Incorporating the Thiazolo[5,4-d]thiazole Unit into a Coordination Polymer with Interdigitated Structure


Abstract
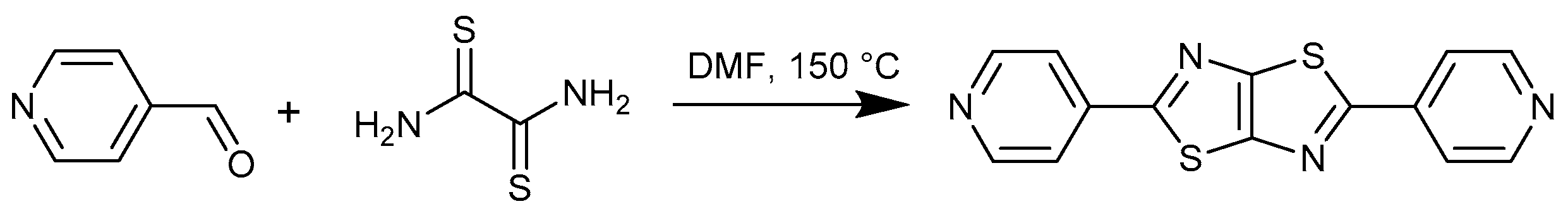
1. Introduction
2. Materials and Methods
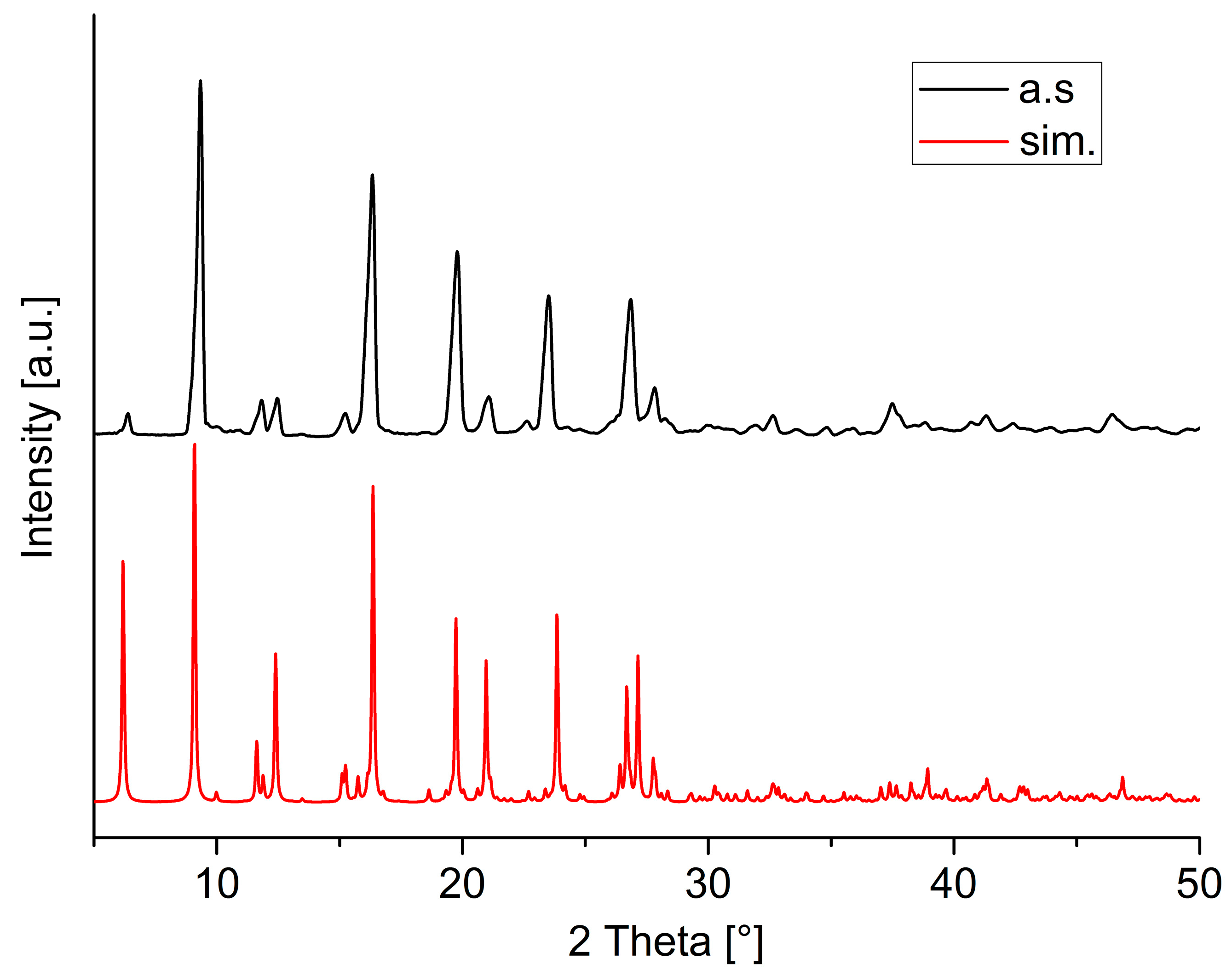
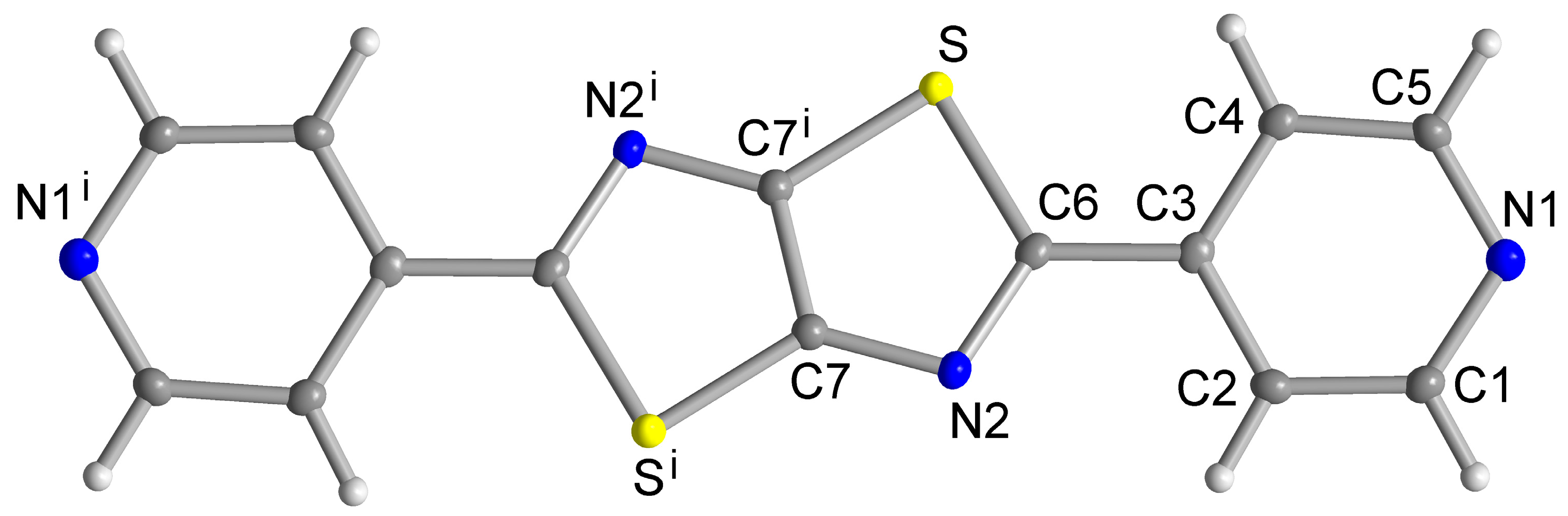
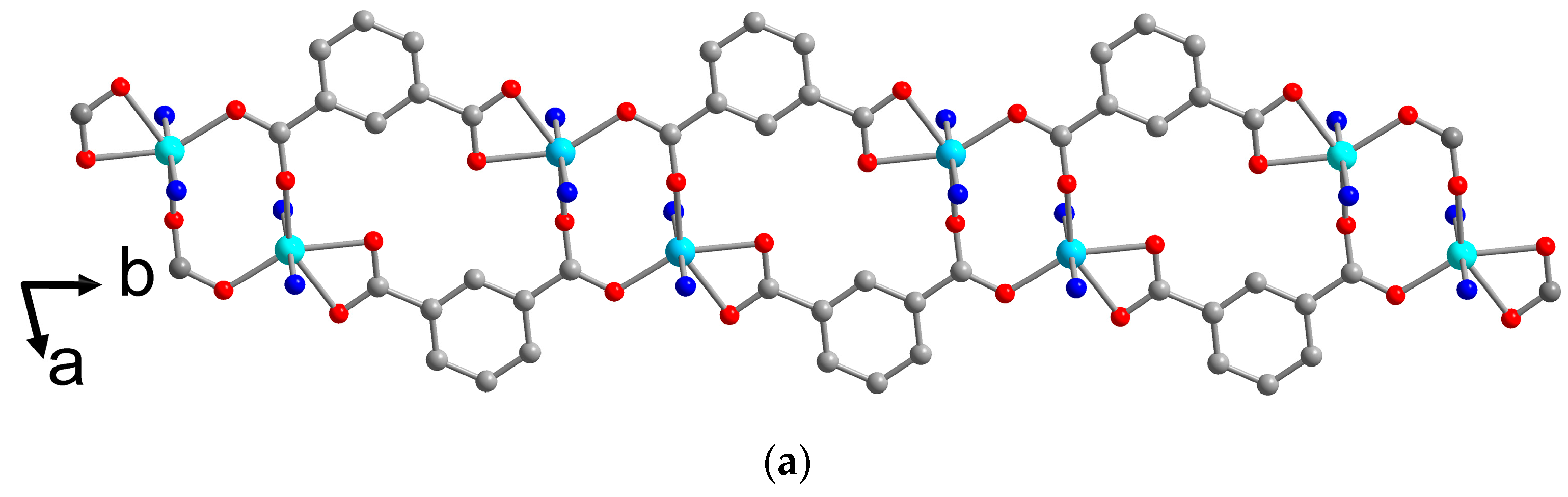
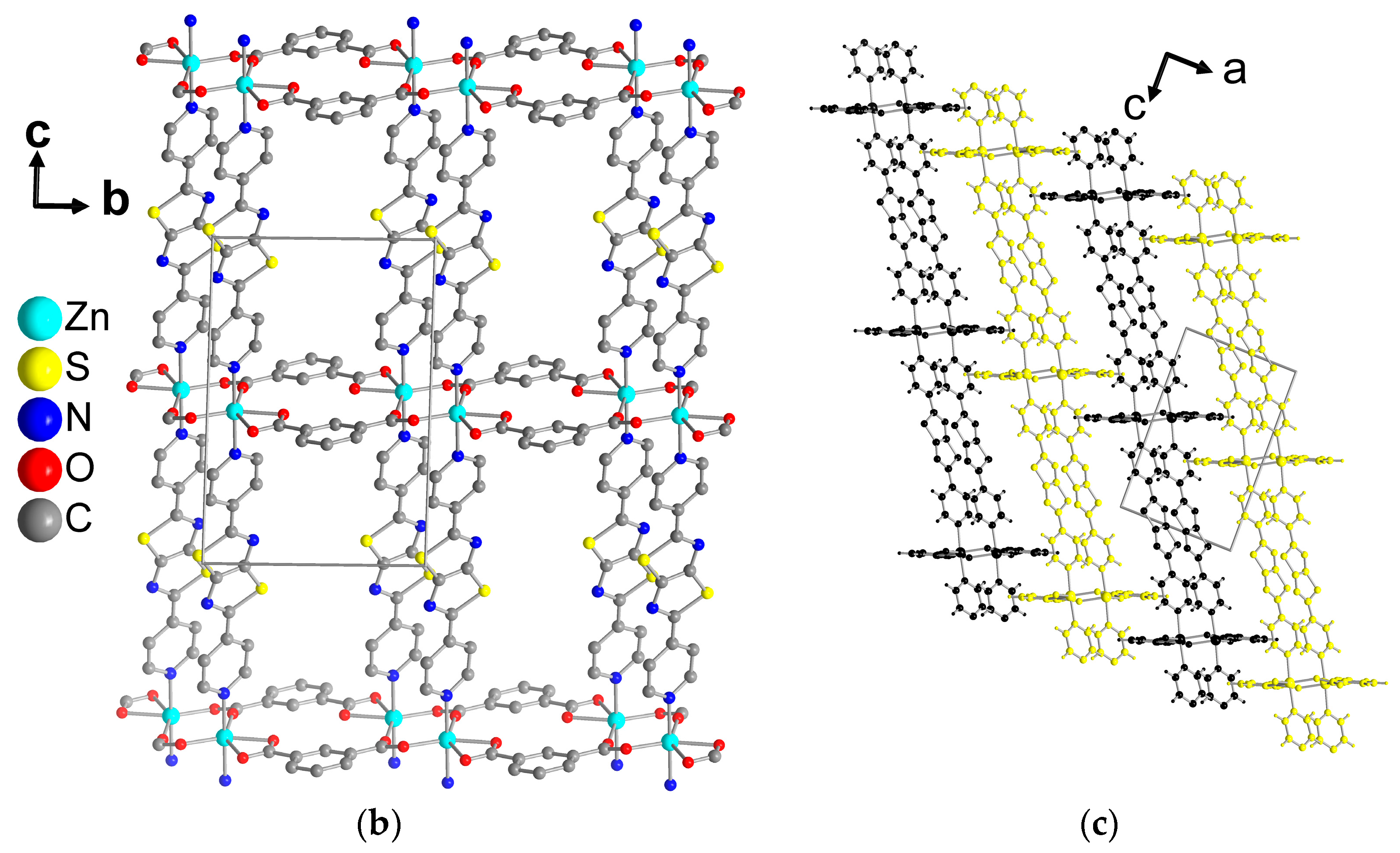
Single Crystal X-ray Structures
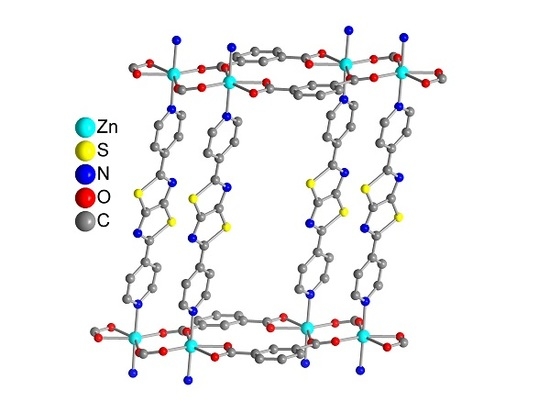
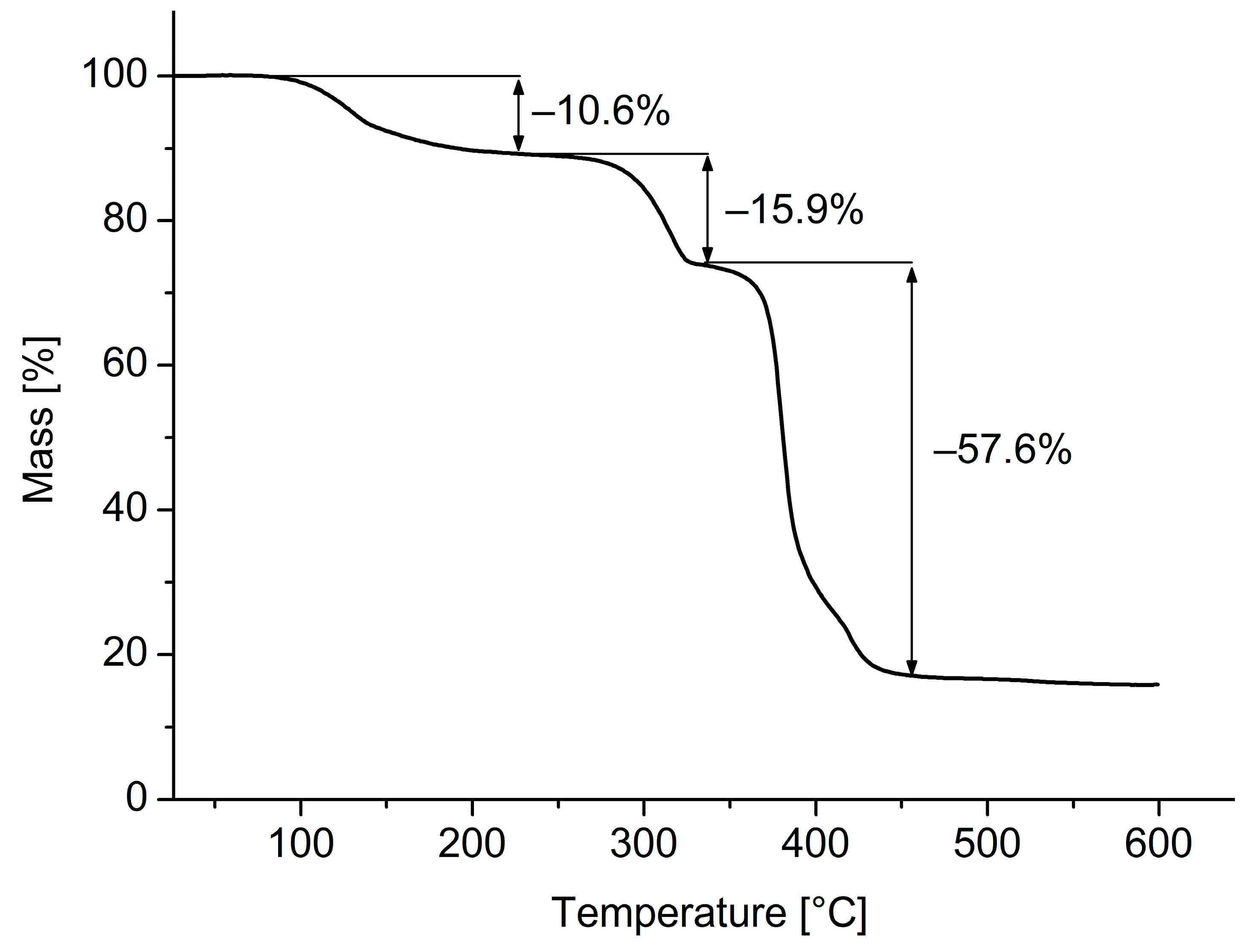
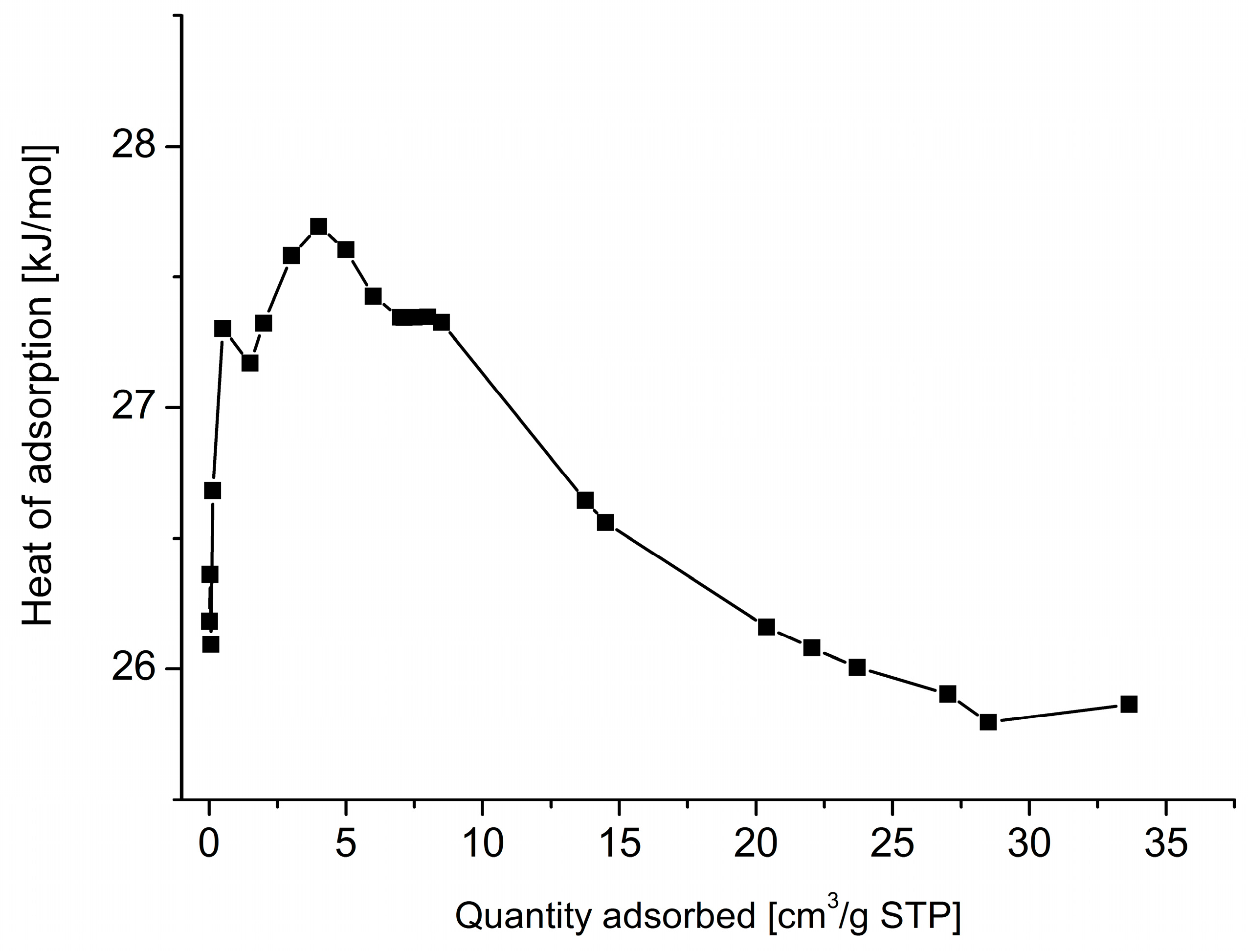
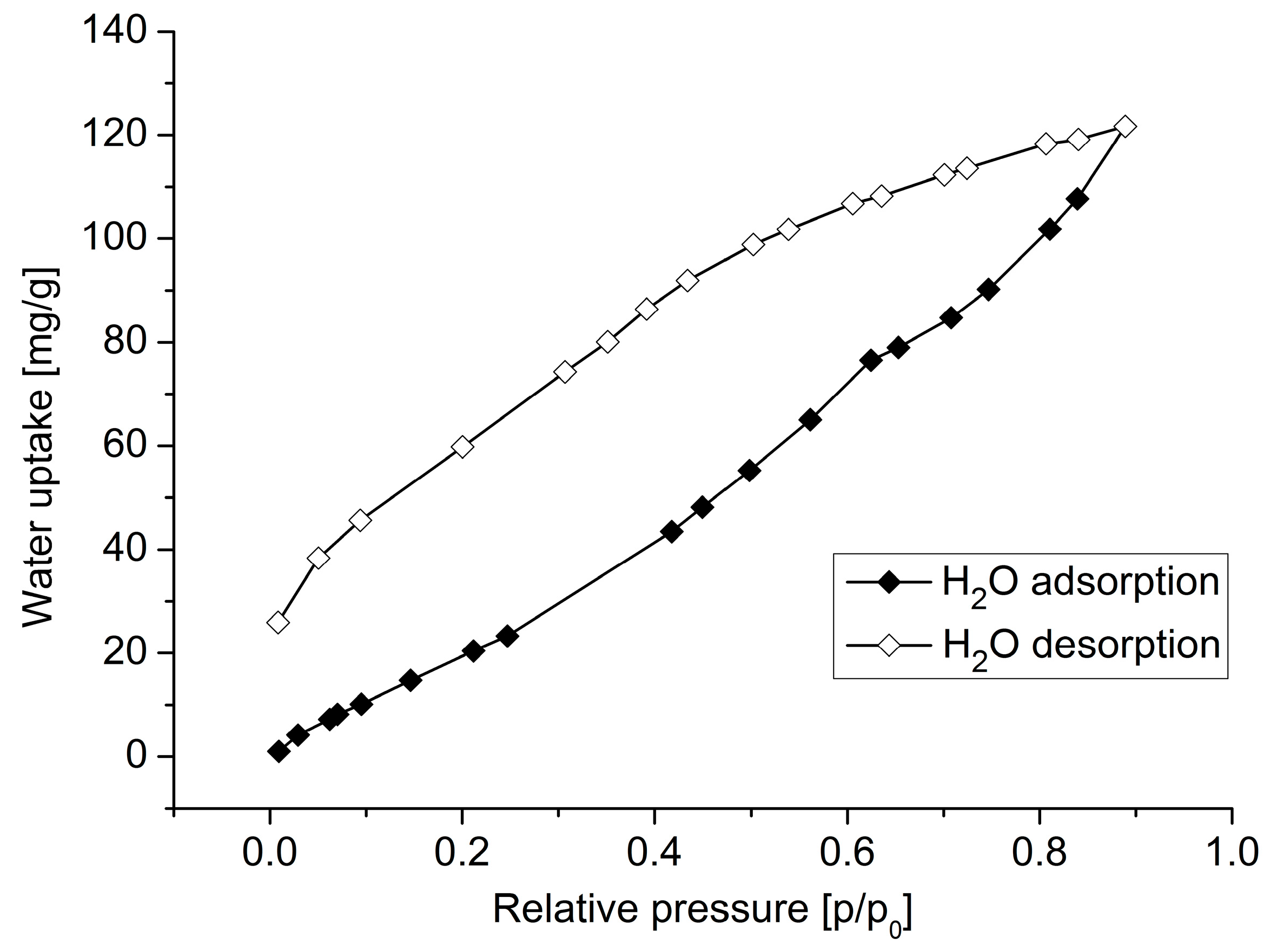
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yaghi, O.M.; O’Keeffe, M.; Ockwig, N.W.; Chae, H.K.; Eddaoudi, M.; Kim, J. Reticular synthesis and the design of new materials. Nature 2003, 423, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Maurin, G.; Serre, C.; Cooper, A.; Férey, G. The new age of MOFs and of their porous-related solids. Chem. Soc. Rev. 2017, 46, 3104–3107. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Ko, N.; Go, Y.B.; Aratani, N.; Choi, S.B.; Choi, E.; Yazaydin, A.Ö.; Snurr, R.Q.; O’Keeffe, M.; Kim, J.; Yaghi, O.M. Ultrahigh porosity in metal-organic frameworks. Science 2010, 329, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Adil, K.; Belmabkhout, Y.; Pillai, R.S.; Cadiau, A.; Bhatt, P.M.; Assen, A.H.; Maurin, G.; Eddaoudi, M. Gas/vapour separation using ultra-microporous metal–organic frameworks: Insights into the structure/separation relationship. Chem. Soc. Rev. 2017, 46, 3402–3430. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Dechnik, J.; Gascon, J.; Doonan, C.J.; Janiak, C.; Sumby, C.J. Mixed-matrix membranes. Angew. Chem. Int. Ed. 2017, 56, 9292–9310. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Farha, O.K.; Roberts, J.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal-organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Janiak, C. MOF catalysts in biomass upgrading towards value-added fine chemicals. CrystEngComm 2017, 19, 4092–4117. [Google Scholar] [CrossRef]
- Kitao, T.; Zhang, Y.; Kitagawa, S.; Wang, B.; Uemura, T. Hybridization of MOFs and polymers. Chem. Soc. Rev. 2017, 46, 3108–3133. [Google Scholar] [CrossRef] [PubMed]
- Lustig, W.P.; Mukherjee, S.; Rudd, N.D.; Desai, A.V.; Li, J.; Ghosh, S.K. Metal–organic frameworks: functional luminescent and photonic materials for sensing applications. Chem. Soc. Rev. 2017, 46, 3242–3285. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Deibert, B.J.; Li, J. Luminescent metal-organic frameworks for chemical sensing and explosive detection. Chem. Soc. Rev. 2014, 43, 5815–5840. [Google Scholar] [CrossRef] [PubMed]
- Gangu, K.K.; Maddila, S.; Mukkamala, S.B.; Jonnalagadda, S.B. A review on contemporary Metal–Organic Framework materials. Inorg. Chim. Acta 2016, 446, 61–74. [Google Scholar] [CrossRef]
- Jeremias, F.; Fröhlich, D.; Janiak, C.; Henninger, S.K. Water and methanol adsorption on MOFs for cycling heat transformation processes. New J. Chem. 2014, 38, 1846–1852. [Google Scholar] [CrossRef]
- Almeida Paz, F.A.; Klinowski, J.; Vilela, S.M.F.; Tome, J.P.C.; Cavaleiro, J.A.S.; Rocha, J. Ligand design for functional metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 1088–1110. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.K.; Cohen, S.M. Postsynthetic modification of metal-organic frameworks—A progress report. Chem. Soc. Rev. 2011, 40, 498–519. [Google Scholar] [CrossRef] [PubMed]
- Islamoglu, T.; Goswami, S.; Li, Z.; Howarth, A.J.; Farha, O.K.; Hupp, J.T. Postsynthetic tuning of metal–organic frameworks for targeted applications. Acc. Chem. Res. 2017, 50, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Biradha, K.; Sarkar, M.; Rajput, L. Crystal engineering of coordination polymers using 4,4′-bipyridine as a bond between transition metal atoms. Chem. Commun. 2006, 4169–4179. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Ghoshal, D. Selective carbon dioxide adsorption by mixed-ligand porous coordination polymers. CrystEngComm 2015, 17, 8388–8413. [Google Scholar] [CrossRef]
- Haldar, R.; Maji, T.K. Metal-organic frameworks (MOFs) based on mixed linker systems: Structural diversities towards functional materials. CrystEngComm 2013, 15, 9276–9295. [Google Scholar] [CrossRef]
- Bae, Y.-S.; Mulfort, K.L.; Frost, H.; Ryan, P.; Punnathanam, S.; Broadbelt, L.J.; Hupp, J.T.; Snurr, R.Q. Separation of CO2 from CH4 using mixed-ligand metal–organic frameworks. Langmuir 2008, 24, 8592–8598. [Google Scholar] [CrossRef] [PubMed]
- Henke, S.; Schneemann, A.; Wütscher, A.; Fischer, R.A. Directing the breathing behavior of pillared-layered metal–organic frameworks via a systematic library of functionalized linkers bearing flexible substituents. J. Am. Chem. Soc. 2012, 134, 9464–9474. [Google Scholar] [CrossRef] [PubMed]
- Glomb, S.; Woschko, D.; Makhloufi, G.; Janiak, C. Metal–organic frameworks with internal urea-functionalized dicarboxylate linkers for SO2 and NH3 adsorption. ACS Appl. Mater. Interfaces 2017, 9, 37419–37434. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Martínez, V.M.; Furukawa, S.; Kondo, M.; Shimomura, S.; Uehara, H.; Nakahama, M.; Sugimoto, K.; Kitagawa, S. Molecular decoding using luminescence from an entangled porous framework. Nat. Commun. 2011, 2, 168. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Tanaka, D.; Nakagawa, K.; Kitagawa, S. Selective guest sorption in an interdigitated porous framework with hydrophobic pore surfaces. Chem. Commun. 2007, 3395–3397. [Google Scholar] [CrossRef]
- Tanaka, D.; Nakagawa, K.; Higuchi, M.; Horike, S.; Kubota, Y.; Kobayashi, T.C.; Takata, M.; Kitagawa, S. Kinetic gate-opening process in a flexible porous coordination polymer. Angew. Chem. Int. Ed. 2008, 47, 3914–3918. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Horike, S.; Inubushi, Y.; Nakagawa, K.; Kubota, Y.; Takata, M.; Kitagawa, S. Solid solutions of soft porous coordination polymers: Fine-tuning of gas adsorption properties. Angew. Chem. 2010, 122, 4930–4934. [Google Scholar] [CrossRef]
- Nakagawa, K.; Tanaka, D.; Horike, S.; Shimomura, S.; Higuchi, M.; Kitagawa, S. Enhanced selectivity of CO2 from a ternary gas mixture in an interdigitated porous framework. Chem. Commun. 2010, 46, 4258–4260. [Google Scholar] [CrossRef] [PubMed]
- Hijikata, Y.; Horike, S.; Sugimoto, M.; Sato, H.; Matsuda, R.; Kitagawa, S. Relationship between channel and sorption properties in coordination polymers with interdigitated structures. Chem. Eur. J. 2011, 17, 5138–5144. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.; Horike, S.; Nakagawa, K.; Kitagawa, S. Synthesis and adsorption properties of azulene-containing porous interdigitated framework. Chem. Lett. 2012, 41, 425–426. [Google Scholar] [CrossRef]
- Johnson, J.R.; Ketcham, R. Thiazolothiazoles. I. The reaction of aromatic aldehydes with dithiooxamide. J. Am. Chem. Soc. 1960, 82, 2719–2724. [Google Scholar] [CrossRef]
- Bevk, D.; Marin, L.; Lutsen, L.; Vanderzande, D.; Maes, W. Thiazolo[5,4-d]thiazoles - promising building blocks in the synthesis of semiconductors for plastic electronics. RSC Adv. 2013, 3, 11418–11431. [Google Scholar] [CrossRef]
- Reginato, G.; Mordini, A.; Zani, L.; Calamante, M.; Dessi, A. Photoactive compounds based on the thiazolo[5,4-d]thiazole core and their application in organic and hybrid photovoltaics. Eur. J. Org. Chem. 2016, 233–251. [Google Scholar] [CrossRef]
- Zampese, J.A.; Keene, F.R.; Steel, P.J. Diastereoisomeric dinuclear ruthenium complexes of 2,5-di(2-pyridyl)thiazolo[5,4-d]thiazole. Dalton Trans. 2004, 4124–4129. [Google Scholar] [CrossRef] [PubMed]
- Falcão, E.H.L.; Naraso; Feller, R.K.; Wu, G.; Wudl, F.; Cheetham, A.K. Hybrid organic–inorganic framework structures: Influence of cation size on metal–oxygen–metal connectivity in the alkaline earth thiazolothiazoledicarboxylates. Inorg. Chem. 2008, 47, 8336–8342. [Google Scholar]
- Aprea, A.; Colombo, V.; Galli, S.; Masciocchi, N.; Maspero, A.; Palmisano, G. Thiazolo[5,4-d]thiazole-2,5-dicarboxylic acid, C6H2N2O4S2, and its coordination polymers. Solid State Sci. 2010, 12, 795–802. [Google Scholar] [CrossRef]
- Rizzuto, F.J.; Faust, T.B.; Chan, B.; Hua, C.; D’Alessandro, D.M.; Kepert, C.J. Experimental and computational studies of a multi-electron donor–acceptor ligand containing the thiazolo[5,4-d]thiazole core and its incorporation into a metal–organic framework. Chem. Eur. J. 2014, 20, 17597–17605. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Rizzuto, F.J.; Zhang, X.; Tuna, F.; Collison, D.; D’Alessandro, D.M. Spectroelectrochemical properties of a Ru(II) complex with a thiazolo[5,4-d]thiazole triarylamine ligand. New J. Chem. 2017, 41, 108–114. [Google Scholar] [CrossRef]
- Zhu, X.; Tian, C.; Jin, T.; Wang, J.; Mahurin, S.M.; Mei, W.; Xiong, Y.; Hu, J.; Feng, X.; Liu, H. Thiazolothiazole-linked porous organic polymers. Chem. Commun. 2014, 50, 15055–15058. [Google Scholar] [CrossRef] [PubMed]
- APEX2. SAINT, Data Reduction and Frame Integration Program for the CCD Area-Detector System, Bruker Analytical X-ray Systems; Data Collection Program for the CCD Area-Detector System: Madison, WI, USA, 1997–2006.
- Sheldrick, G.M. SADABS: Area-Detector Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHElXL. Acta Crystallogr. Sect A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K. DIAMOND, version 4.4; Crystal and Molecular Structure Visualization; Crystal Impact—K. Brandenburg & H. Putz Gbr: Bonn, Germany, 2009–2017.
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D—Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Knighton, R.C.; Hallett, A.J.; Kariuki, B.M.; Pope, S.J.A. A one-step synthesis towards new ligands based on aryl-functionalised thiazolo[5,4-d]thiazole chromophores. Tetrahedron Lett. 2010, 51, 5419–5422. [Google Scholar] [CrossRef]
- Hisamatsu, S.; Masu, H.; Azumaya, I.; Takahashi, M.; Kishikawa, K.; Kohmoto, S. U-shaped aromatic ureadicarboxylic acids as versatile building blocks: Construction of ladder and zigzag networks and channels. Cryst. Growth Des. 2011, 11, 5387–5395. [Google Scholar] [CrossRef]
- Das, A.; D’Alessandro, D.M. Tuning the functional sites in metal-organic frameworks to modulate CO2 heats of adsorption. CrystEngComm 2015, 17, 706–718. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, H.; Emge, T.J.; Gong, Q.; Nijem, N.; Chabal, Y.J.; Kong, L.; Langreth, D.C.; Liu, H.; Zeng, H.; Li, J. Enhancing gas adsorption and separation capacity through ligand functionalization of microporous metal–organic framework structures. Chem. Eur. J. 2011, 17, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Marshall, M.; Chaffee, A.L. CO2 adsorption-based separation by metal organic framework (Cu-BTC) versus zeolite (13X). Energy Fuels 2009, 23, 2785–2789. [Google Scholar] [CrossRef]
- Caskey, S.R.; Wong-Foy, A.G.; Matzger, A.J. Dramatic tuning of carbon dioxide uptake via metal substitution in a coordination polymer with cylindrical pores. J. Am. Chem. Soc. 2008, 130, 10870–10871. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, P.L.; Bourrelly, S.; Serre, C.; Vimont, A.; Daturi, M.; Hamon, L.; De Weireld, G.; Chang, J.-S.; Hong, D.-Y.; Kyu Hwang, Y.; et al. High uptakes of CO2 and CH4 in mesoporous metal-organic frameworks MIL-100 and MIL-101. Langmuir 2008, 24, 7245–7250. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.M.; D’Alessandro, D.M.; Krishna, R.; Long, J.R. Enhanced carbon dioxide capture upon incorporation of N,N′-dimethylethylenediamine in the metal-organic framework CuBTTri. Chem. Sci. 2011, 2, 2022–2028. [Google Scholar] [CrossRef]
- Bourrelly, S.; Llewellyn, P.L.; Serre, C.; Millange, F.; Loiseau, T.; Férey, G. Different adsorption behaviors of methane and carbon dioxide in the isotypic nanoporous metal terephthalates MIL-53 and MIL-47. J. Am. Chem. Soc. 2005, 127, 13519–13521. [Google Scholar] [CrossRef] [PubMed]
- Pera-Titus, M.; Farrusseng, D. Guest-induced gate opening and breathing phenomena in soft porous crystals: Building thermodynamically consistent isotherms. J. Phys. Chem. C 2012, 116, 1638–1649. [Google Scholar] [CrossRef]












{kind=link}
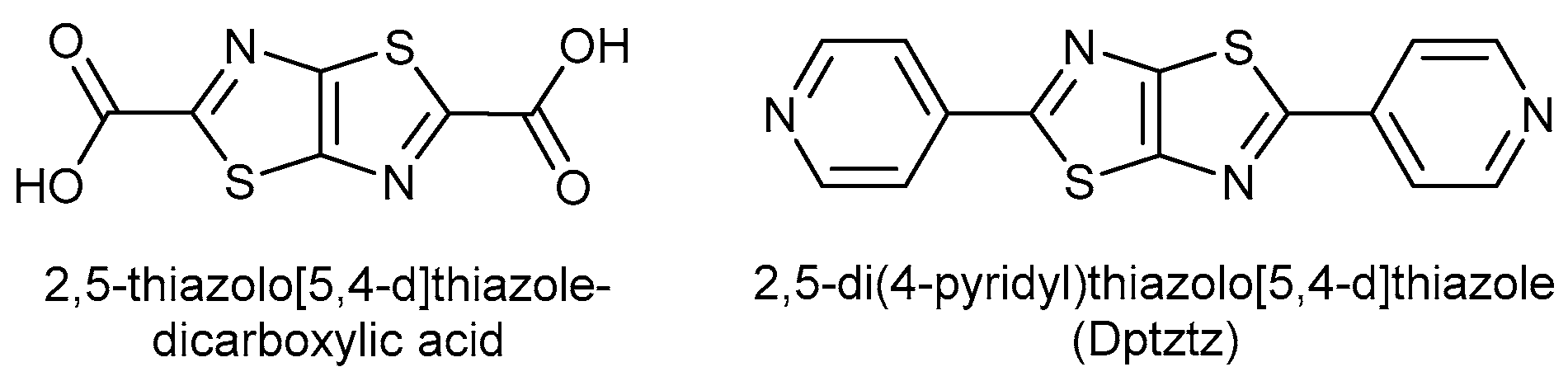{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dptztz | [Zn(1,3 BDC)Dptztz]·DMF | |
|---|---|---|
| Chemical formula | C14H8N4S2 | C22H12N4O4S2Zn·C3H7NO |
| Mr | 296.36 | 598.94 |
| Crystal system, space group | Monoclinic, P21/c | Triclinic, |
| Temperature (K) | 100 | 100 |
| a (Å) | 8.3873 (5) | 9.1388 (6) |
| b (Å) | 6.3140 (3) | 10.0354 (7) |
| c (Å) | 11.7170 (6) | 14.2804 (11) |
| α (°) | 90 | 88.417 (4) |
| β (°) | 93.699 (3) | 88.236 (5) |
| γ (°) | 90 | 75.636 (4) |
| V (Å3) | 619.21 (6) | 1267.86 (16) |
| Z | 2 | 2 |
| μ (mm−1) | 0.423 | 1.181 |
| Crystal size (mm) | 0.10 × 0.05 × 0.05 | 0.10 × 0.05 × 0.01 |
| Absorption correction | Multi-scan, wR2(int) was 0.1649 before and 0.0771 after correction. The Ratio of minimum to maximum transmission is 0.8473. The λ/2 correction factor is 0.0015. | Multi-scan, wR2(int) was 0.1533 before and 0.0488 after correction. The Ratio of minimum to maximum transmission is 0.9318. The λ/2 correction factor is 0.0015. |
| Tmin, Tmax | 0.6330, 0.7471 | 0.6951, 0.7460 |
| No. of measured, independent and observed reflections | 6837, 965, 847 [I > 2σ(I)] | 17151, 4743, 3696 [I > 2σ(I)] |
| Rint | 0.049 | 0.045 |
| (sin θ/λ)max (Å−1) | 0.639 | 0.612 |
| R, wR(F2), S [F2 > 2σ (F2)] R, wR(F2), S [all data] | 0.0284, 0.0675, 1.067 0.359, 0.0699, 1.067 | 0.0400, 0.0849, 1.055 0.0609, 0.0916, 1.055 |
| No. of reflections | 965 | 4743 |
| No. of parameters | 91 | 364 |
| Δρmax, Δρmin (e·Å−3) | 0.238, -0.182 | 0.645, –0.581 |
| Zn–O1 | 2.0532 (18) | Zn–O4iii | 2.2269 (19) |
| Zn–O2i | 2.0218 (18) | Zn–N1 | 2.166 (3) |
| Zn–O3iii | 2.1569 (19) | Zn–N4ii | 2.151 (3) |
| O1–Zn–O2i | 119.30 (7) | O2i–Zn–N4ii | 89.34 (9) |
| O1–Zn–O3iii | 88.78 (7) | O3iii–Zn–O4iii | 60.01 (7) |
| O1–Zn–O4iii | 148.39 (7) | O3iii–Zn–N1 | 90.18 (9) |
| O1–Zn–N1 | 89.84 (9) | O3iii–Zn–N4ii | 90.56 (9) |
| O1–Zn‒N4ii | 86.00 (9) | O4iii–Zn–N1 | 94.77 (8) |
| O2i–Zn–O3iii | 151.83 (7) | O4iii–Zn–N4ii | 89.21 (8) |
| O2i–Zn–O4iii | 91.82 (7) | N1–Zn–N4ii | 175.76 (8) |
| O2i–Zn–N1 | 91.99 (9) |
| Quantity Adsorbed (cm3/g, mmol/g, wt %) | Total Pore Volume (cm3/g) | |
|---|---|---|
| 195 K | 138, 6.16, 27.1% | 0.246 1 |
| 273 K | 51.9, 2.32, 10.2% | 0.092 2 |
| 293 K | 35.5, 1.59, 7.0% | 0.061 3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millan, S.; Makhloufi, G.; Janiak, C. Incorporating the Thiazolo[5,4-d]thiazole Unit into a Coordination Polymer with Interdigitated Structure. Crystals 2018, 8, 30. https://doi.org/10.3390/cryst8010030
Millan S, Makhloufi G, Janiak C. Incorporating the Thiazolo[5,4-d]thiazole Unit into a Coordination Polymer with Interdigitated Structure. Crystals. 2018; 8(1):30. https://doi.org/10.3390/cryst8010030
Chicago/Turabian StyleMillan, Simon, Gamall Makhloufi, and Christoph Janiak. 2018. "Incorporating the Thiazolo[5,4-d]thiazole Unit into a Coordination Polymer with Interdigitated Structure" Crystals 8, no. 1: 30. https://doi.org/10.3390/cryst8010030
APA StyleMillan, S., Makhloufi, G., & Janiak, C. (2018). Incorporating the Thiazolo[5,4-d]thiazole Unit into a Coordination Polymer with Interdigitated Structure. Crystals, 8(1), 30. https://doi.org/10.3390/cryst8010030