerted Multiple Phenyl Embraces between Ph4P+ Cations Are Attractive and Ubiquitous. Chem. Eur. J. 1996, 2, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-F.; You, J.-M.; Xu, X.-H.; Li, X.-J. A Zn(II) Metal-organic Framework Consisting of Hexagonal Cavities Constructed by Benzyl-functionalized 5-Aminoisophthalate. Chin. J. Struct. Chem. 2015, 34, 562–568. [Google Scholar] [CrossRef]
- Gupta, M.; Ahmad, M.; Singh, R.; Mishra, R.; Sahu, J.; Gupta, A.K. Zn(II)/Cd(II) based coordination polymers synthesized from a semi-flexible dicarboxylate ligand and their emission studies. Polyhedron 2015, 101, 86–92. [Google Scholar] [CrossRef]
- Chucholowski, A.; Fingerle, J.; Iberg, N.; Marki, H.P.; Muller, R.; Pech, M.; Rouge, M.; Schmid, G.; Tschopp, T.; Wessel, H.P. Sulfuric Acid Esters of Sugar Alcohols. U.S. Patent US5,521,160, 28 May 1996. [Google Scholar]
- Gibson, H.W.; Wang, H.; Niu, Z.; Slebodnick, C.; Zhakharov, L.N.; Rheingold, A.L. Rotaxanes from Tetraclams. Macromolecules 2012, 45, 1270–1280. [Google Scholar] [CrossRef]
- Kissel, L.; Pratt, R.H. XDISP Program in WinGX. Acta Crystallogr. 1990, A46, 170–175. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert, version 2.1; Rigaku Americas: The Woodlands, TX, USA, 2015.
- Sheldrick, G.M. SHELXT—Integrated space group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
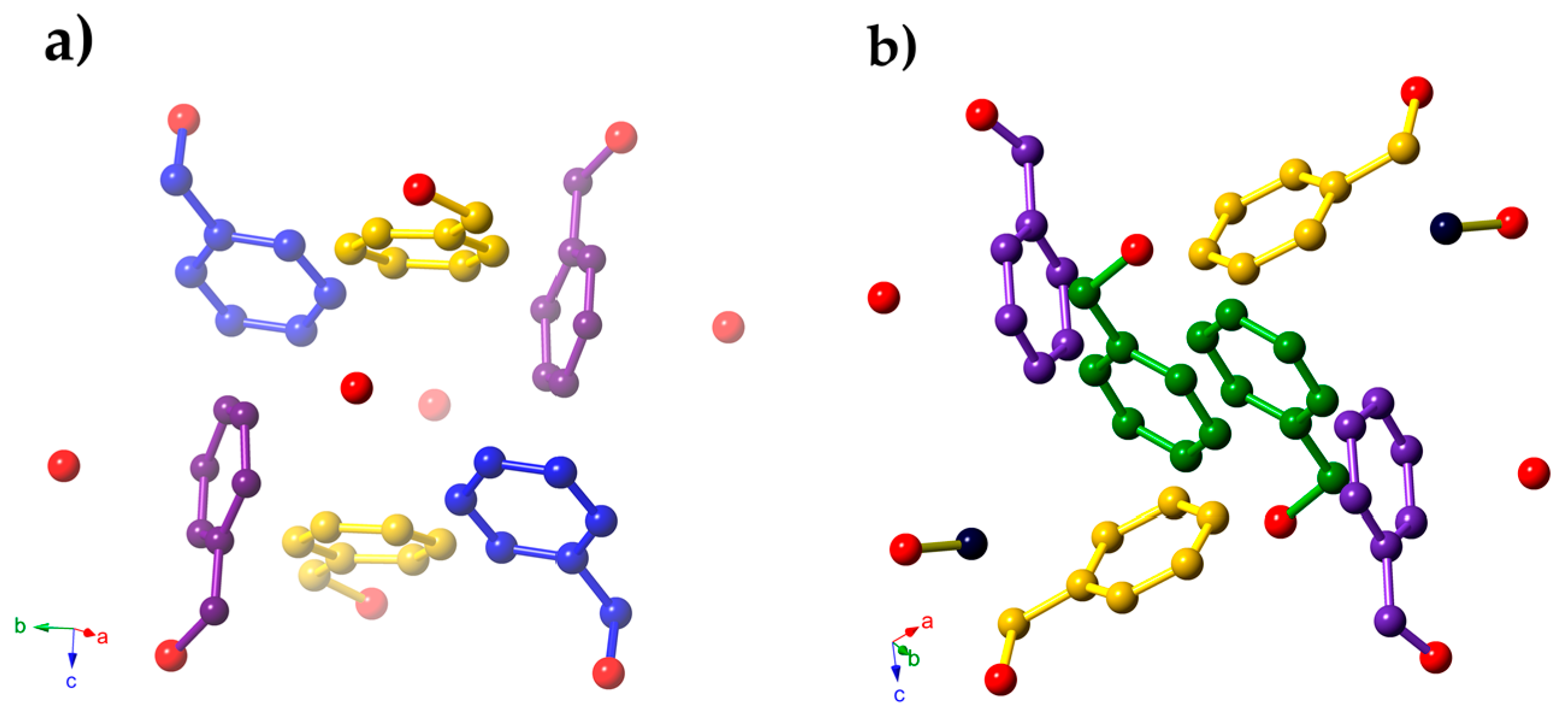

{kind=link}
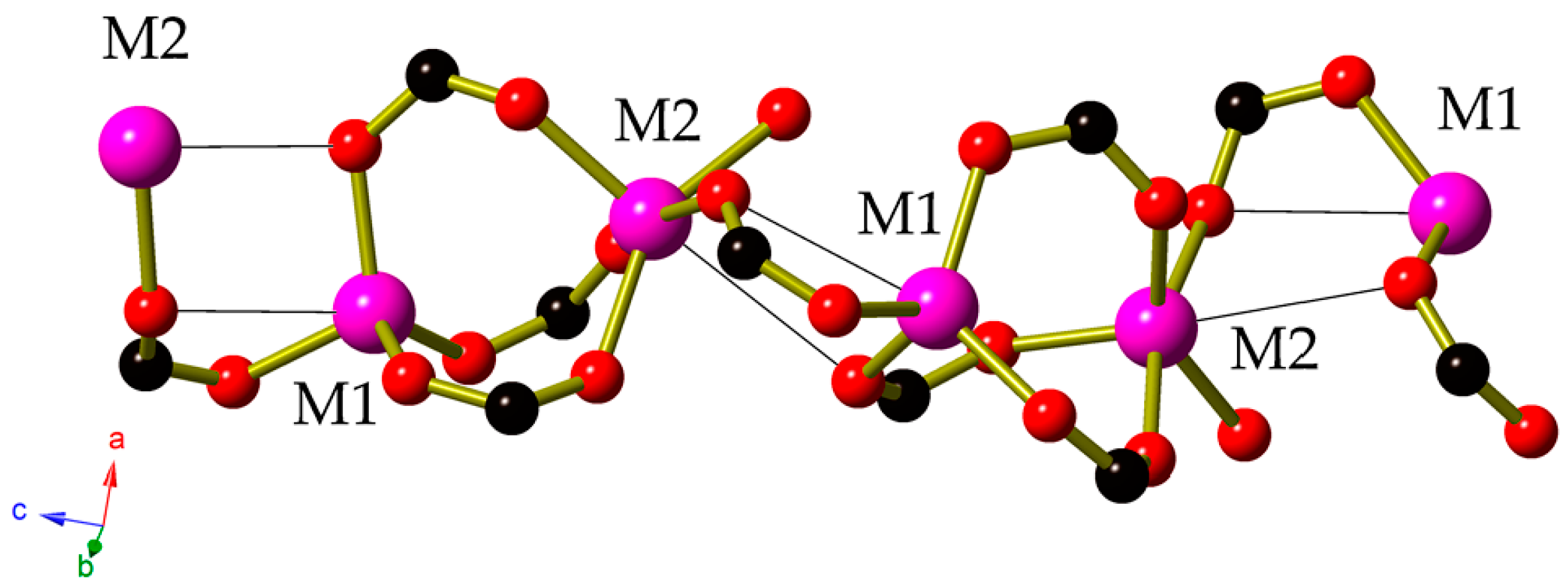
{kind=link}
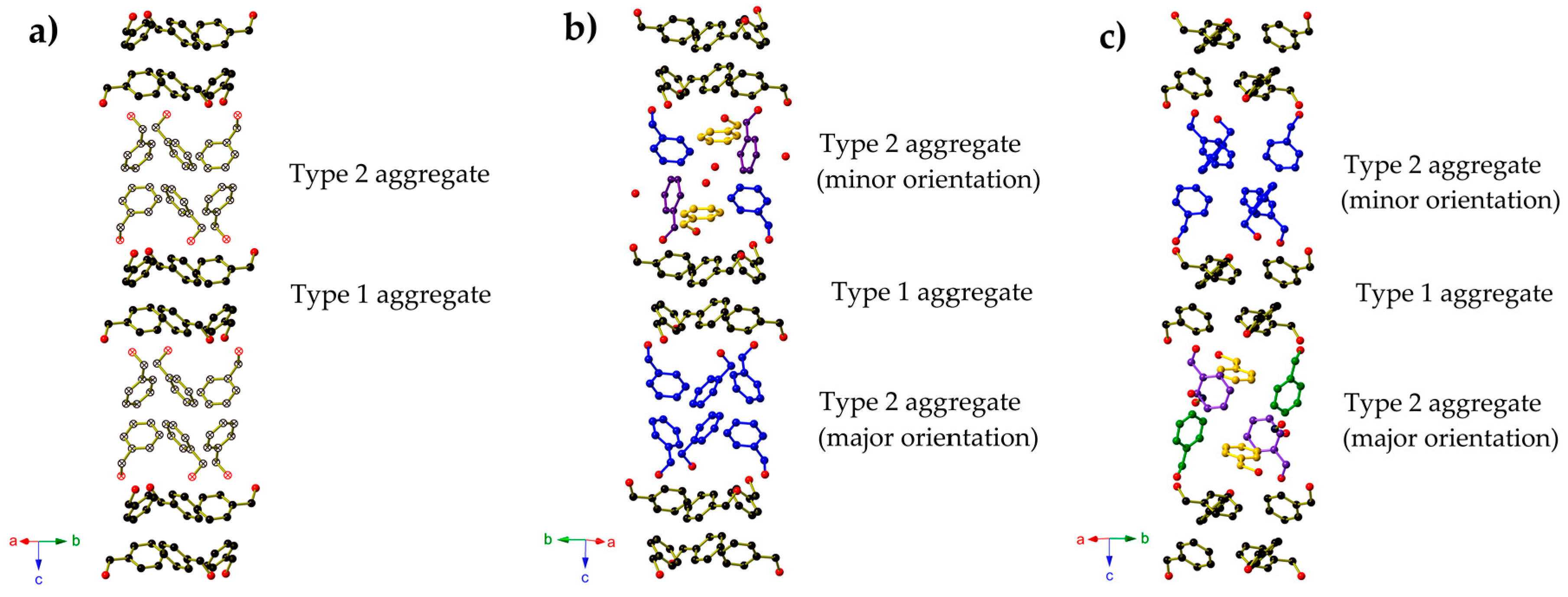{kind=link}
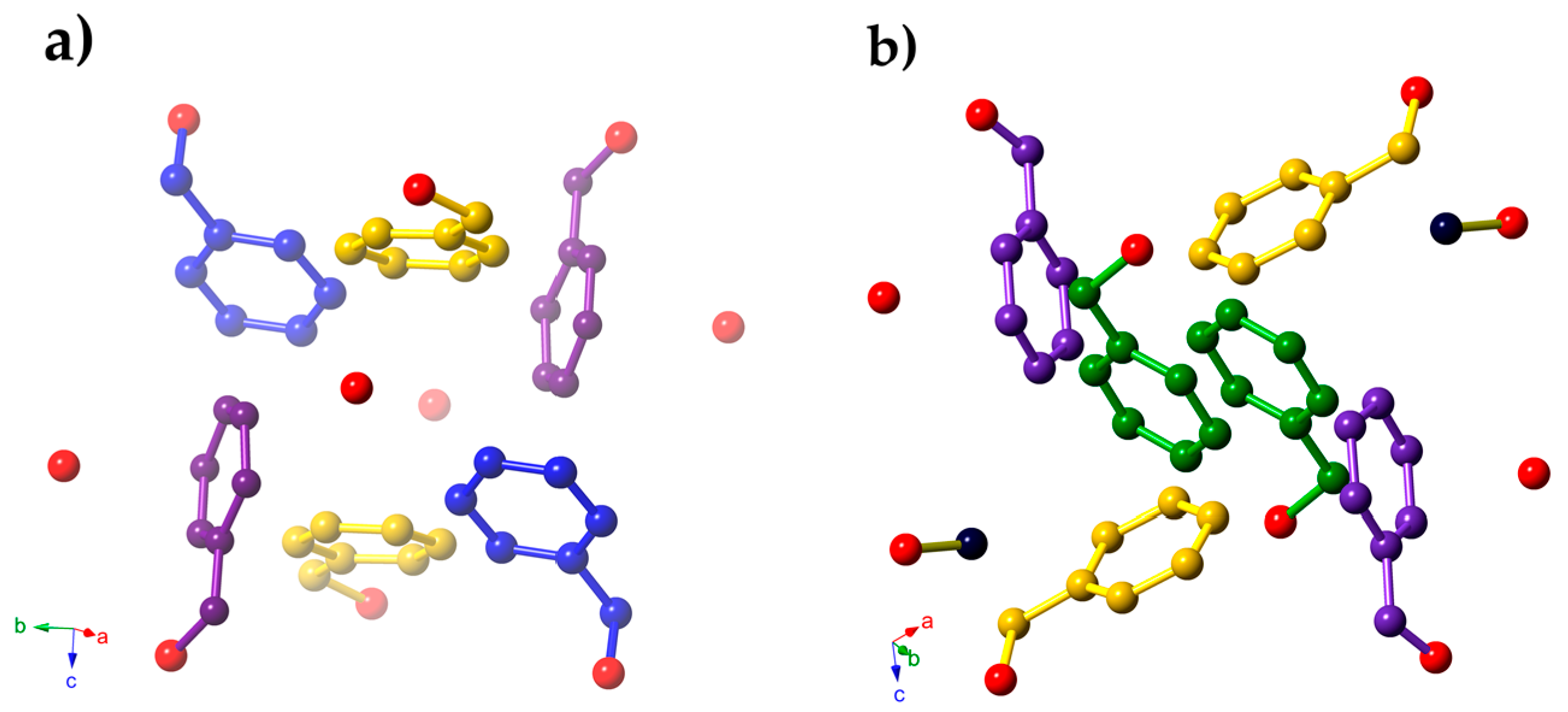{kind=link}
| Zn from MeOH | Zn from EtOH | Zn from iPrOH | Co from MeOH | Co from EtOH | Co from iPrOH | |
|---|---|---|---|---|---|---|
| Molecular Formula | C30.22H23.35O11.45Zn2 | C30H22O11Zn2 | C30H22O11Zn2 | C30.29H23.50Co2O11.58 | C30H22.39Co2O11.19 | C30H22Co2O11 |
| Temperature | 100(2) | 173(2) | 173(2) | 100(2) | 100(2) | 173(2) |
| λ (Å) | 0.7749 | 0.71075 | 0.71075 | 0.7749 | 0.7749 | 0.71075 |
| Crystal System | Trigonal | Trigonal | Trigonal | Trigonal | Trigonal | Trigonal |
| Space Group | R3 | R3 | R3 | R3 | R3 | R3 |
| a (Å) | 27.827(4) | 27.759(3) | 27.6973(18) | 28.013(3) | 27.877(3) | 27.797(4) |
| c (Å) | 18.255(2) | 18.401(3) | 18.2593(18) | 18.1437(19) | 18.192(2) | 18.224(4) |
| V (Å3) | 12242(4) | 12279(3) | 12131(2) | 12330(3) | 12244(3) | 12195(4) |
| Z | 18 | 18 | 18 | 18 | 18 | 18 |
| ρ (g cm−1) | 1.710 | 1.678 | 1.698 | 1.674 | 1.660 | 1.658 |
| μ (mm−1) | 2.309 | 1.822 | 1.844 | 1.609 | 1.617 | 1.282 |
| F(000) | 6413 | 6300 | 6300 | 6334 | 6227 | 6192 |
| GooF | 1.037 | 1.059 | 0.839 | 1.070 | 1.056 | 1.044 |
| Reflections collected/unique/parameters | 29,545/8105/467 | 42,033/6177/394 | 41,948/6193/394 | 29,077/8331/471 | 29,770/8248/441 | 41,642/6174/396 |
| Rint | 0.0712 | 0.0929 | 0.0862 | 0.0595 | 0.0565 | 0.1225 |
| Final R indices (I > 2σ(I)) | R1 = 0.0506 wR2 = 0.1476 | R1 = 0.0385 wR2 = 0.0708 | R1 = 0.0349 wR2 = 0.0575 | R1 = 0.0462 wR2 = 0.1373 | R1 = 0.0405 wR2 = 0.1109 | R1 = 0.0496 wR2 = 0.0960 |
| Final R indices (all data) | R1 = 0.0593 wR2 = 0.1550 | R1 = 0.0732 wR2 = 0.0827 | R1 = 0.0616 wR2 = 0.0629 | R1 = 0.0506 wR2 = 0.1416 | R1 = 0.0471 wR2 = 0.1156 | R1 = 0.1030 wR2 = 0.1167 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCormick, L.J.; Morris, S.A.; Teat, S.J.; Slawin, A.M.Z.; Morris, R.E. Solvent Dependent Disorder in M2(BzOip)2(H2O)·Solvate (M = Co or Zn). Crystals 2018, 8, 6. https://doi.org/10.3390/cryst8010006
McCormick LJ, Morris SA, Teat SJ, Slawin AMZ, Morris RE. Solvent Dependent Disorder in M2(BzOip)2(H2O)·Solvate (M = Co or Zn). Crystals. 2018; 8(1):6. https://doi.org/10.3390/cryst8010006
Chicago/Turabian StyleMcCormick, Laura J., Samuel A. Morris, Simon J. Teat, Alexandra M.Z. Slawin, and Russell E. Morris. 2018. "Solvent Dependent Disorder in M2(BzOip)2(H2O)·Solvate (M = Co or Zn)" Crystals 8, no. 1: 6. https://doi.org/10.3390/cryst8010006
APA StyleMcCormick, L. J., Morris, S. A., Teat, S. J., Slawin, A. M. Z., & Morris, R. E. (2018). Solvent Dependent Disorder in M2(BzOip)2(H2O)·Solvate (M = Co or Zn). Crystals, 8(1), 6. https://doi.org/10.3390/cryst8010006