
Crystallography of Representative MOFs Based on Pillared Cyanonickelate (PICNIC) Architecture


Abstract
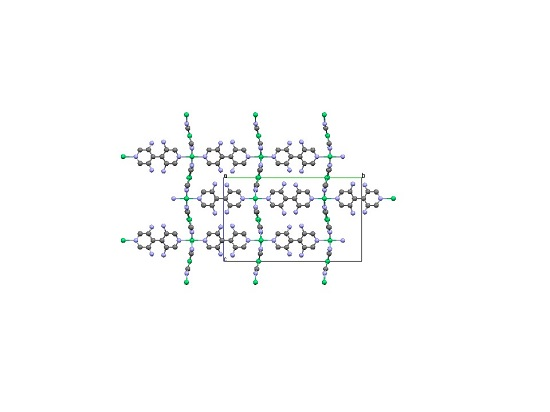
:
1. Introduction
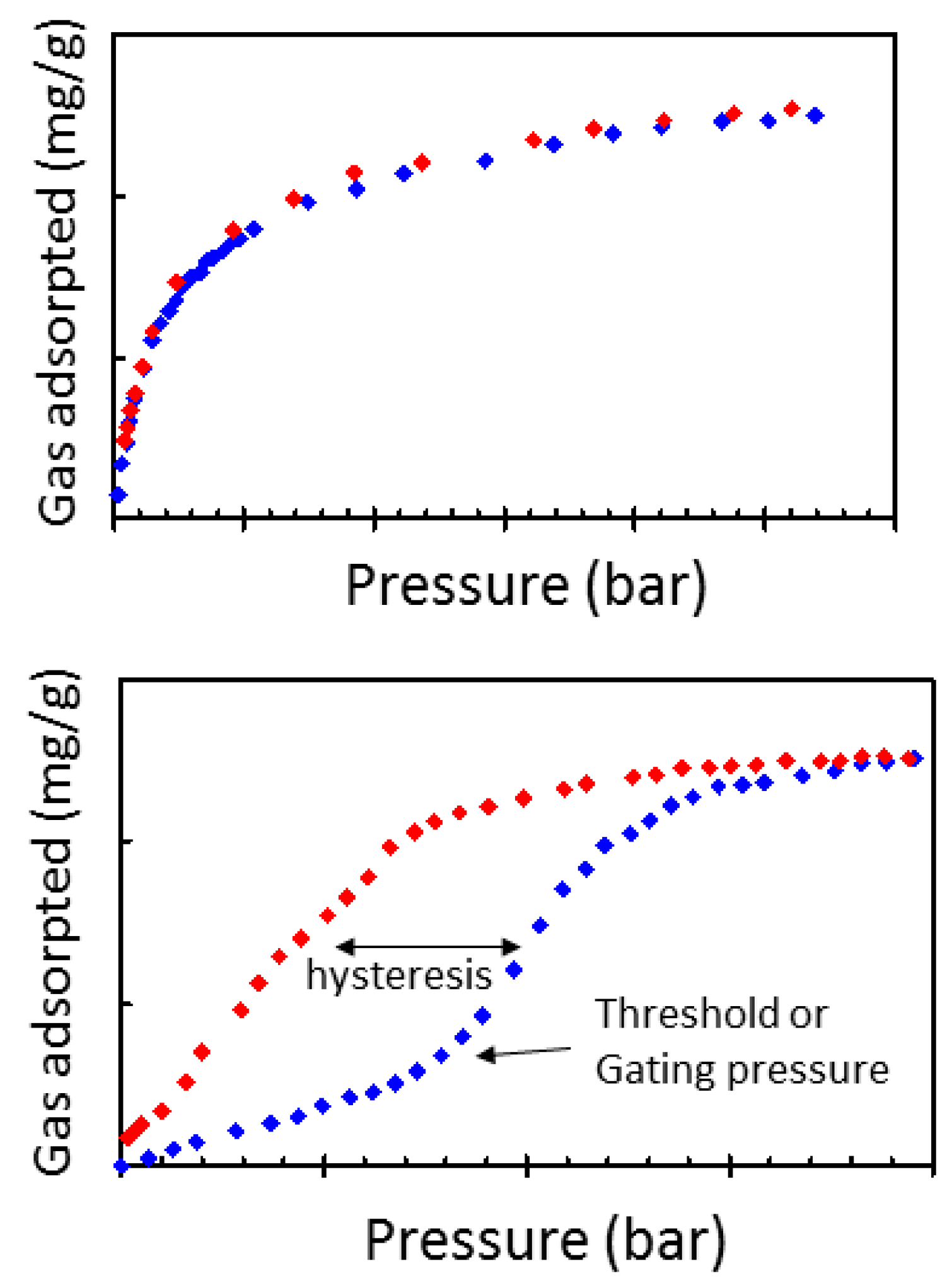
1.1. Flexible Metal Organic Frameworks (MOFs)
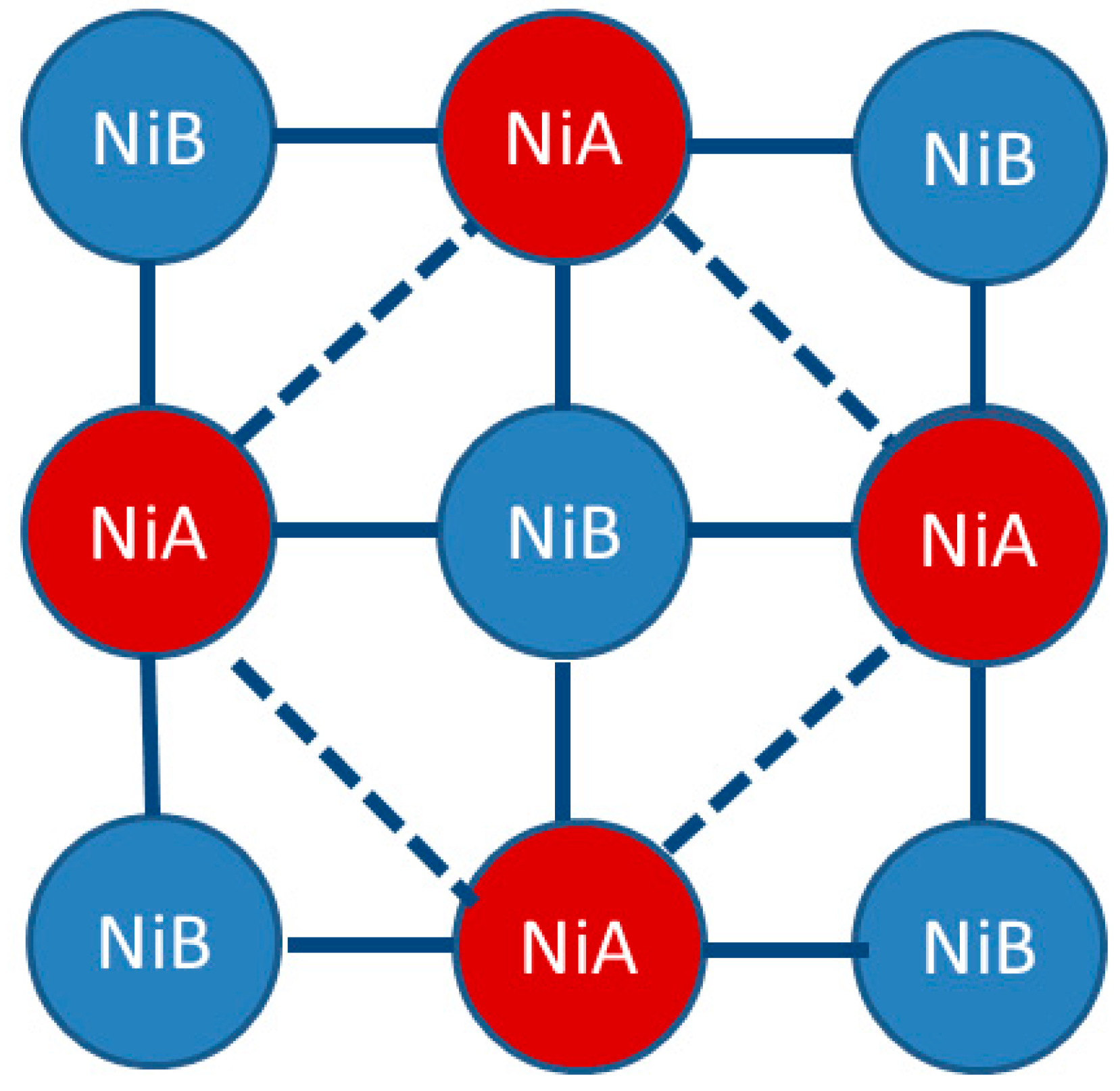
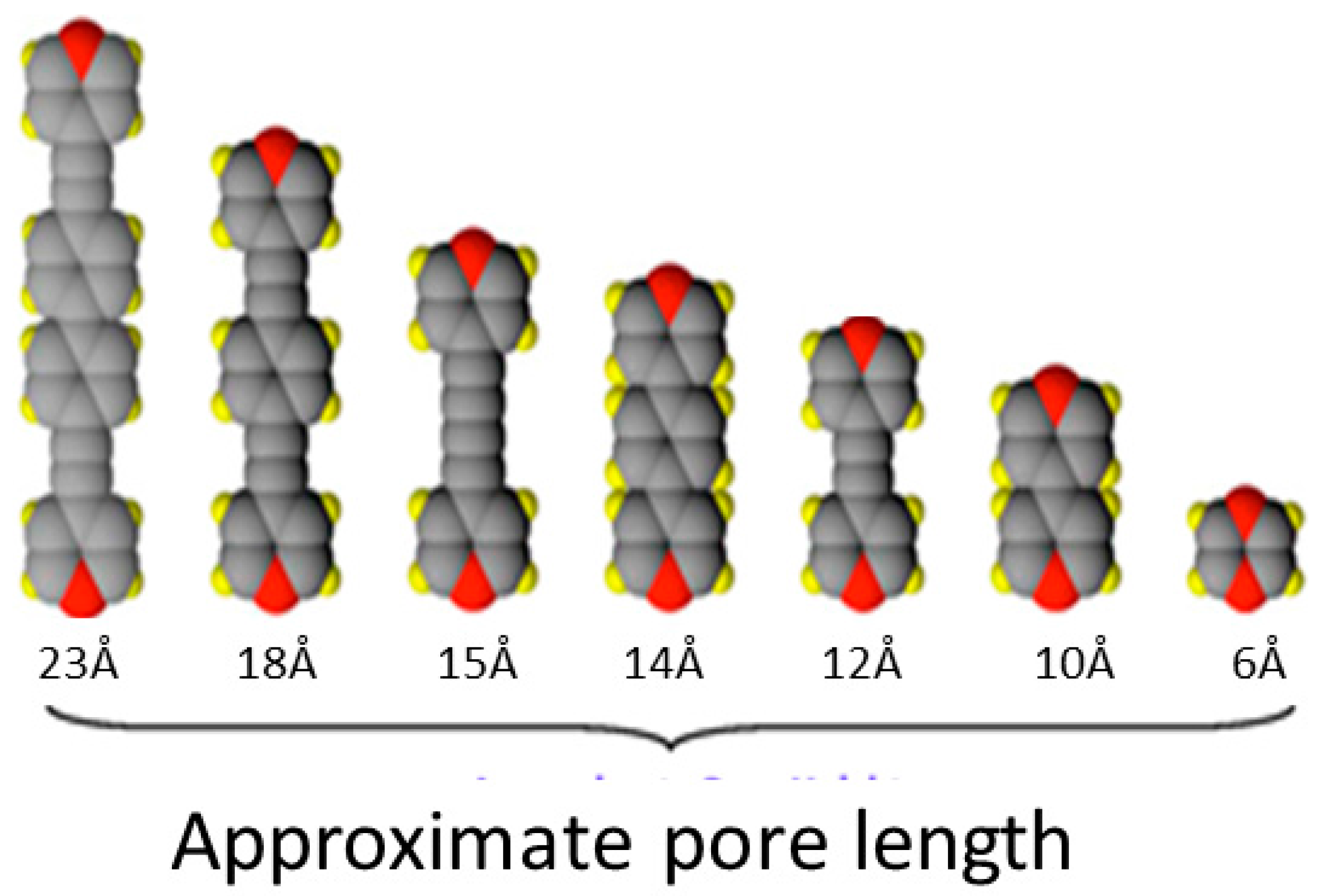
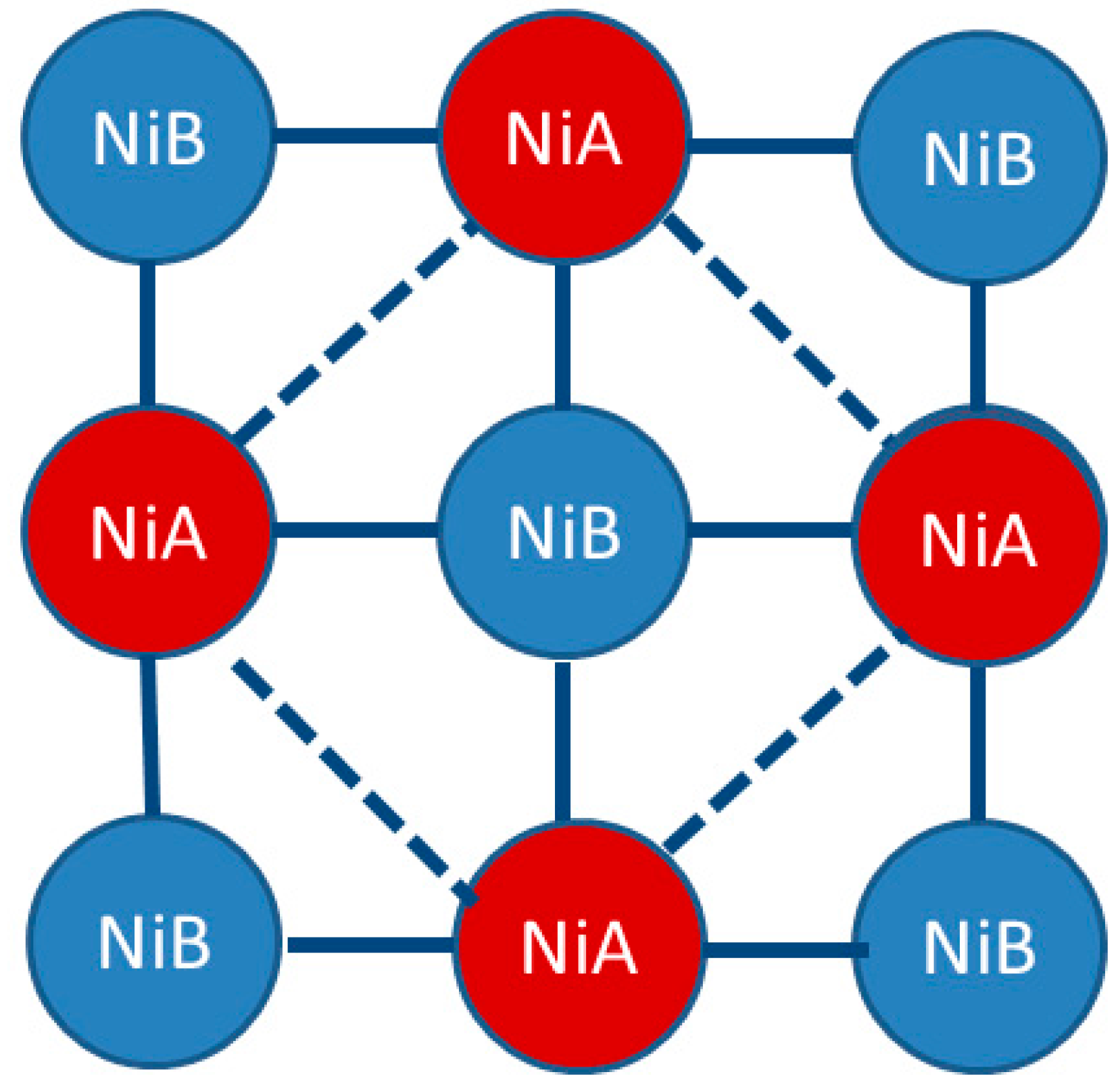
1.2. Ni(CN)4-Based MOFs (PICNICs)
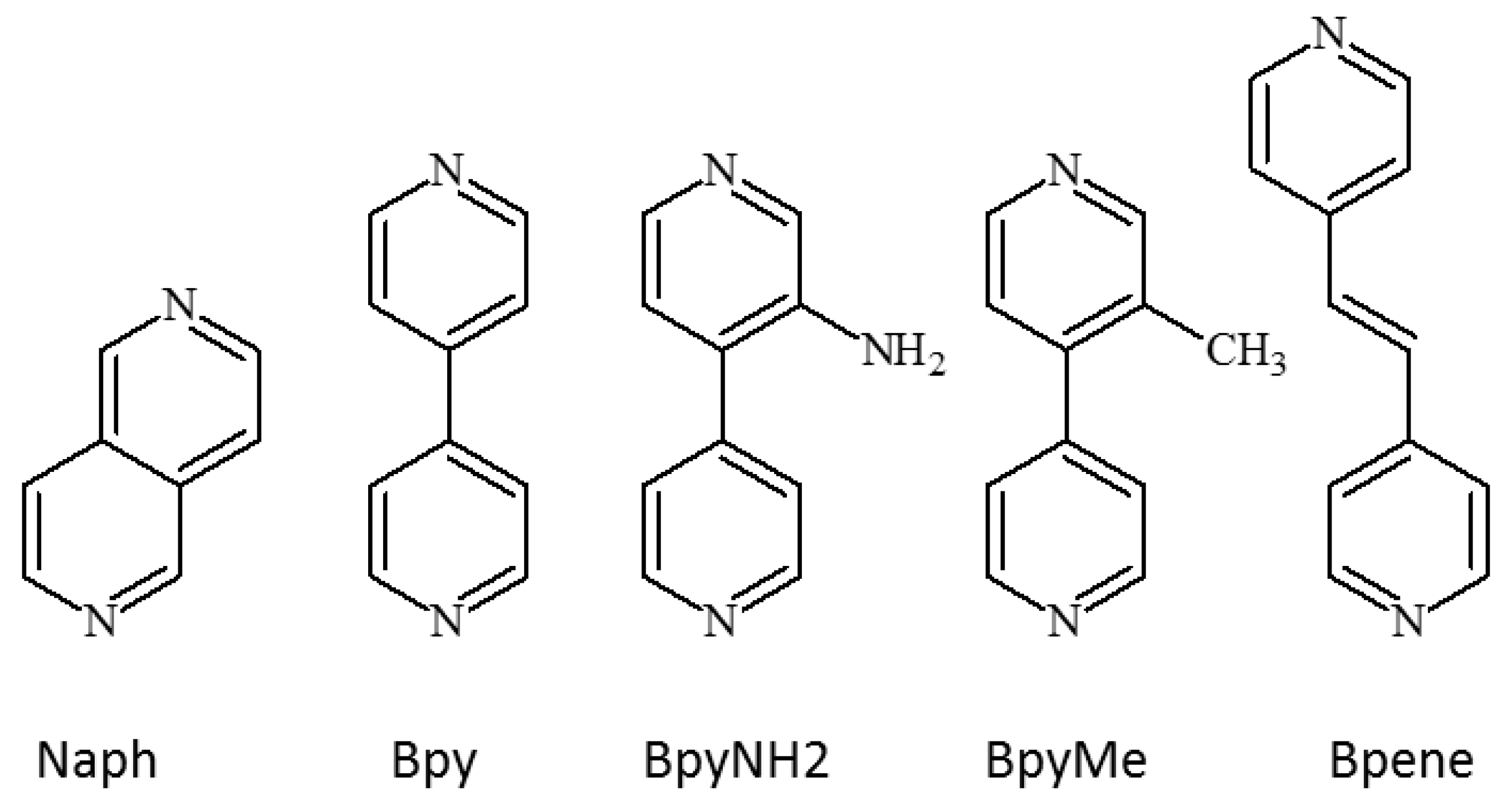
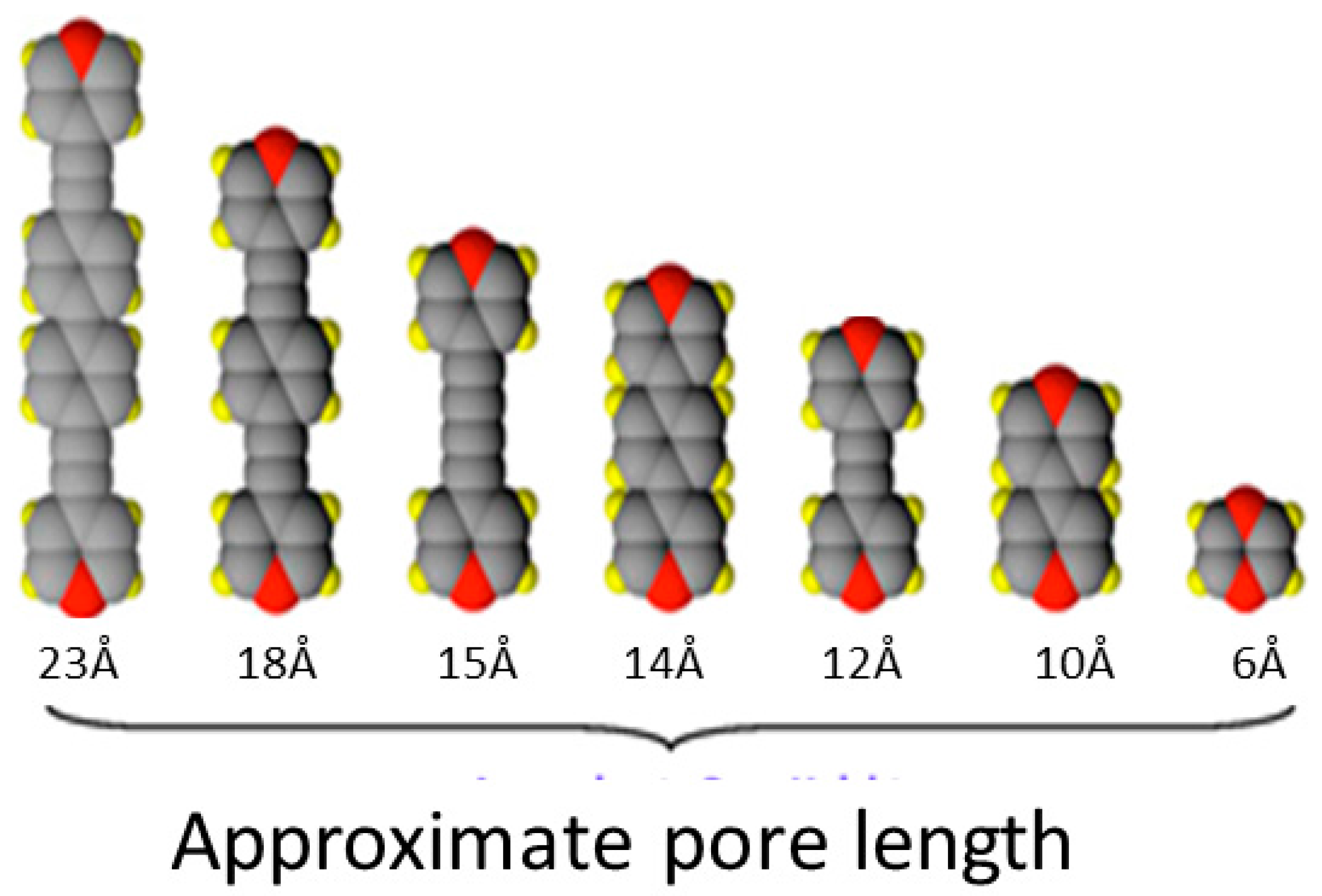
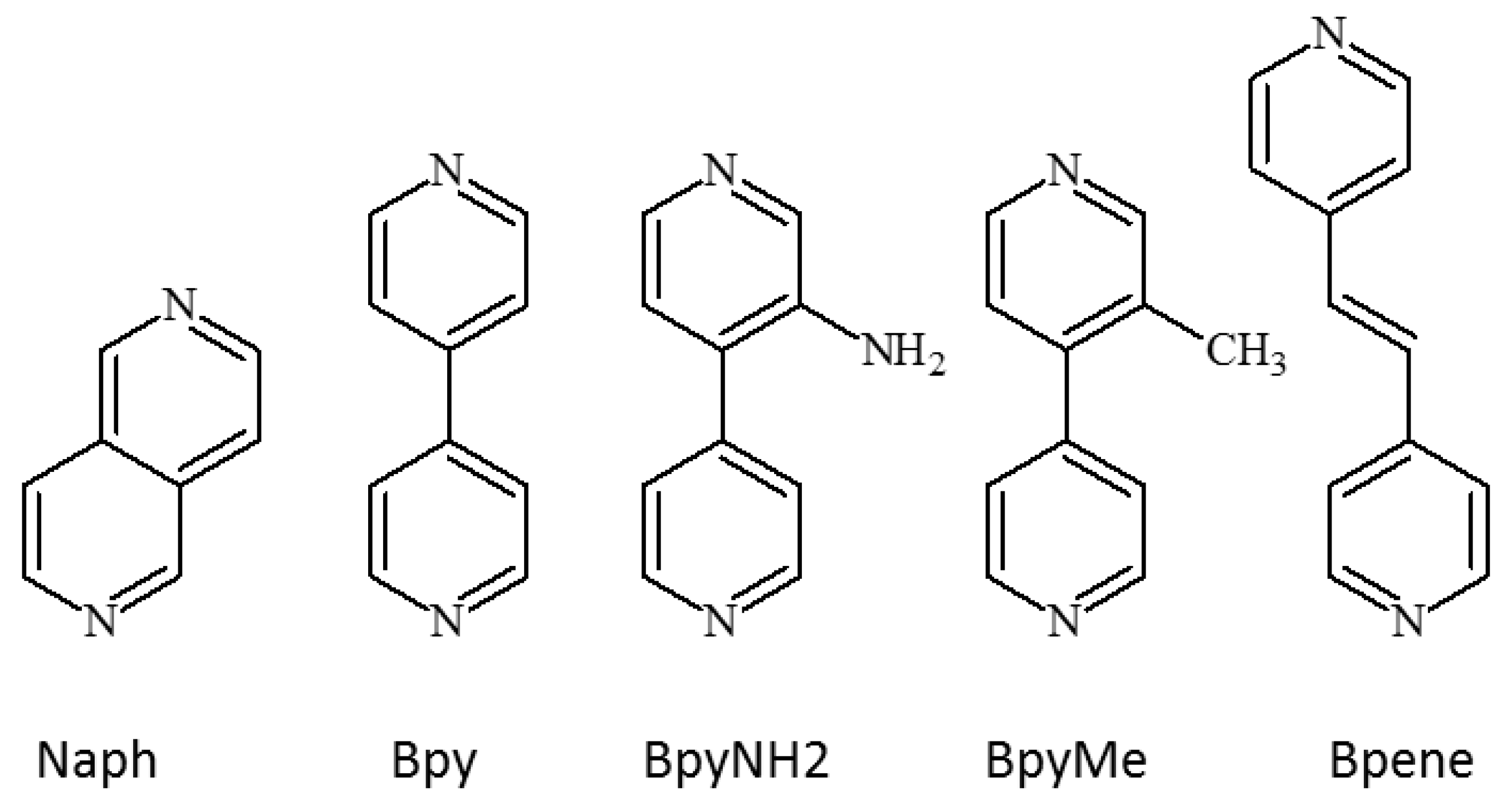
1.3. Five Selected Ni(Cn)4-Based Structural Studies
- 1.
- 1,2-bis(4-pyridyl)ethylene, C12N2H10, [Bpene]
- 2.
- 3-methyl-4,4′-bipyridine, C11N2H10, [BpyMe]
- 3.
- 3-amino-4,4′-bipyridine, C10N3H9, [BpyNH2]
- 4.
- 4,4′-bipyridine, C10N2H8, [Bpy]
- 5.
- 2,6-naphthyridine, C8N2H6, [Naph]
2. Results and Discussion
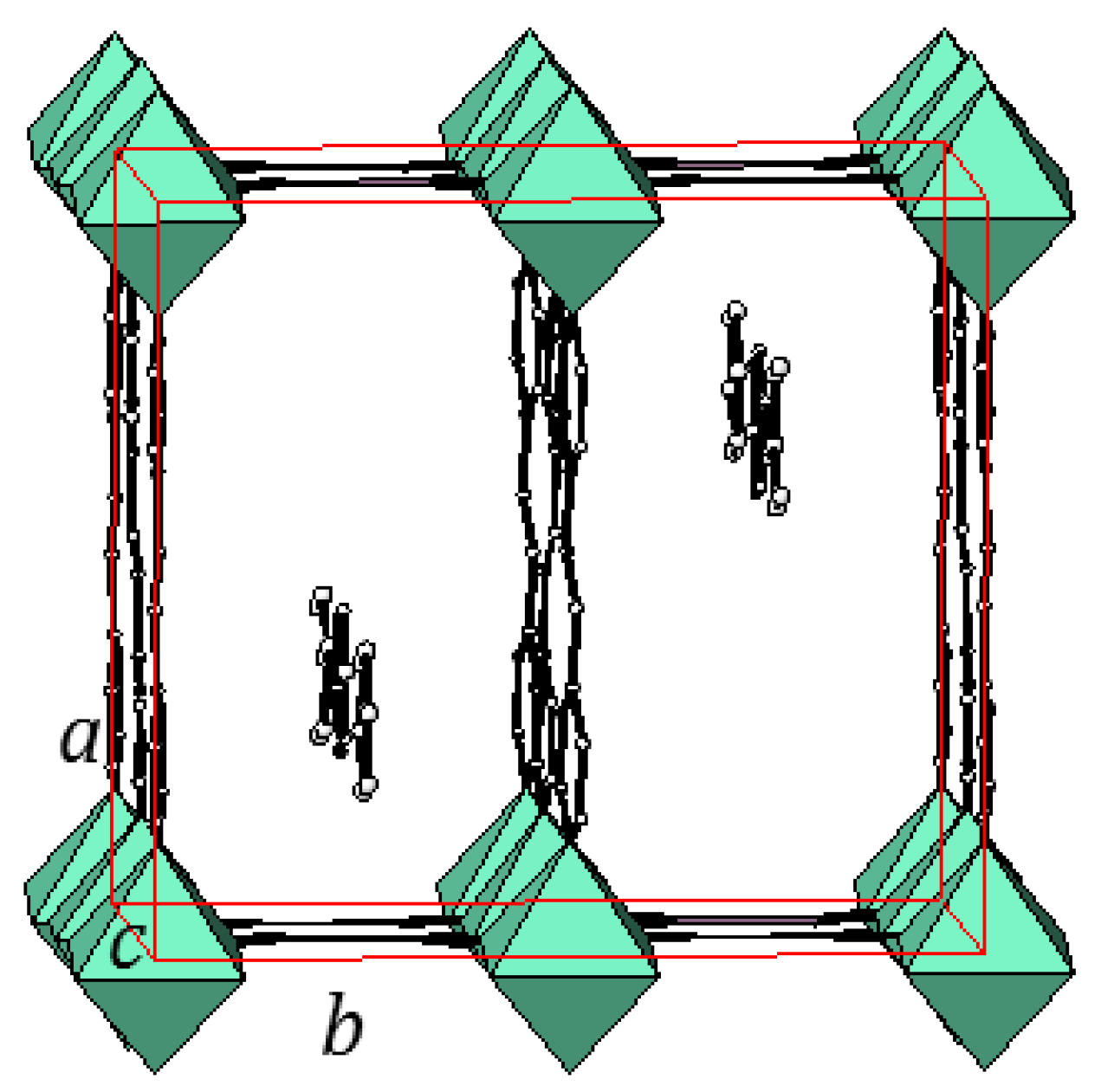
2.1. Three-Dimensional (3-D) Structures
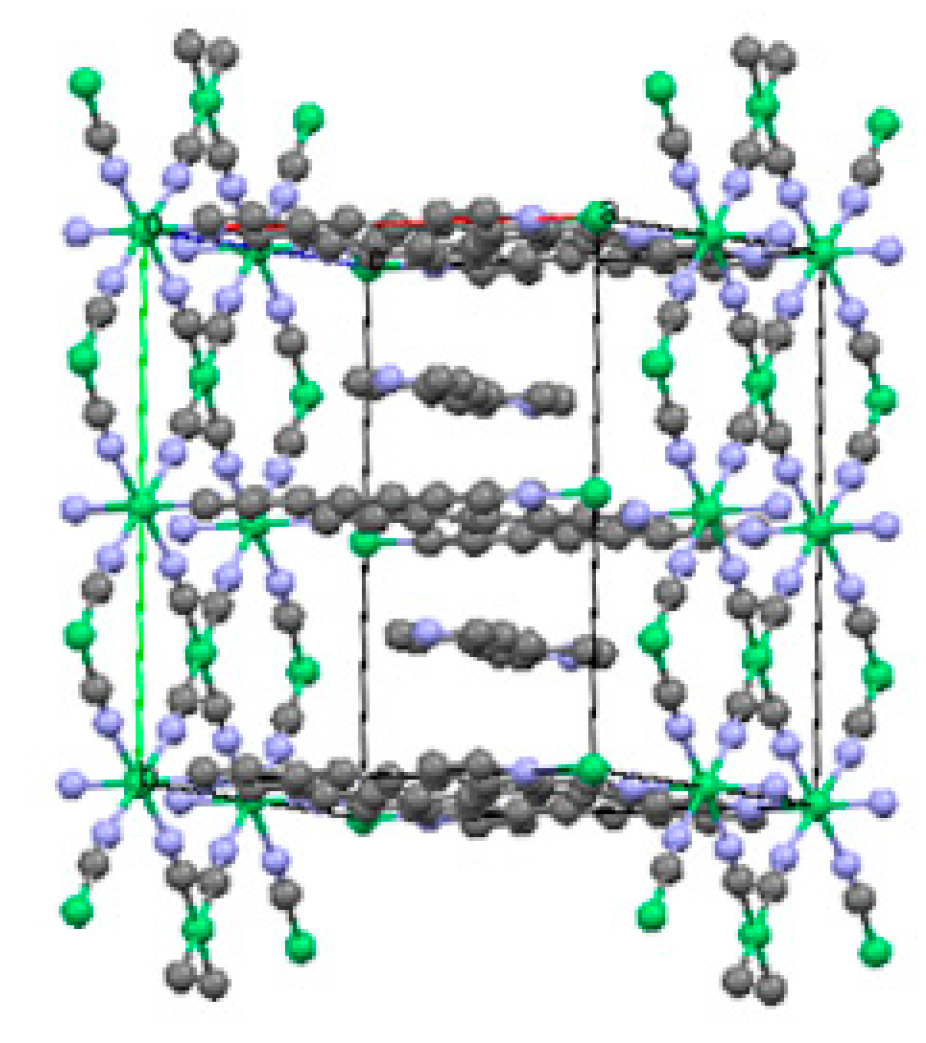
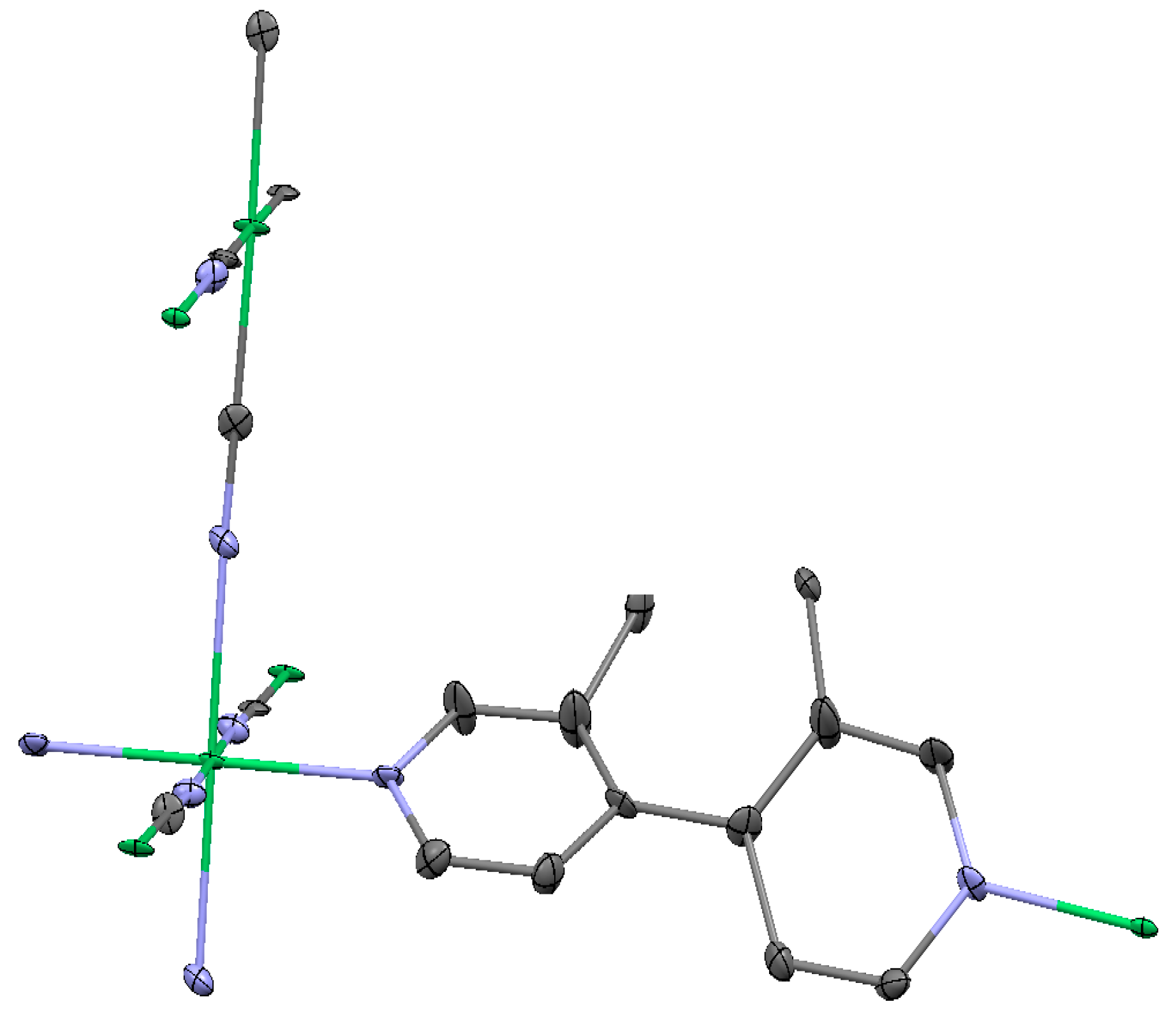
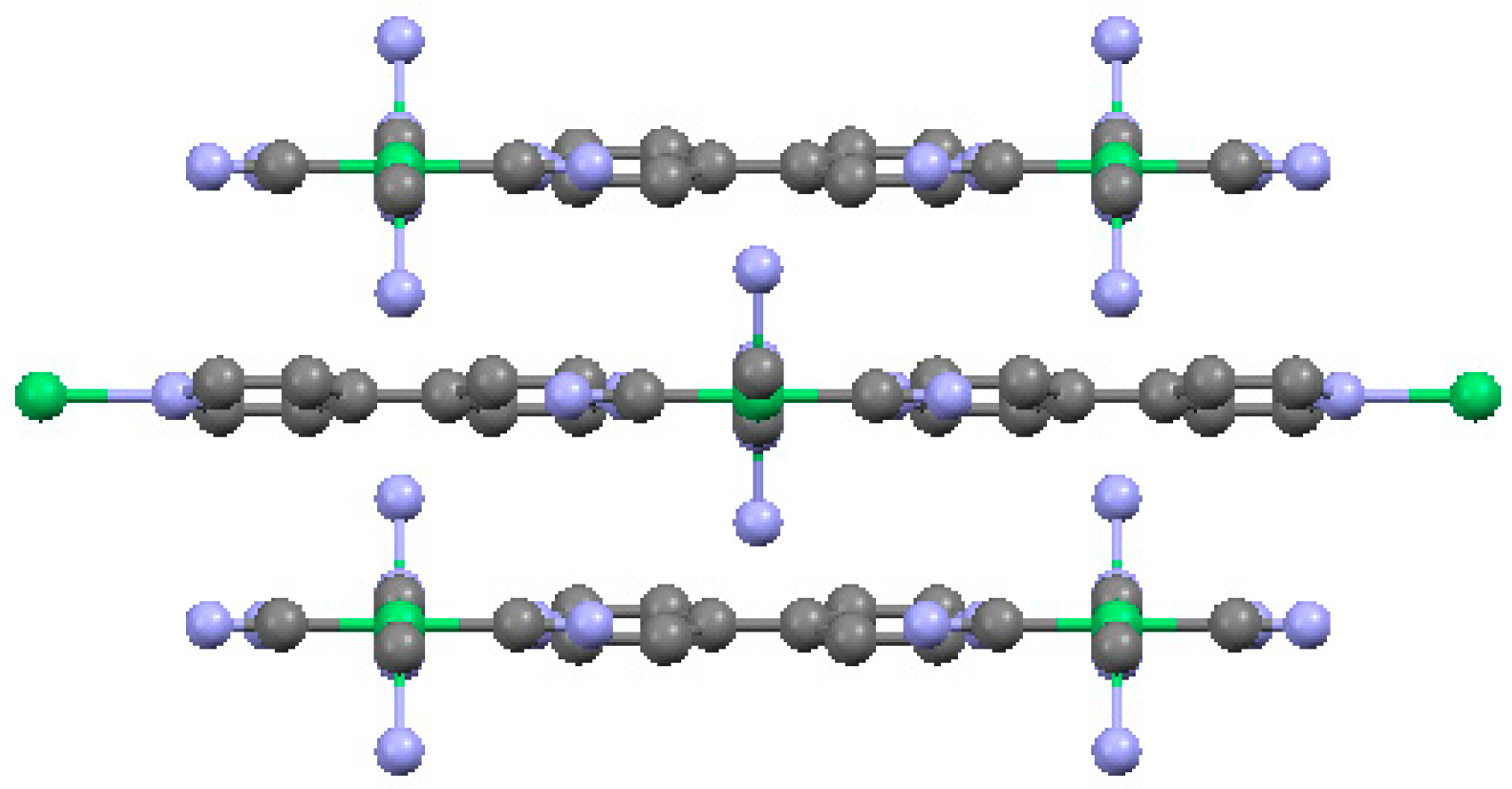
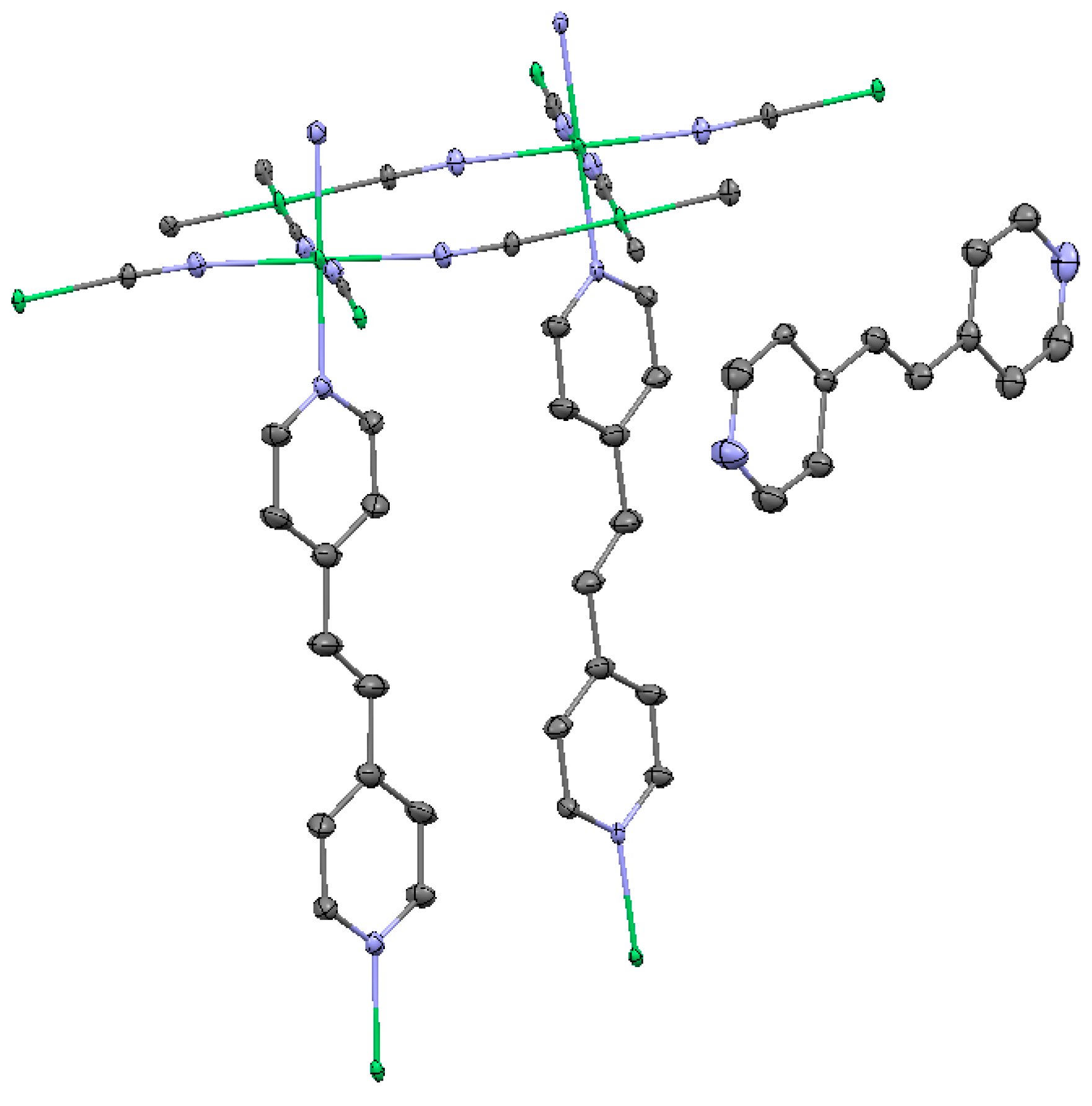
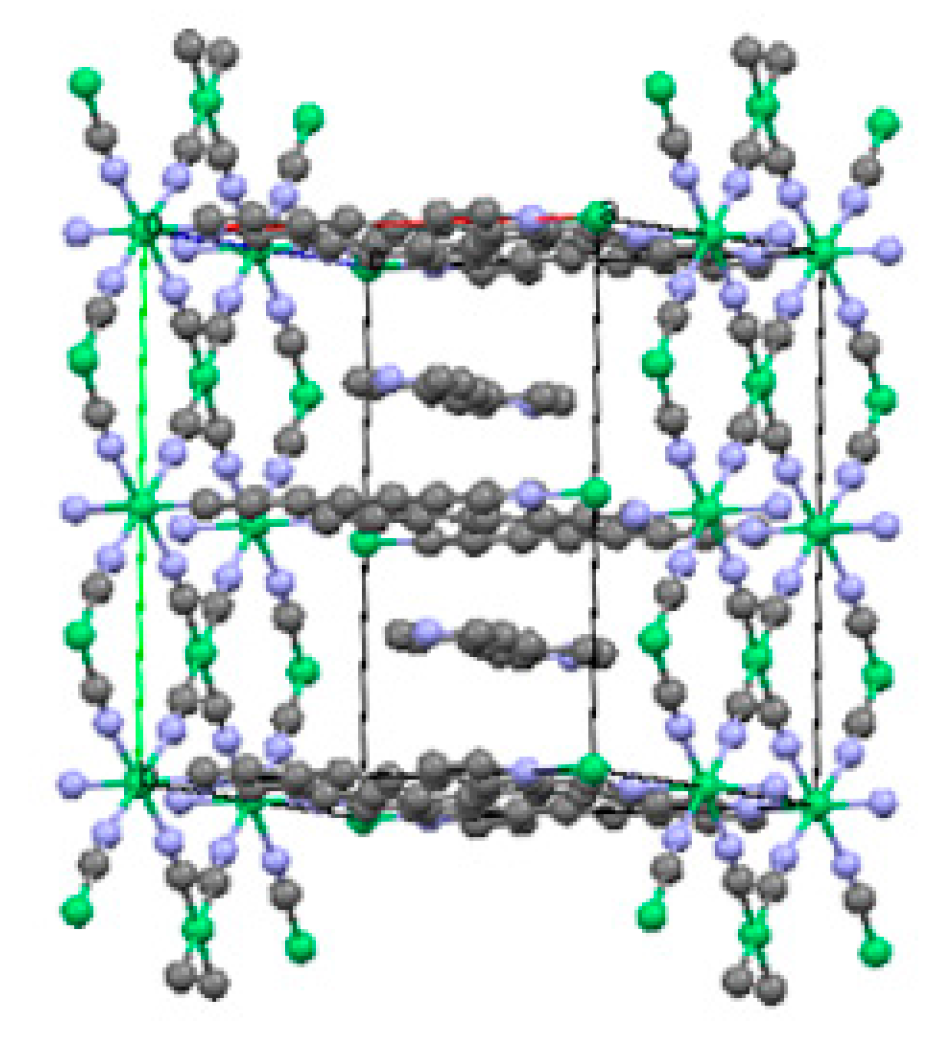
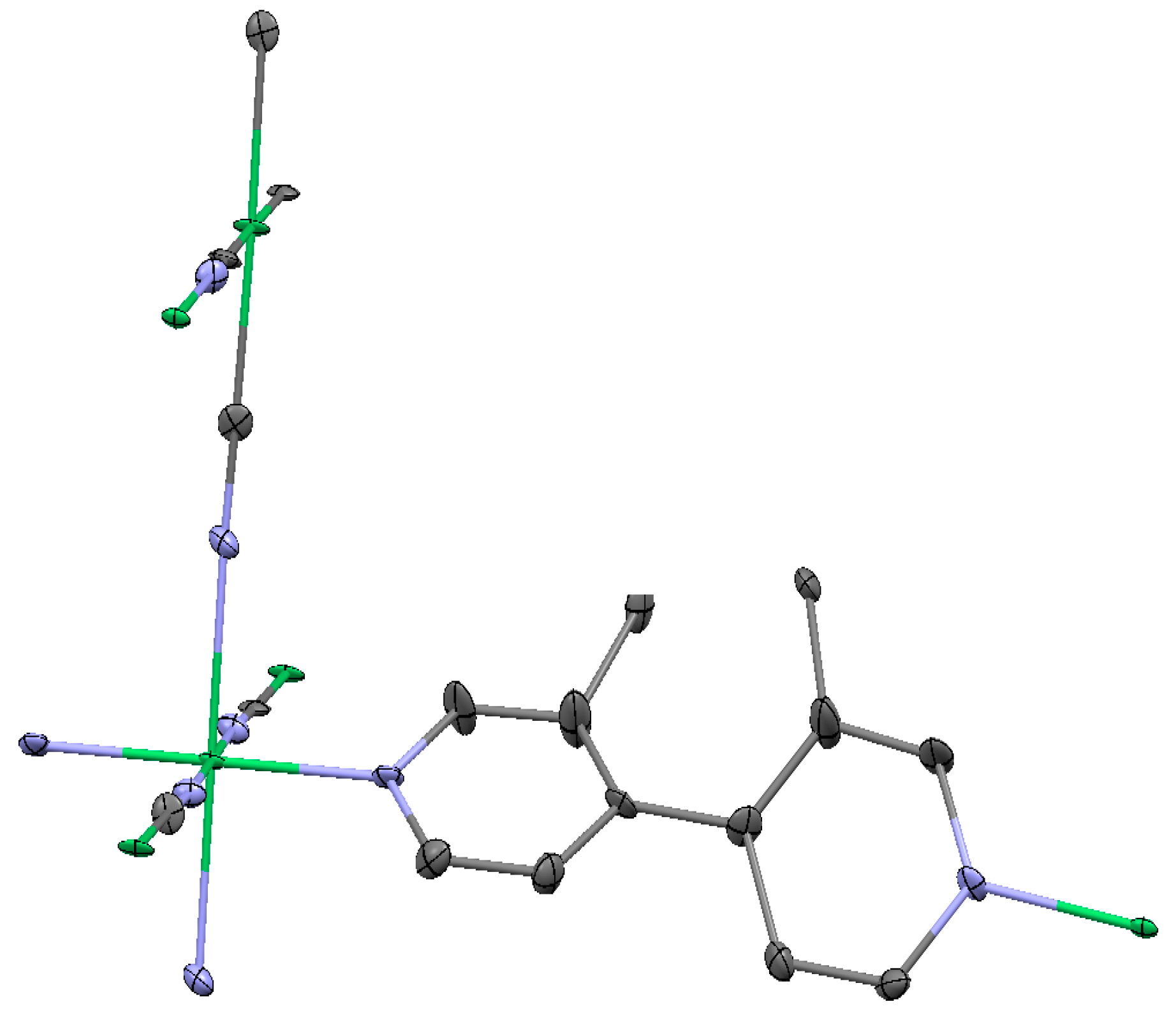
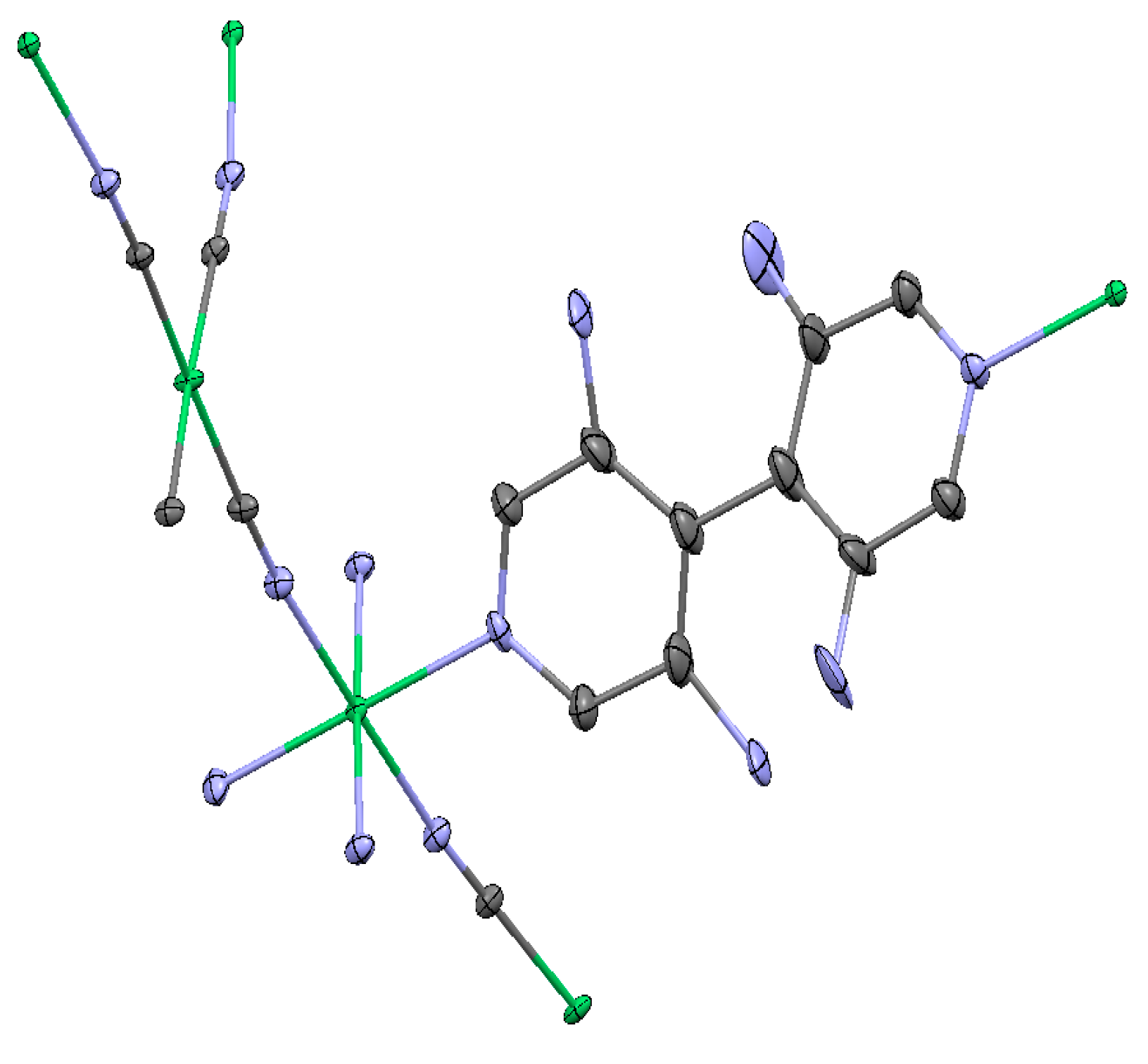
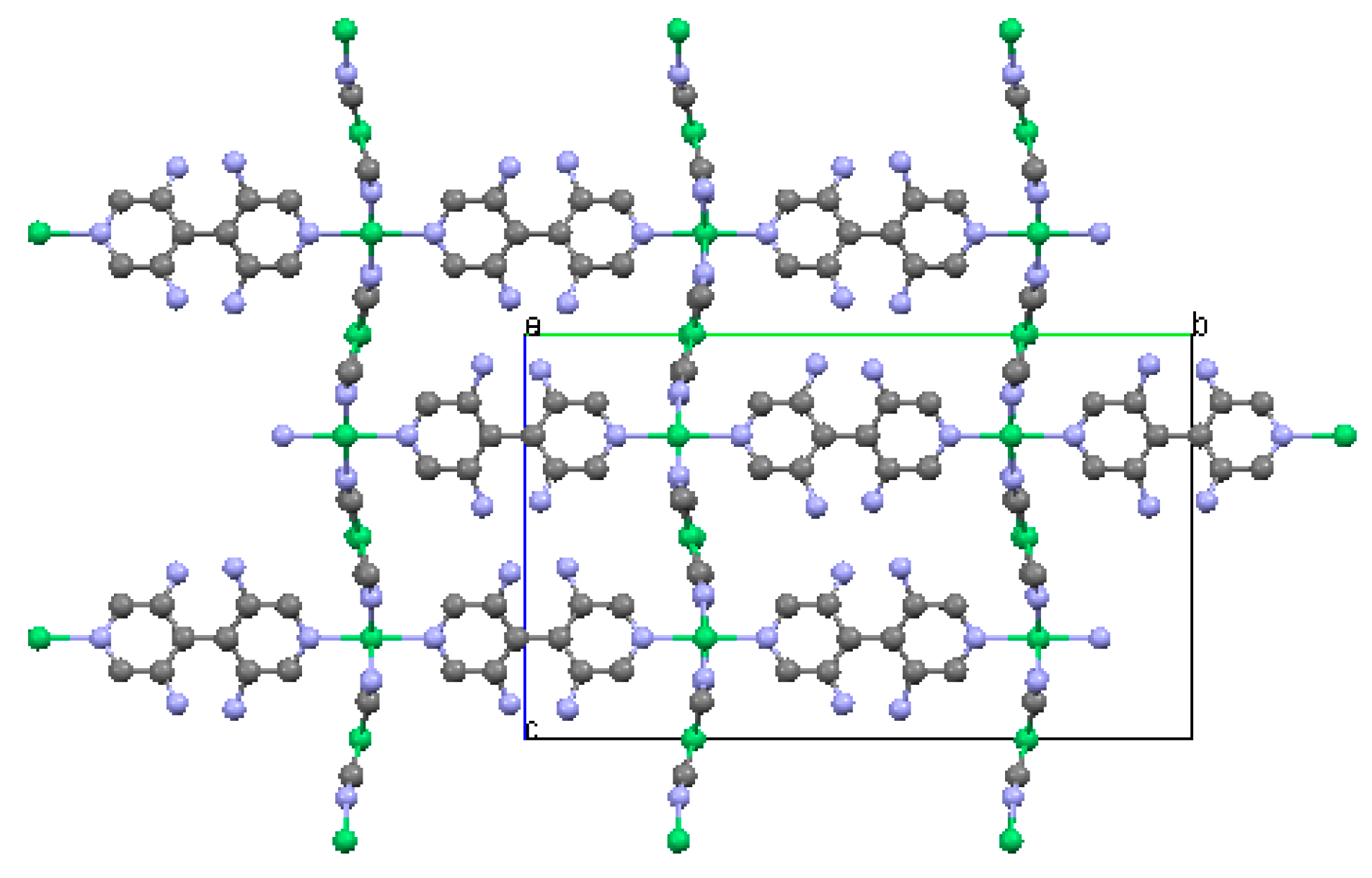
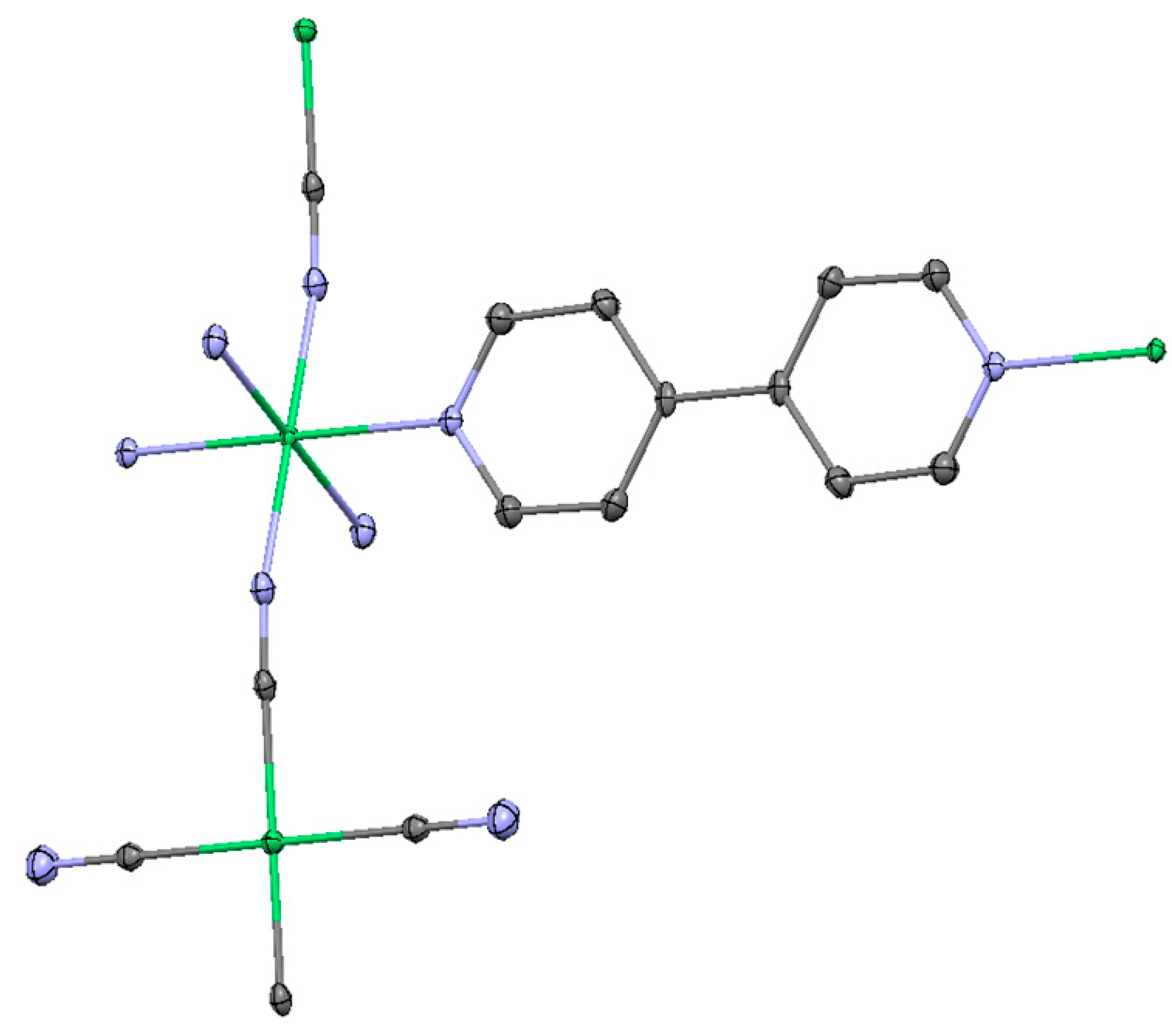
2.1.1. Ni(1,2-bis(4-pyridyl)ethylene)[Ni(CN)4] (Ni-Bpene)
2.1.2. Ni(3-methyl-4,4′-bipyridine)[Ni(CN)4], (Ni-BpyMe)
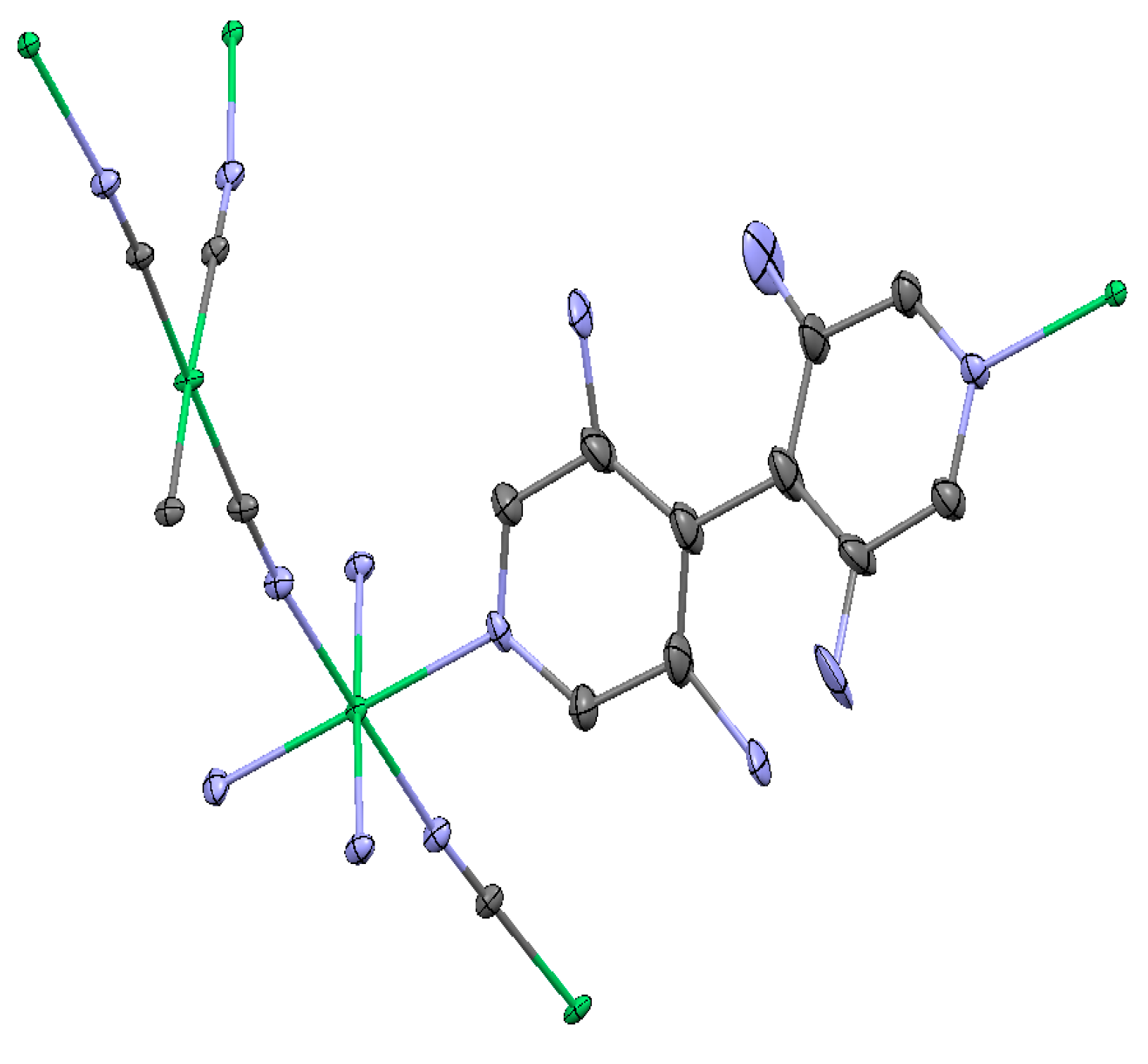
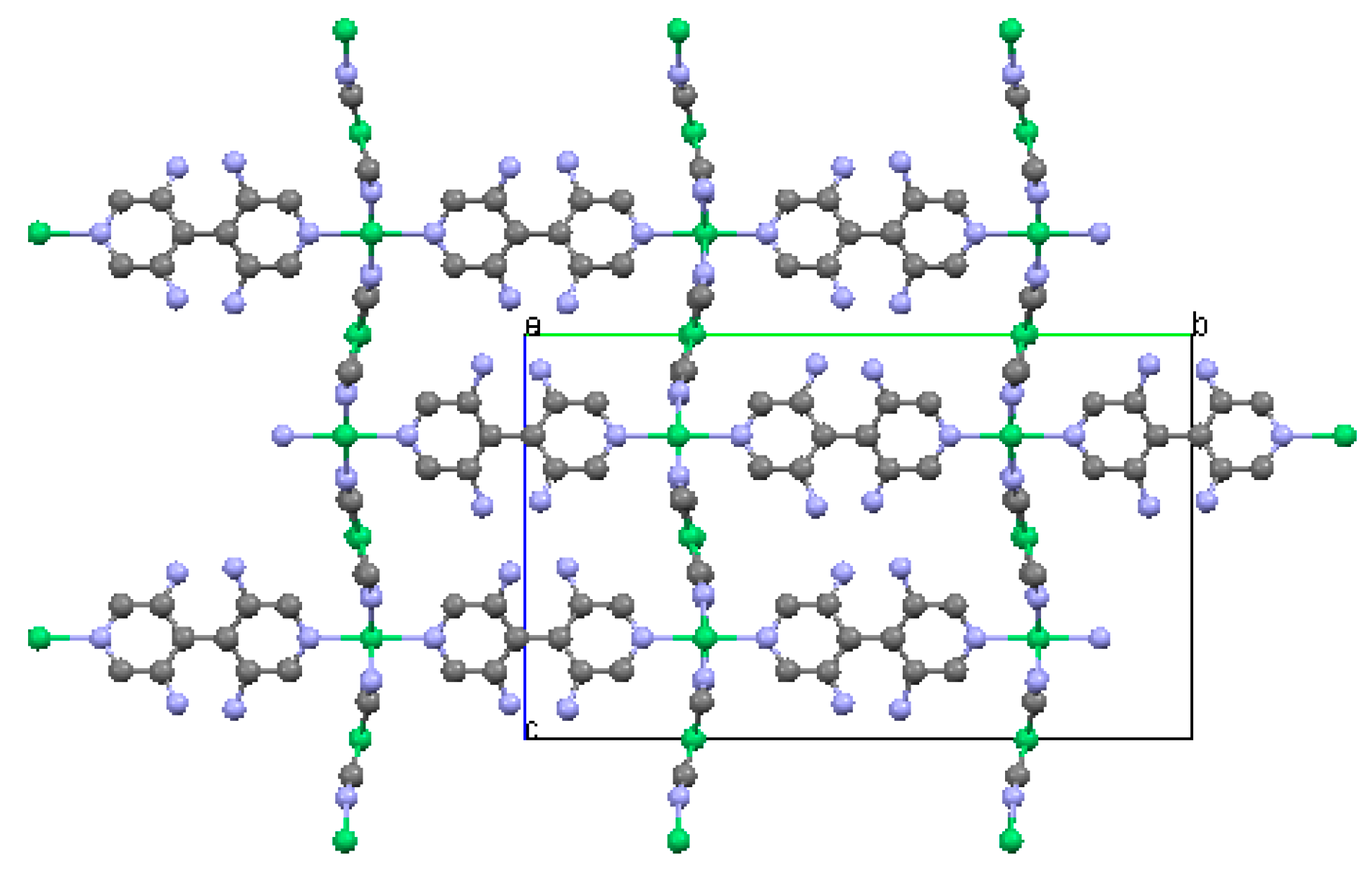
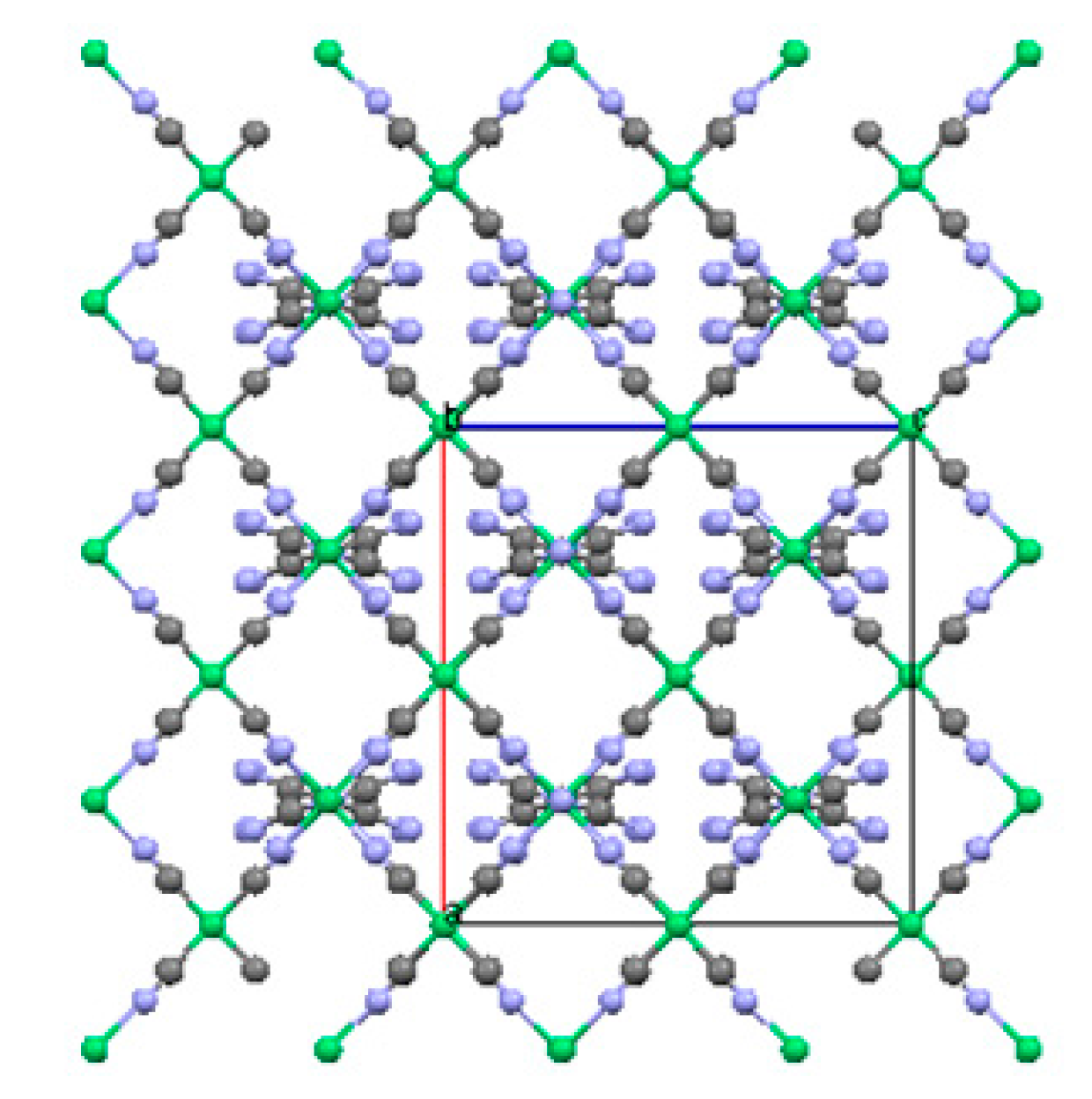
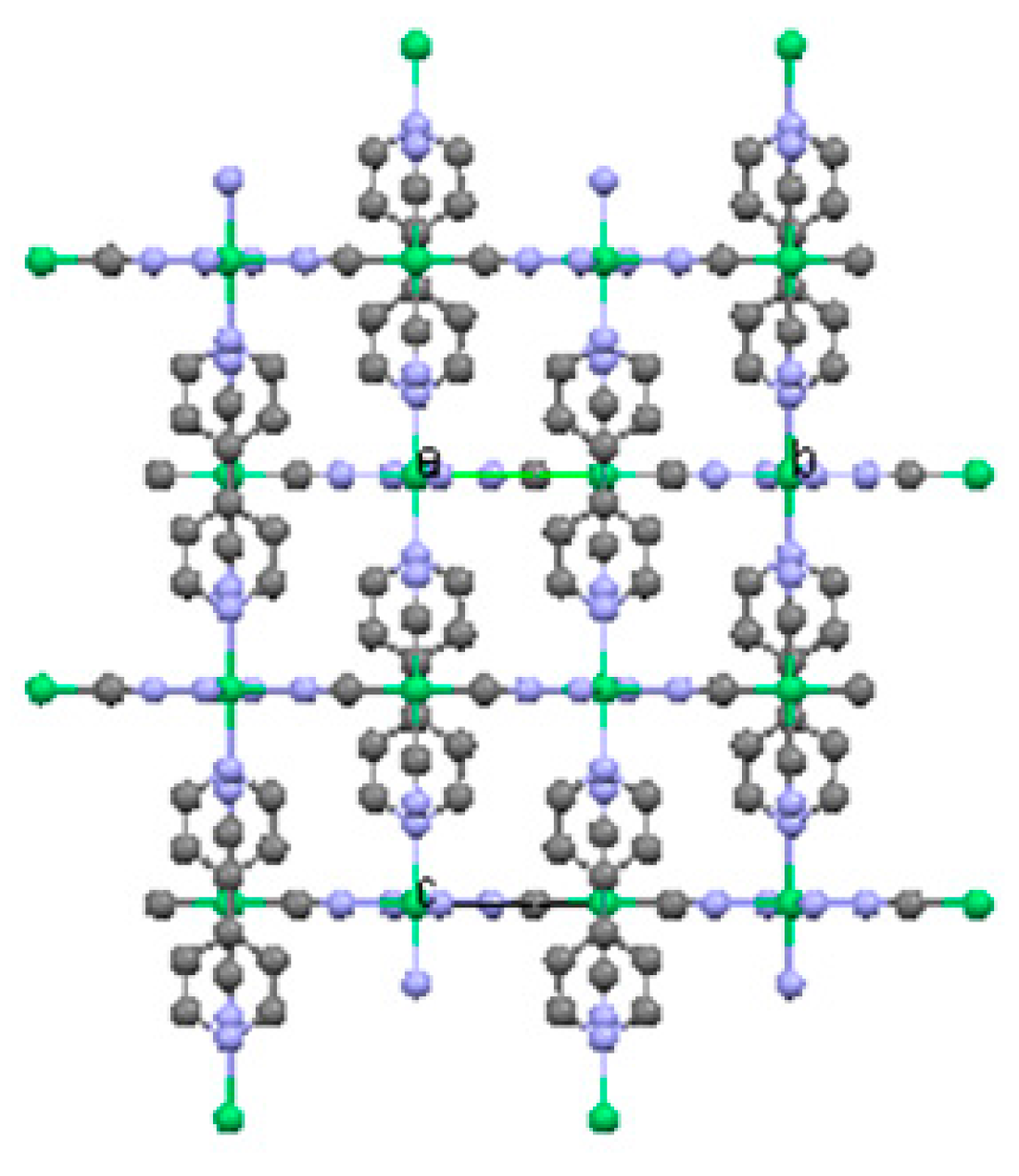
2.1.3. Ni(3-amino-4,4′-bipyridine)[Ni(CN)4], (Ni-BpyNH2)
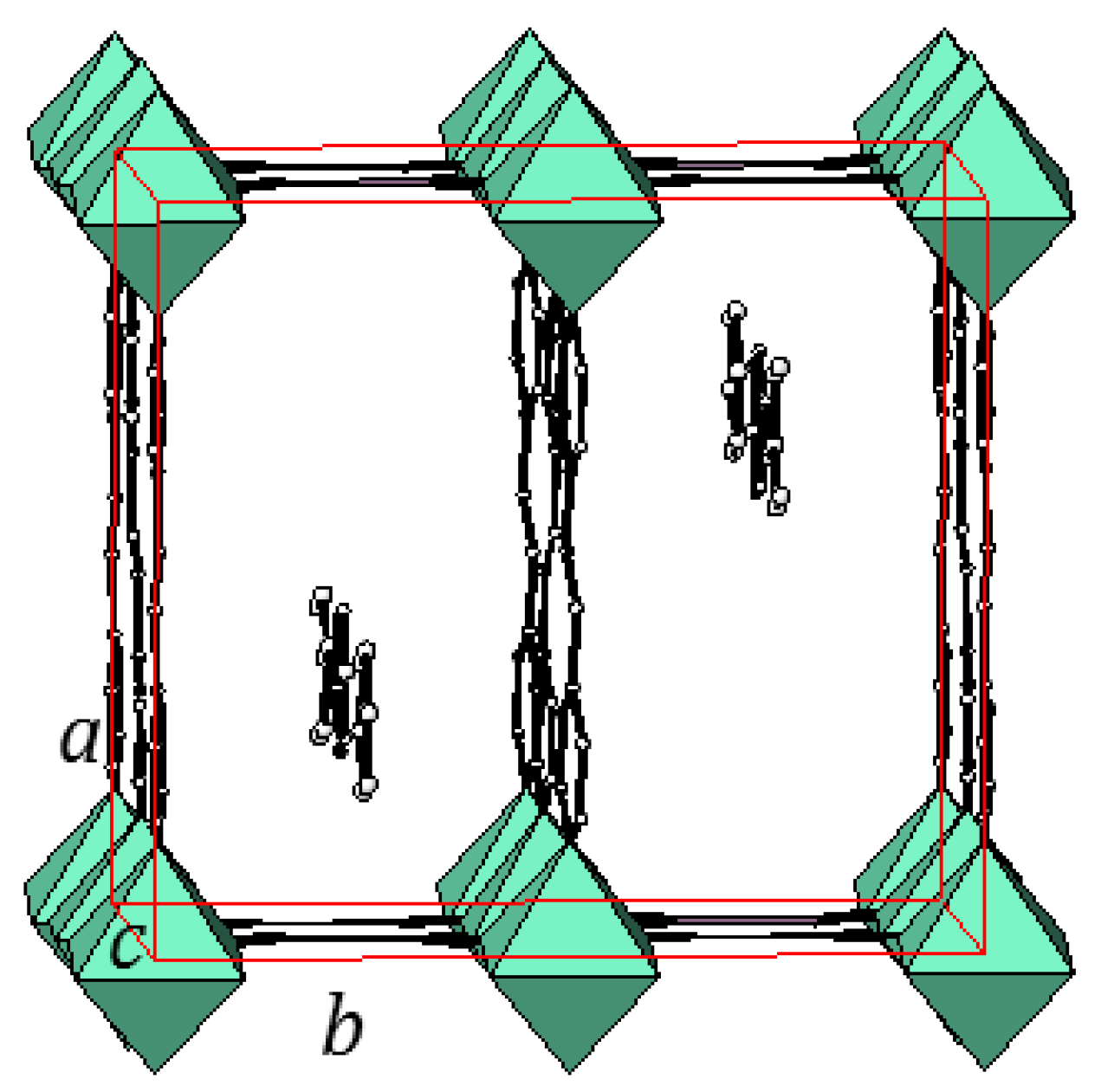
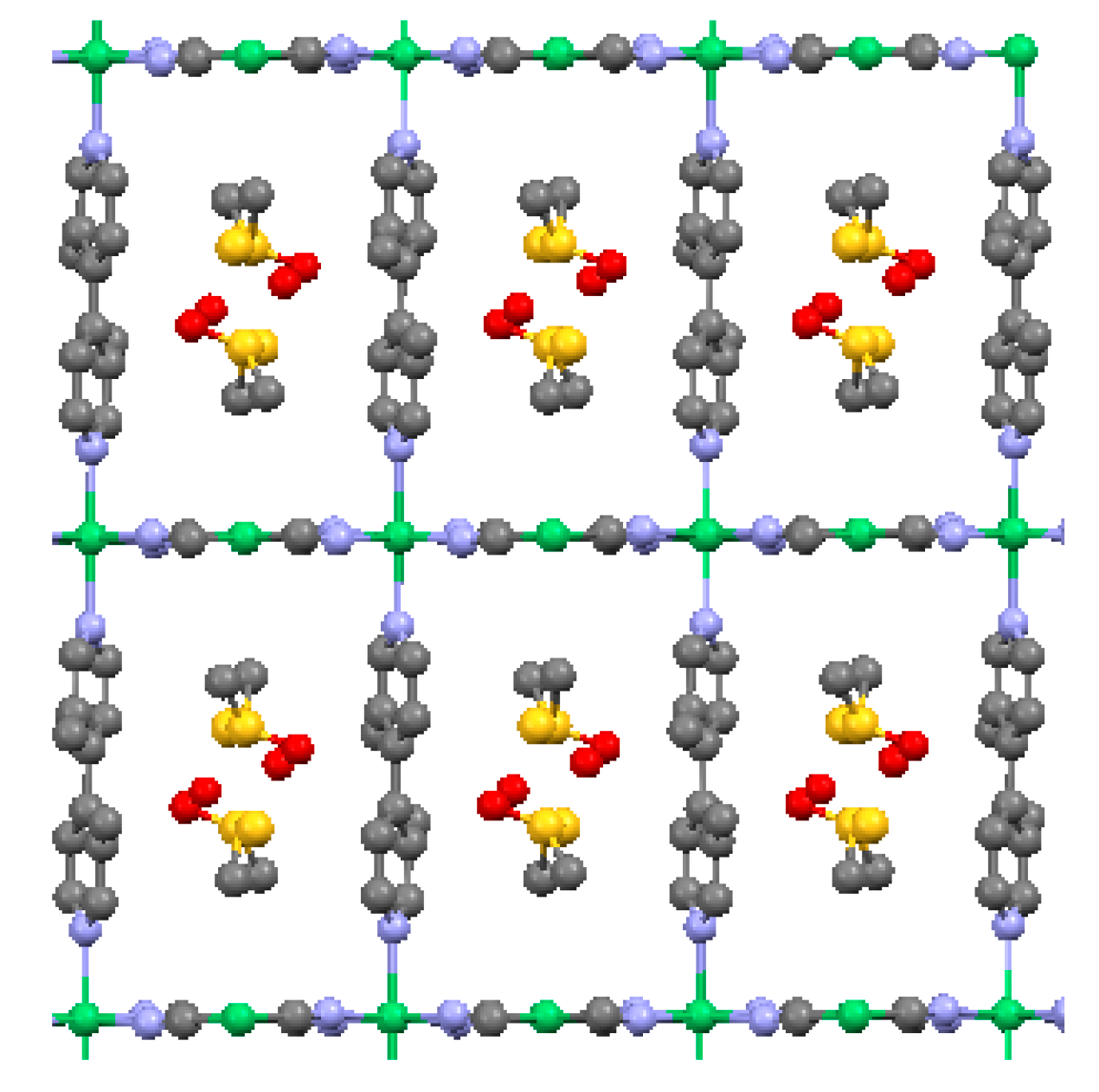
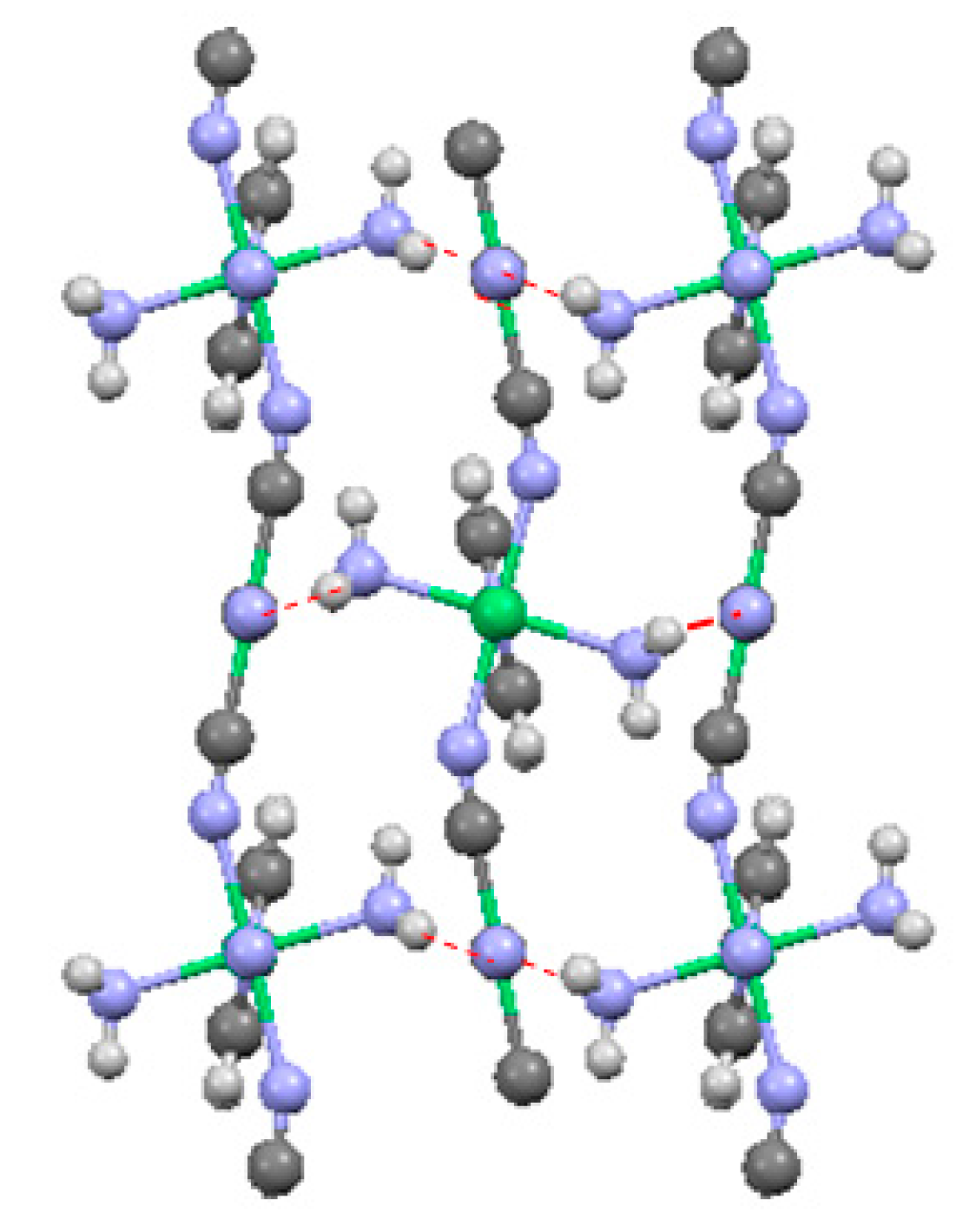
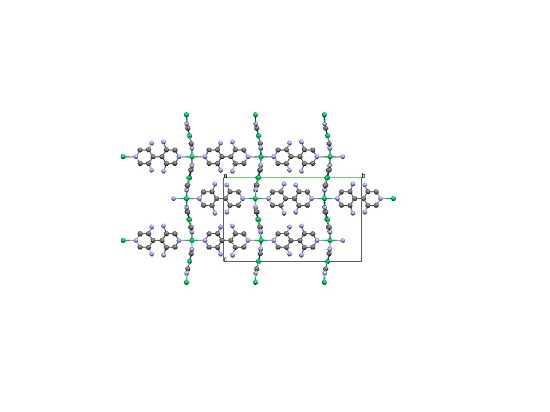
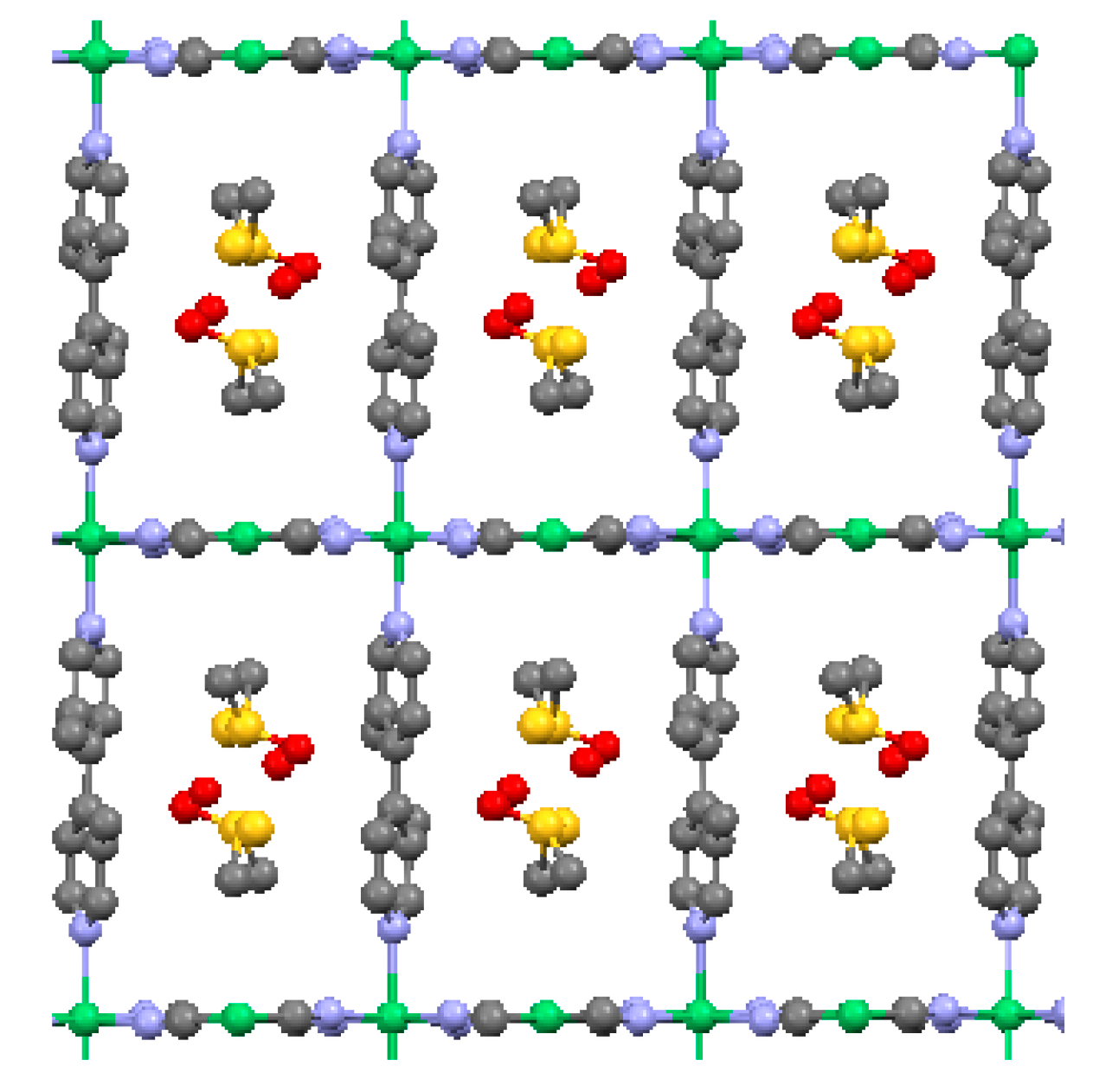
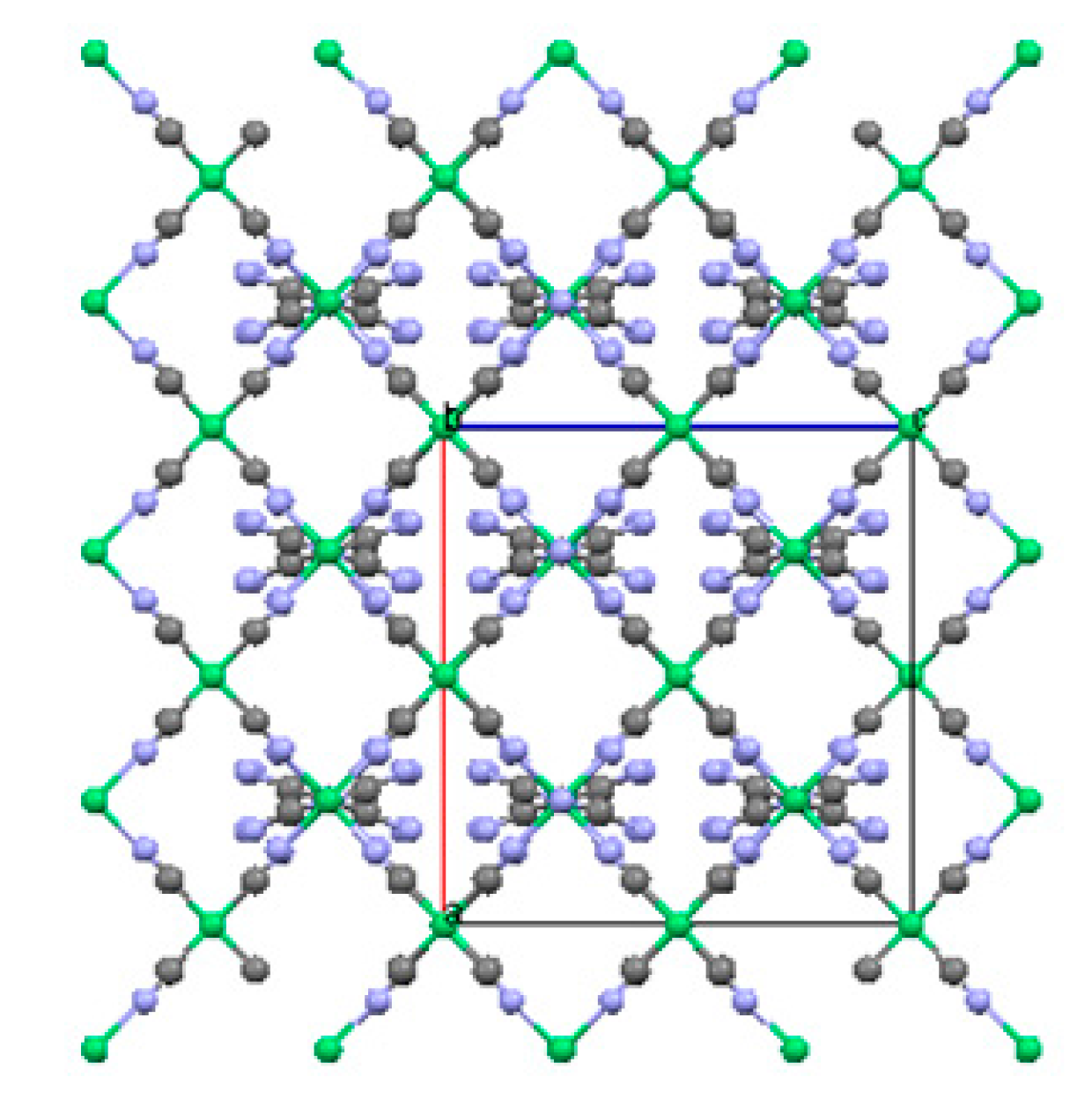
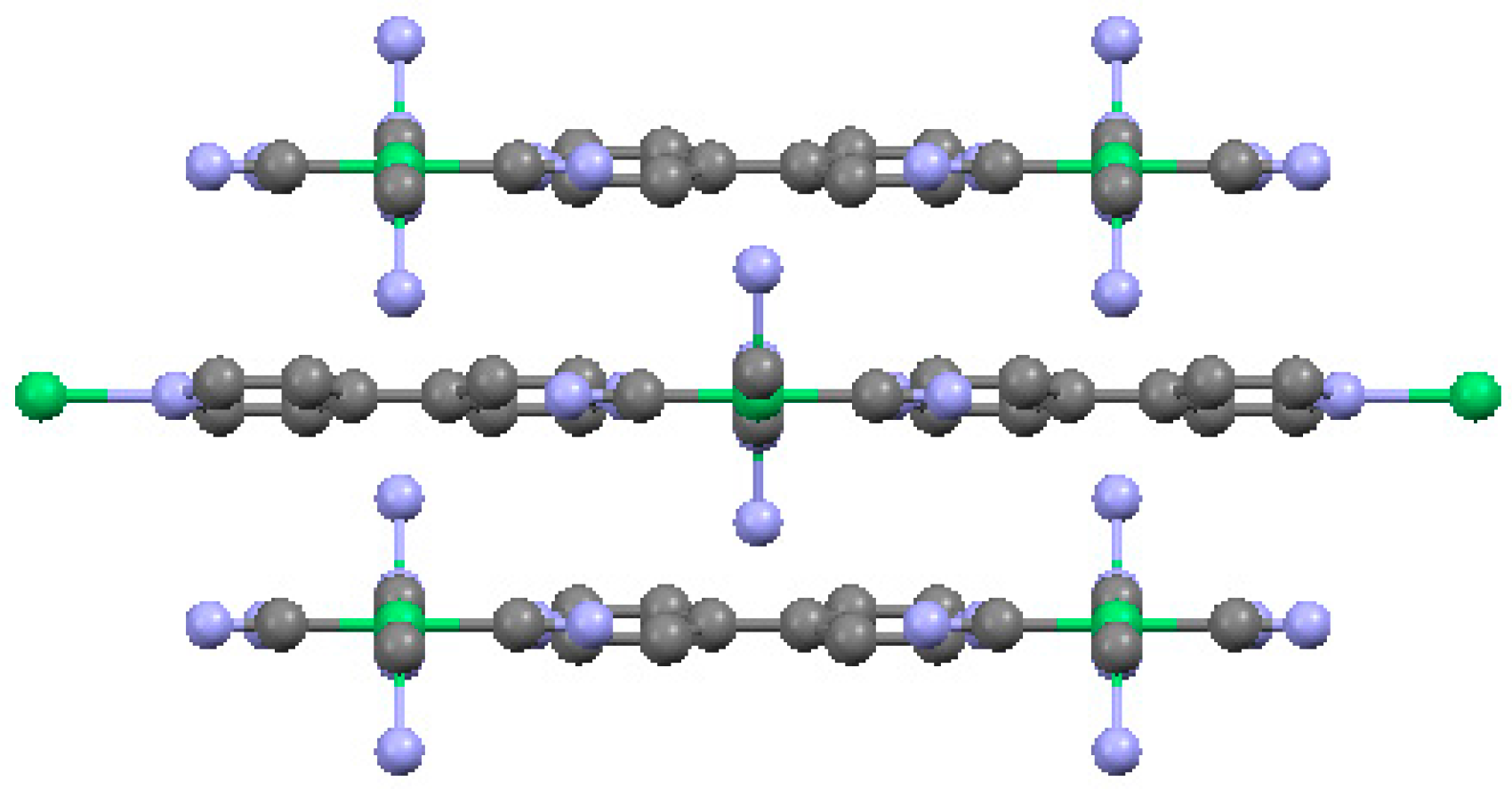
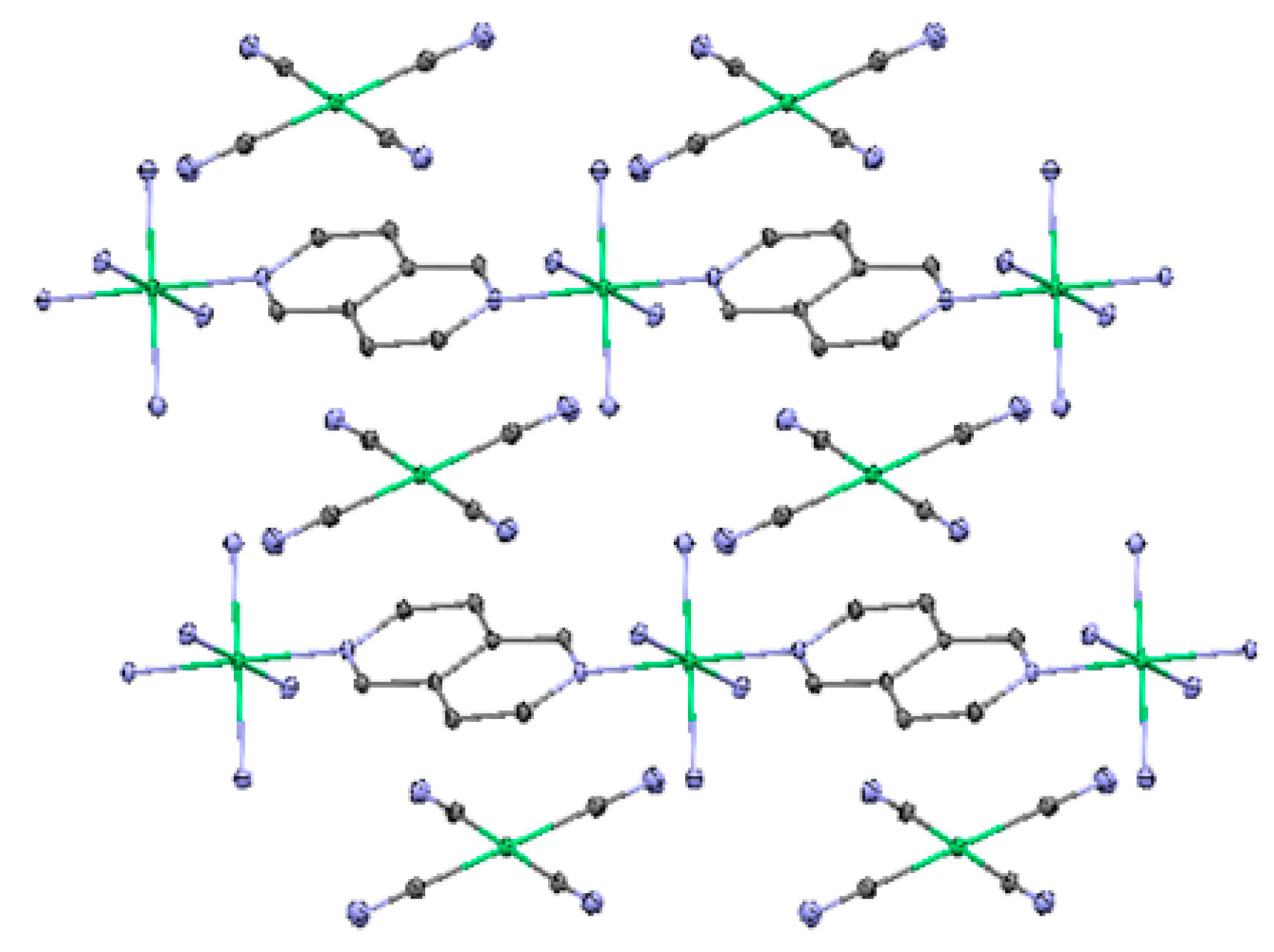
2.2. Two-Dimensional (2-D) Structure
Ni(4,4′-bipyridine)[Ni(CN)4], (Ni-Bpy)
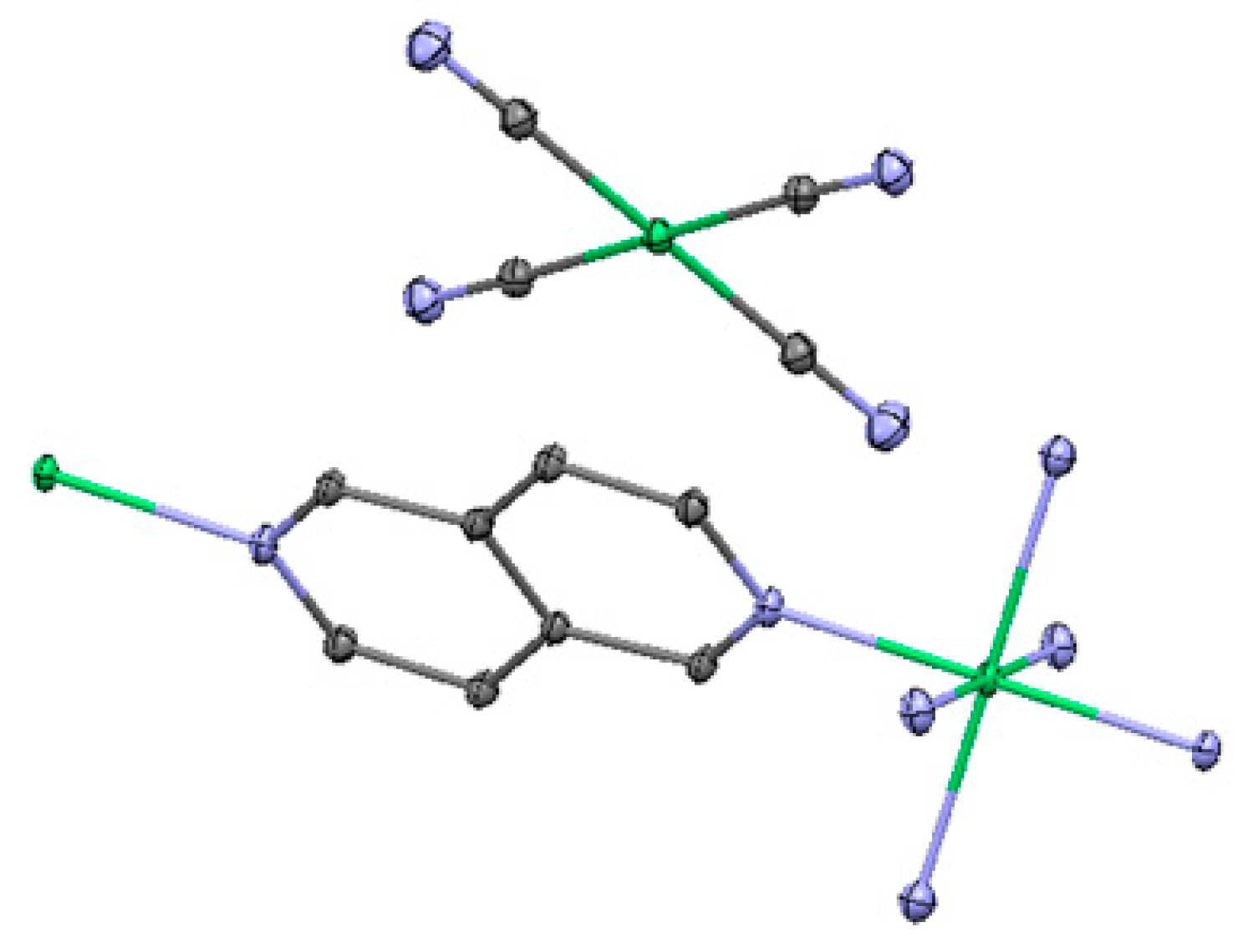
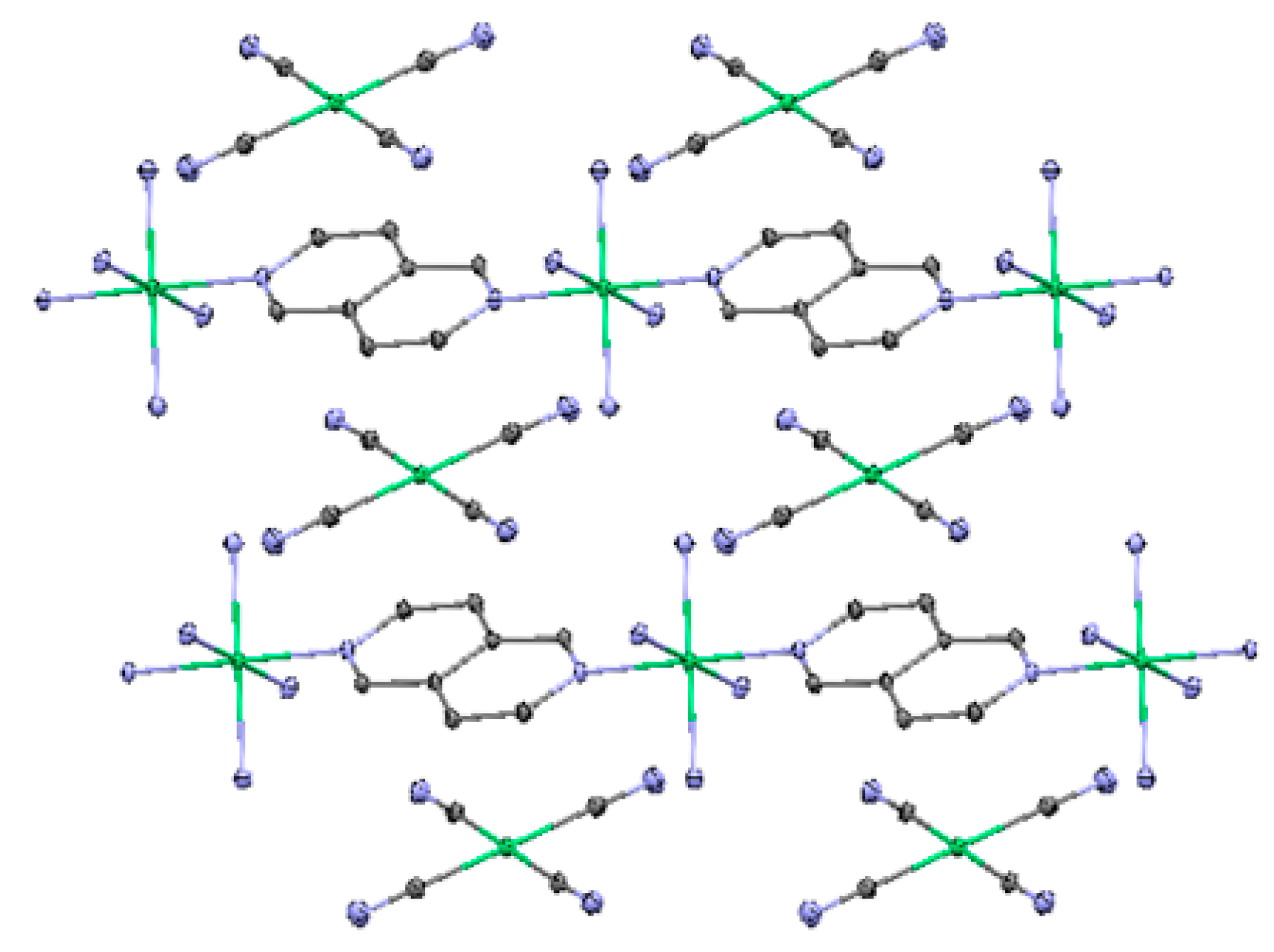
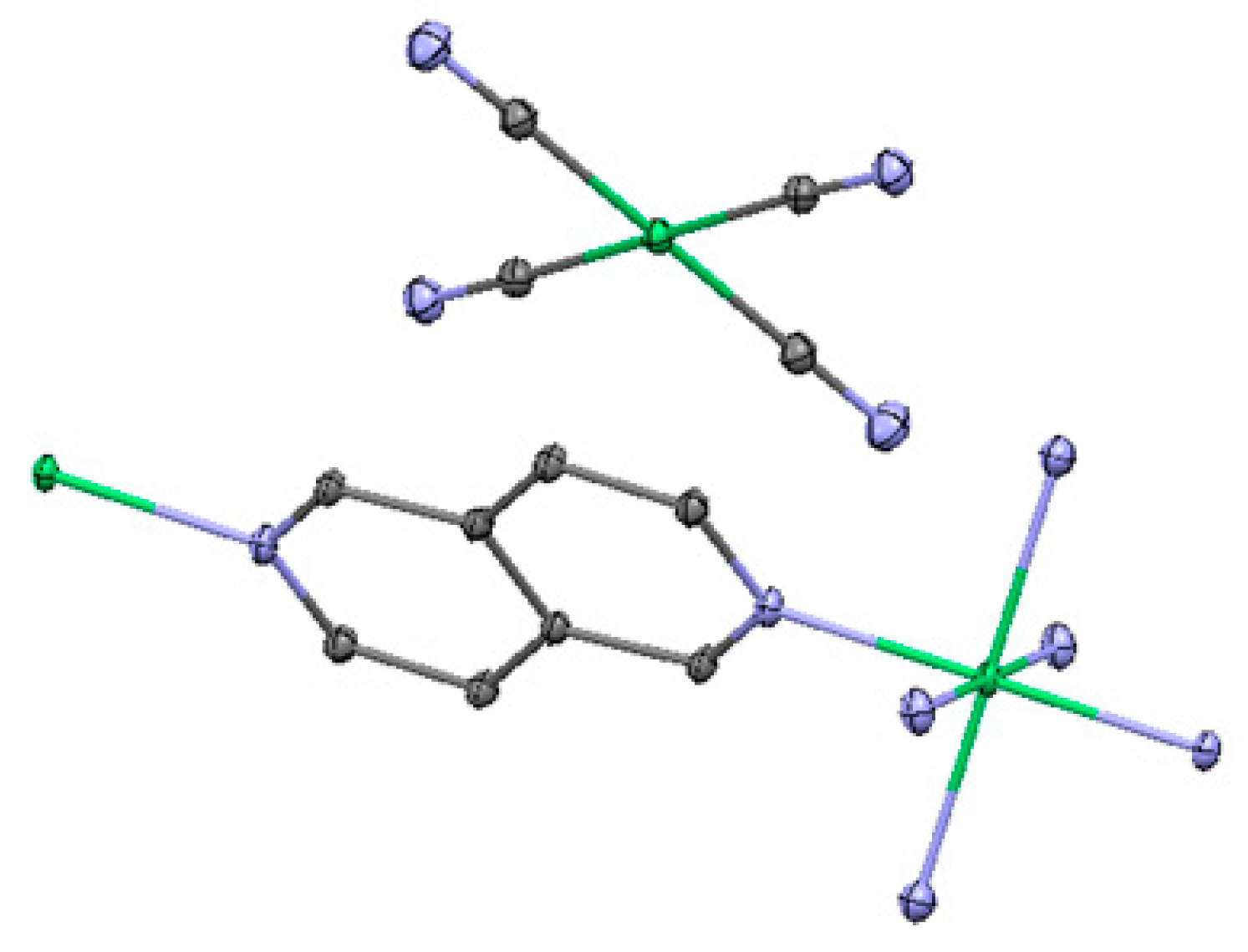
2.3. One-Dimensional (1-D) Structure
(Ni(2,6-naphhyridine)[Ni(CN)4]), (Ni-Naph)
3. Experimental Section
3.1. Material Synthesis and Crystal Growth
3.2. Synchrotron X-Ray Diffraction Experiment
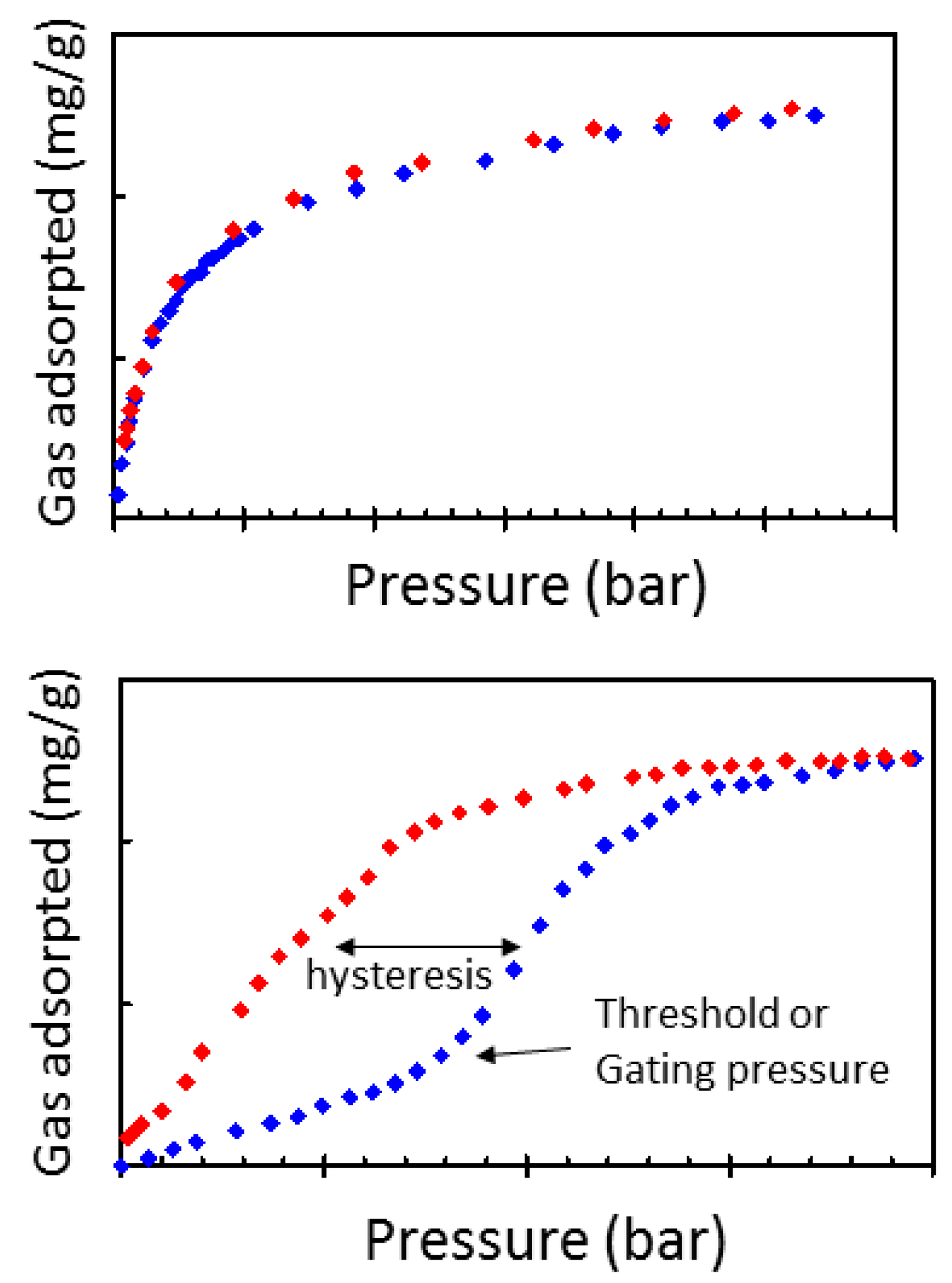
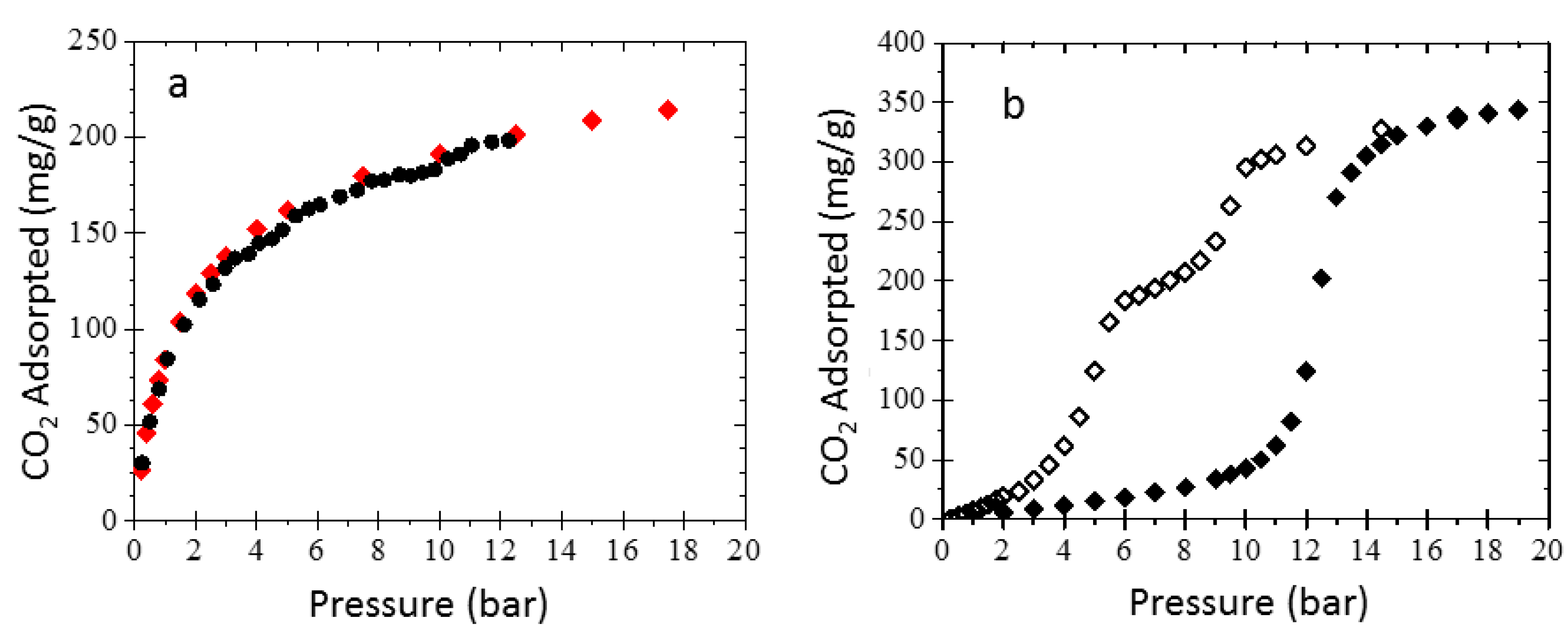
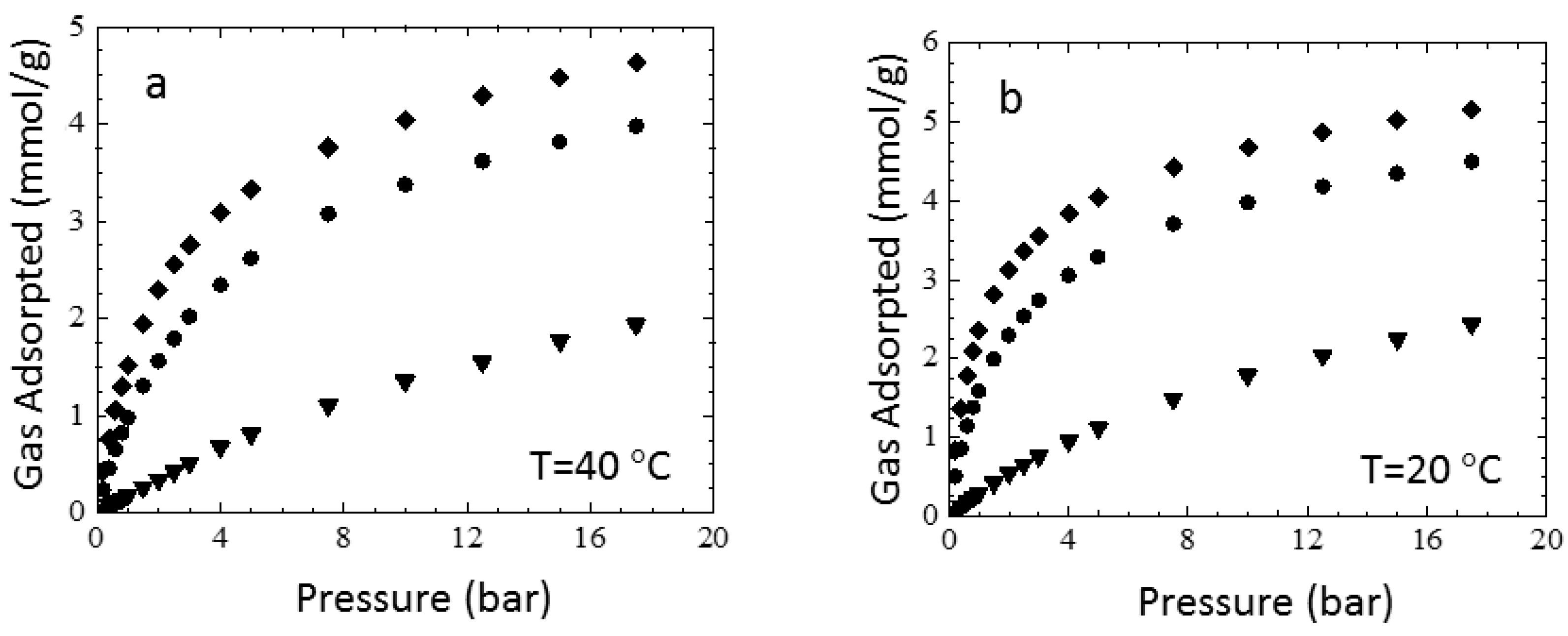
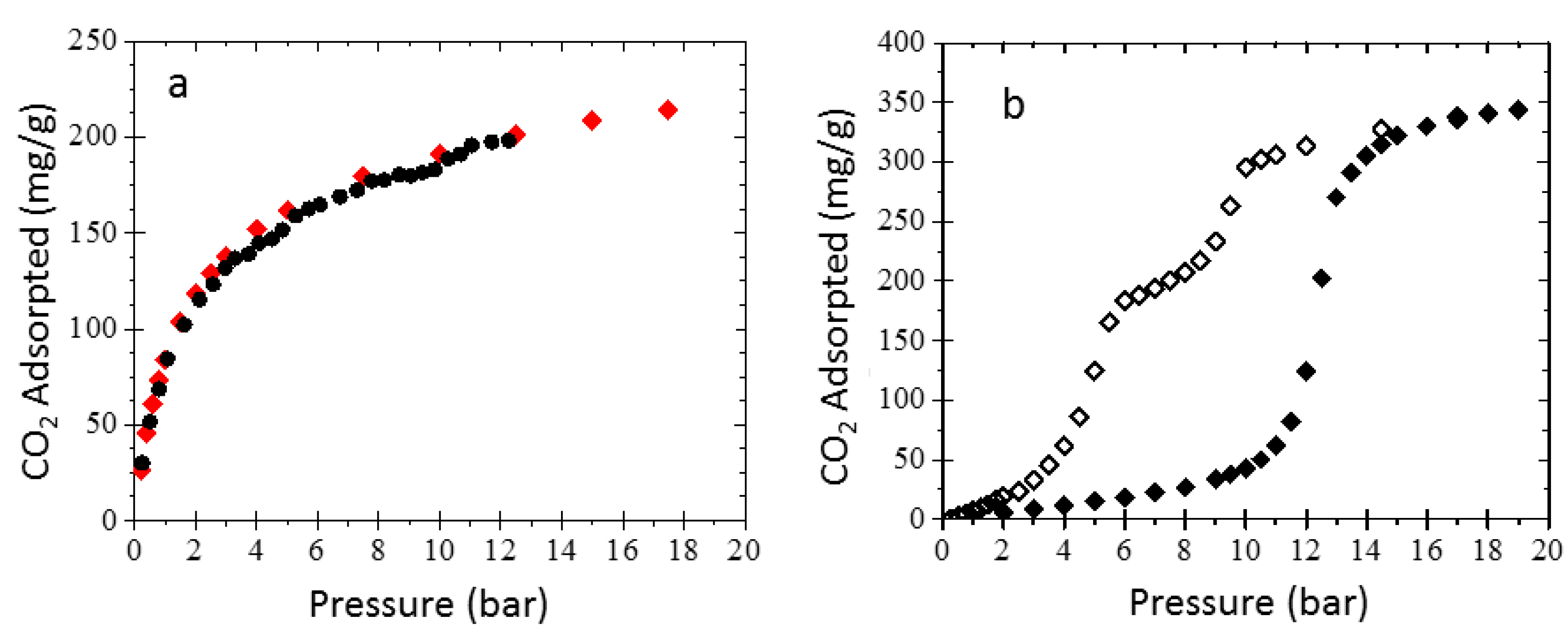
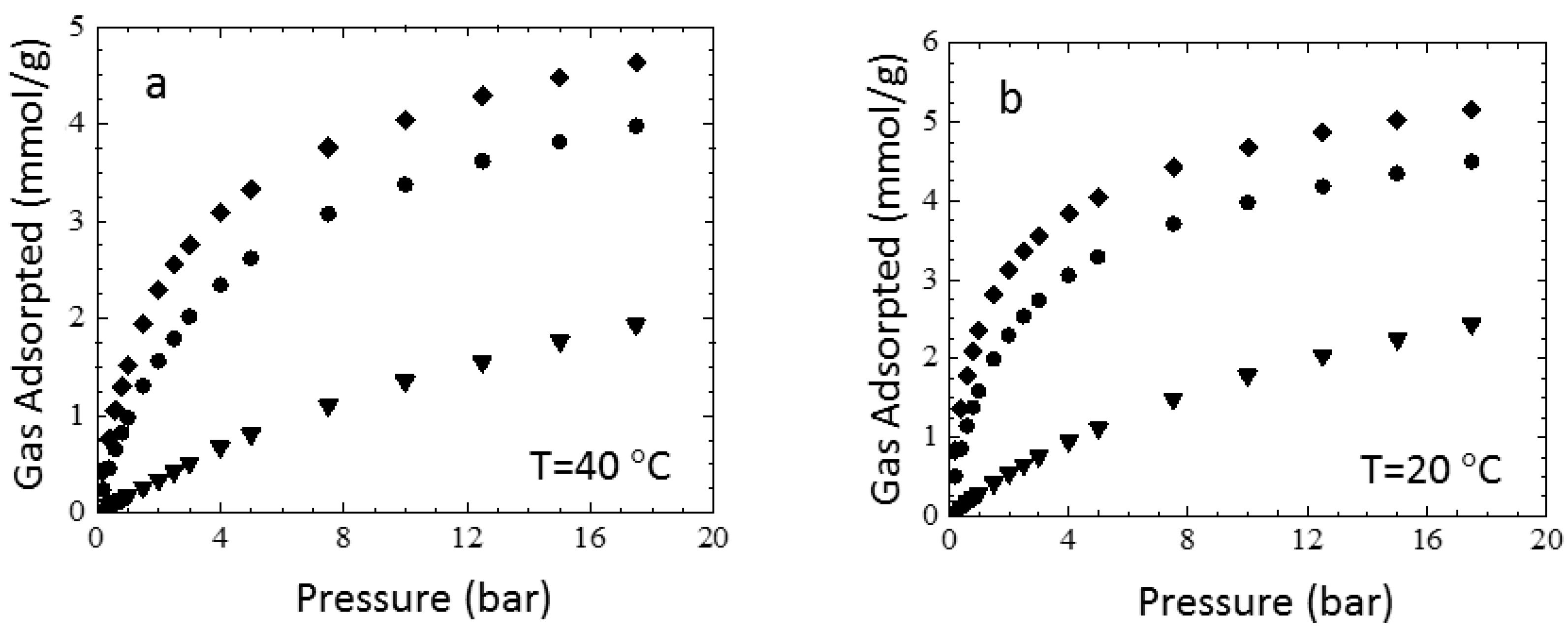
3.3. Sorption Experiments
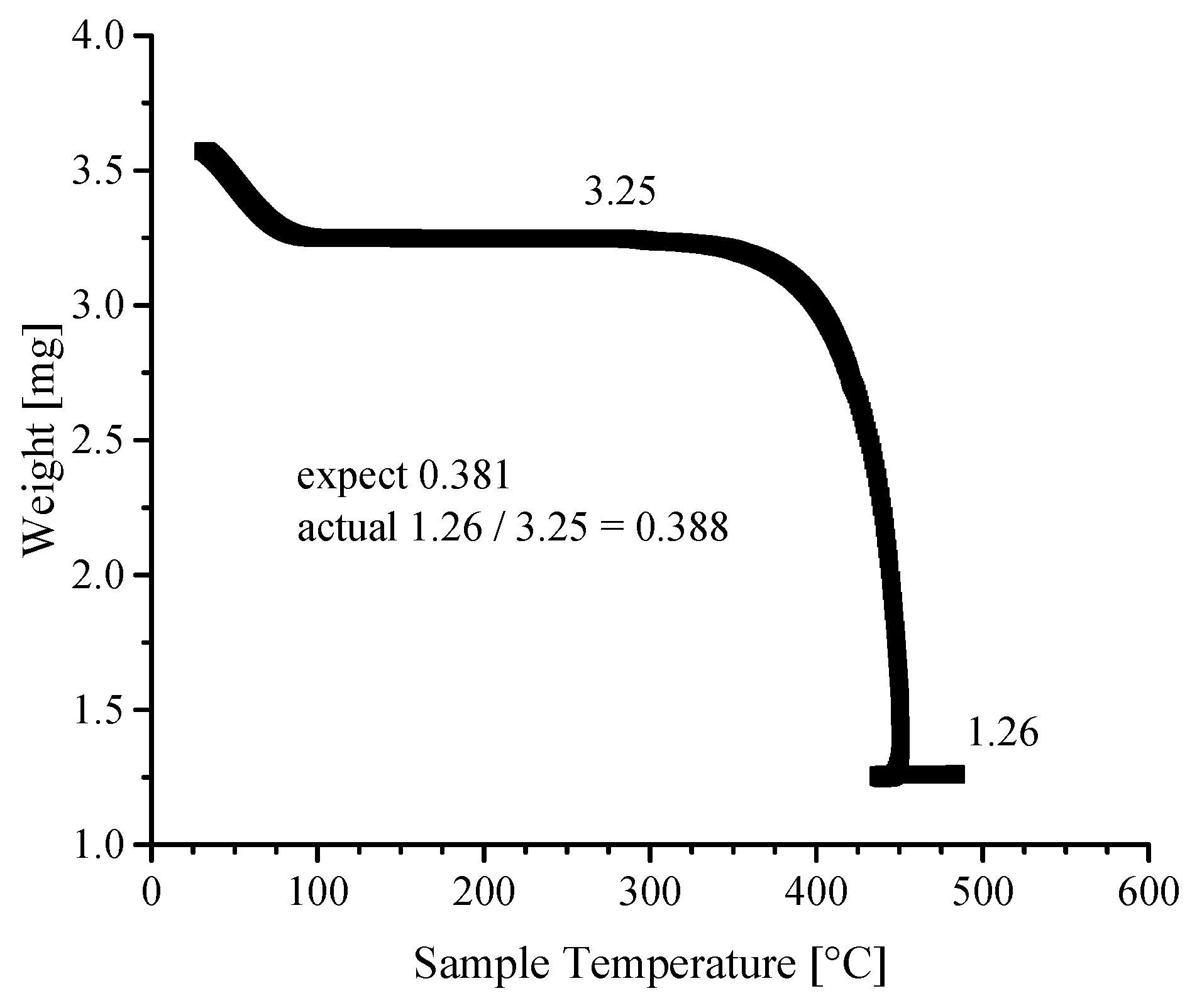
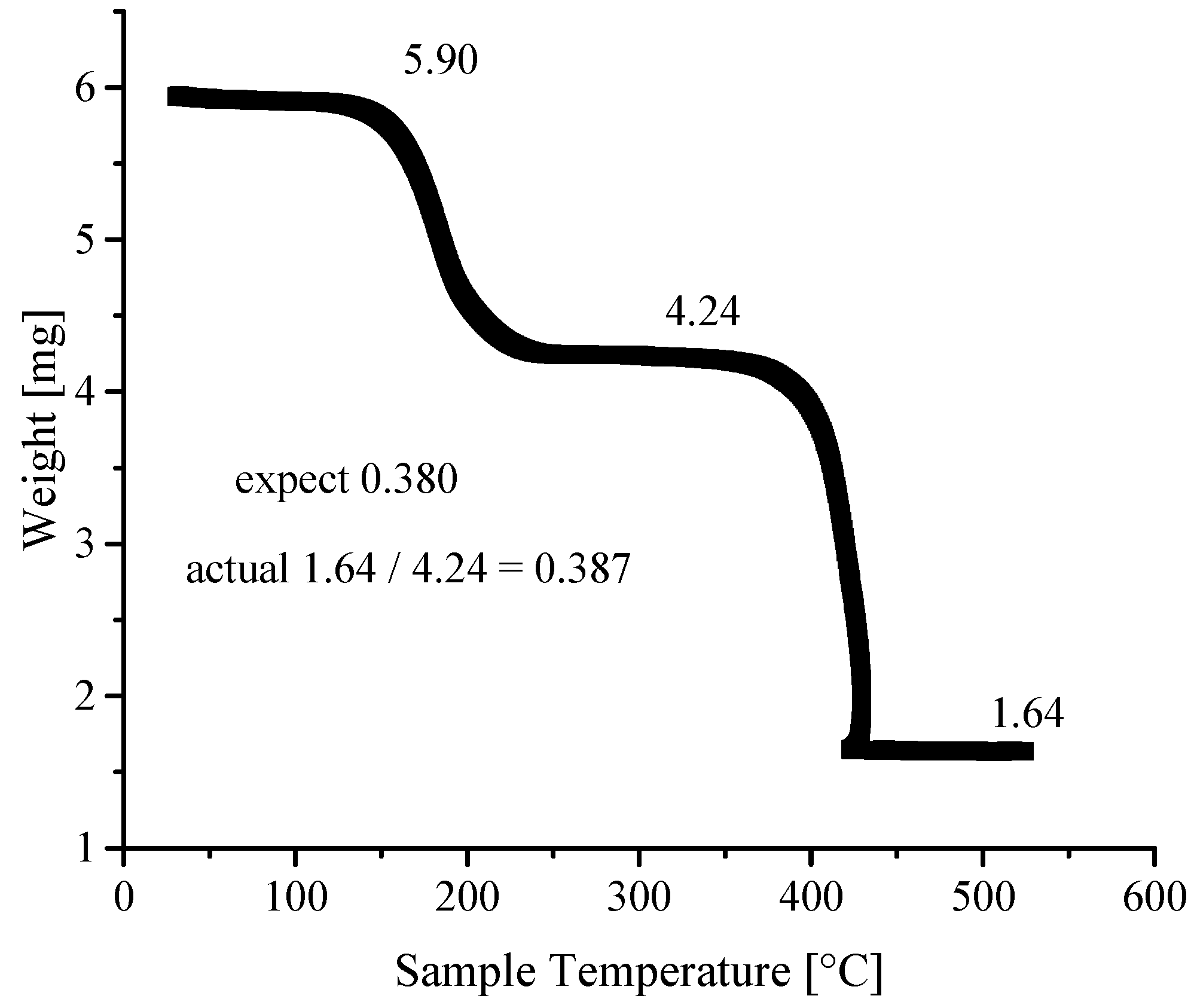
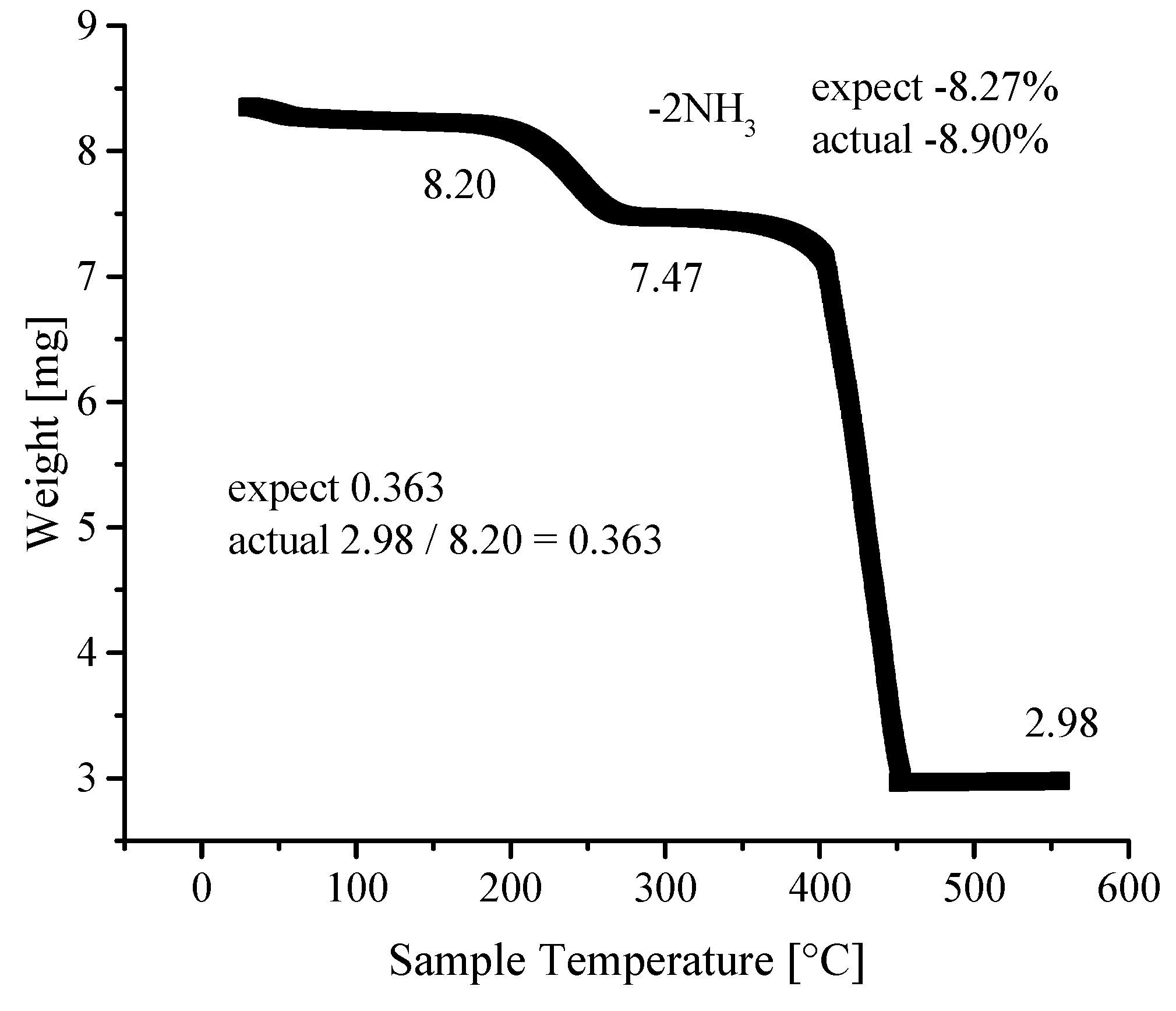
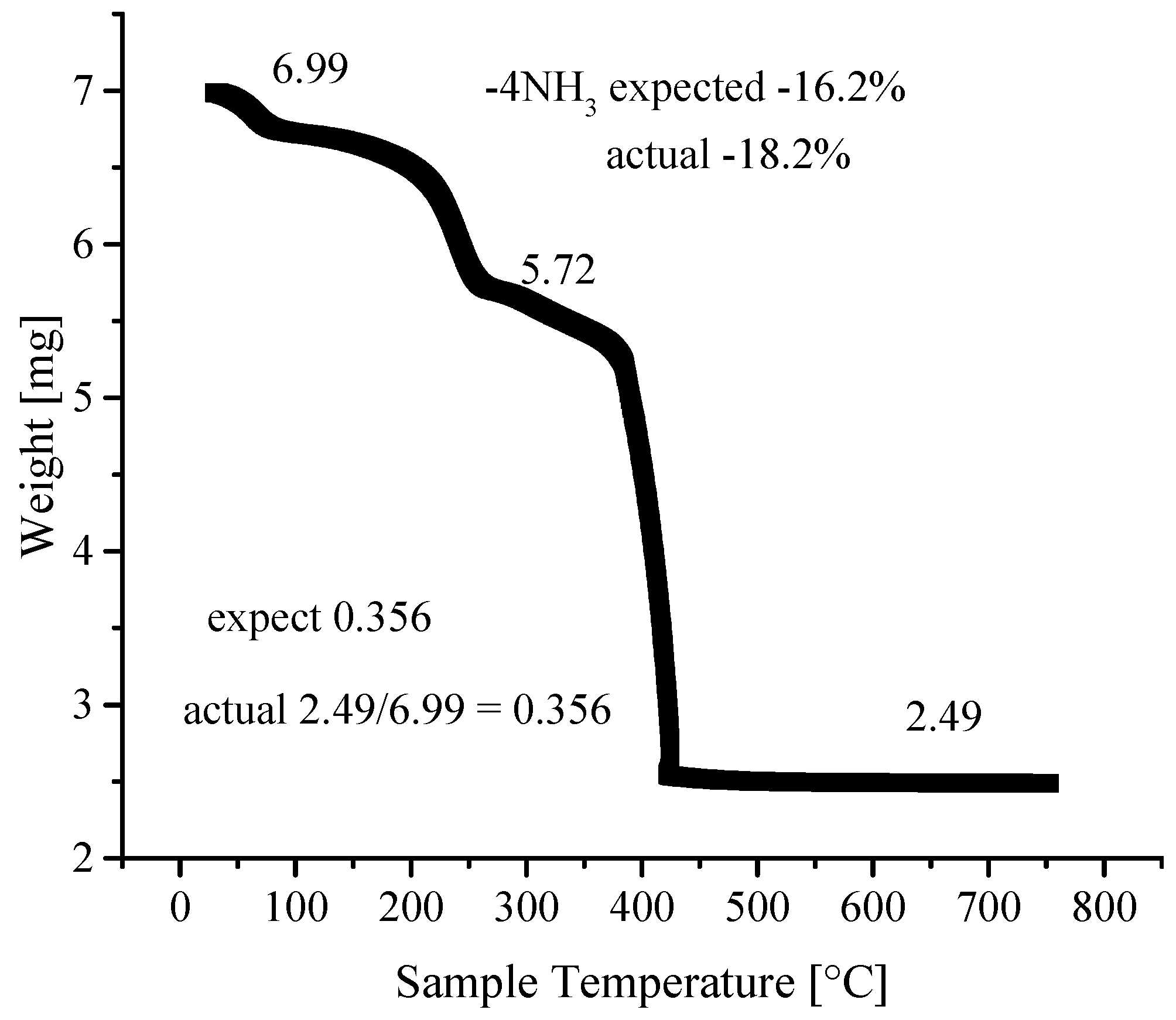
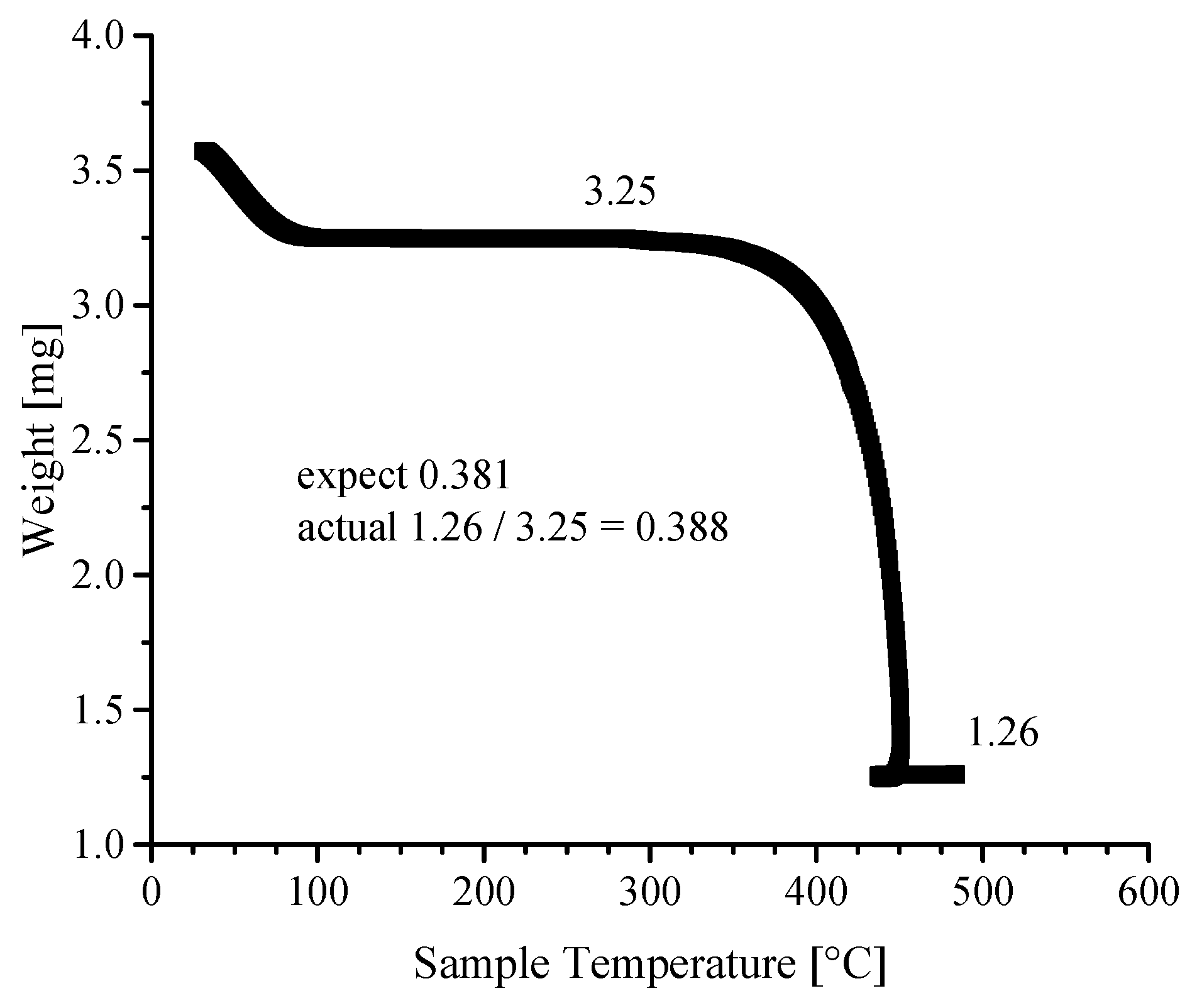
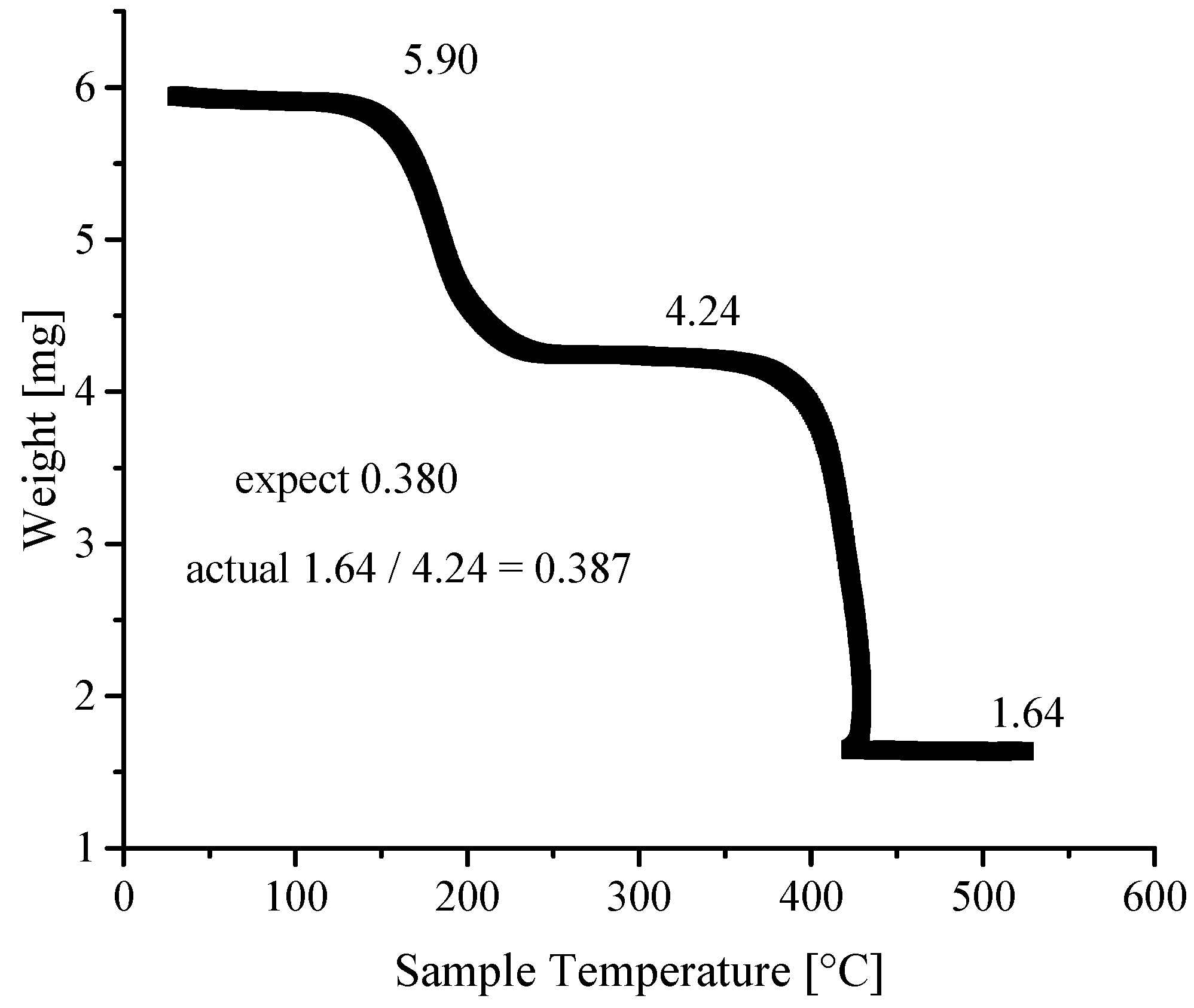
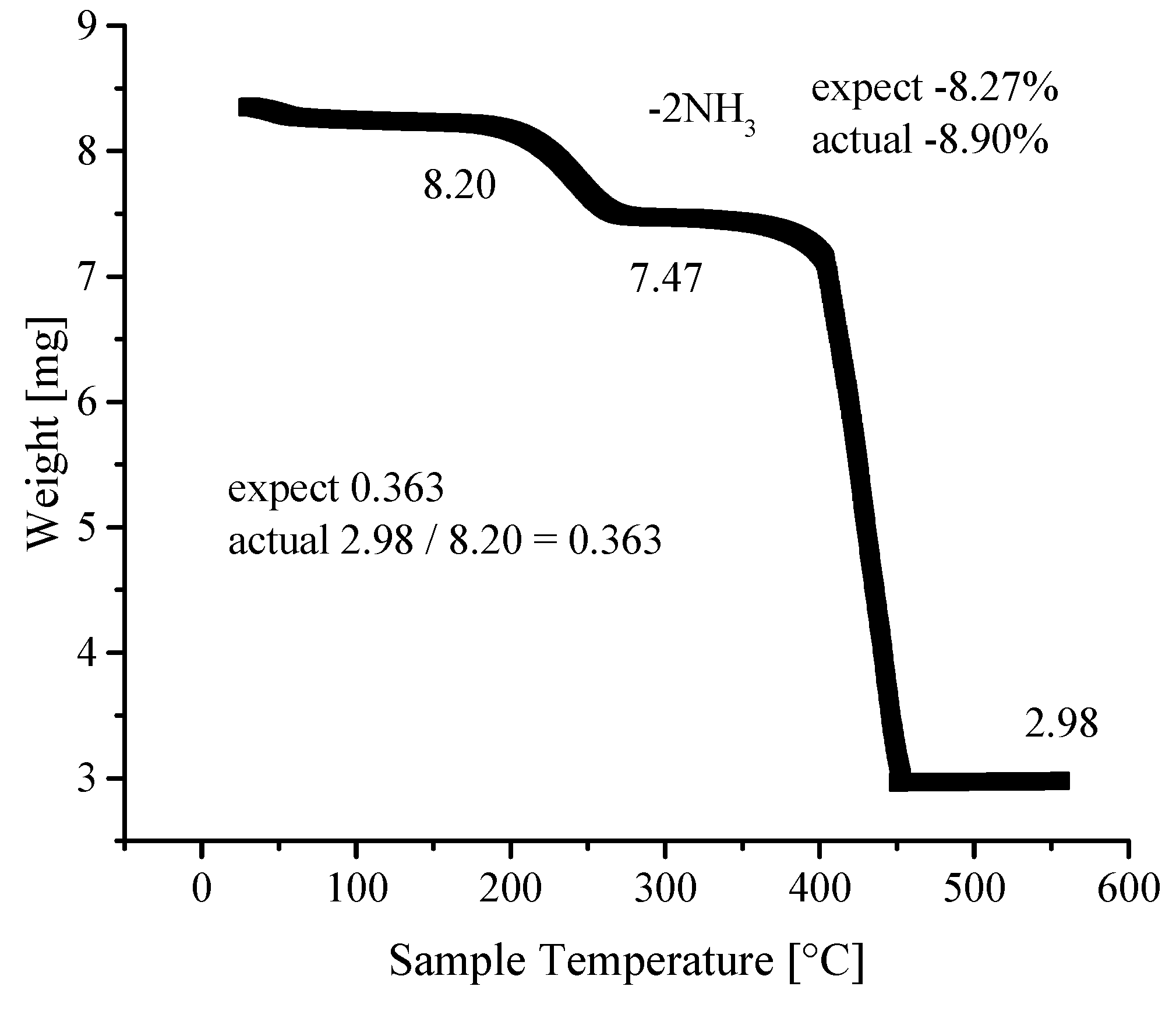
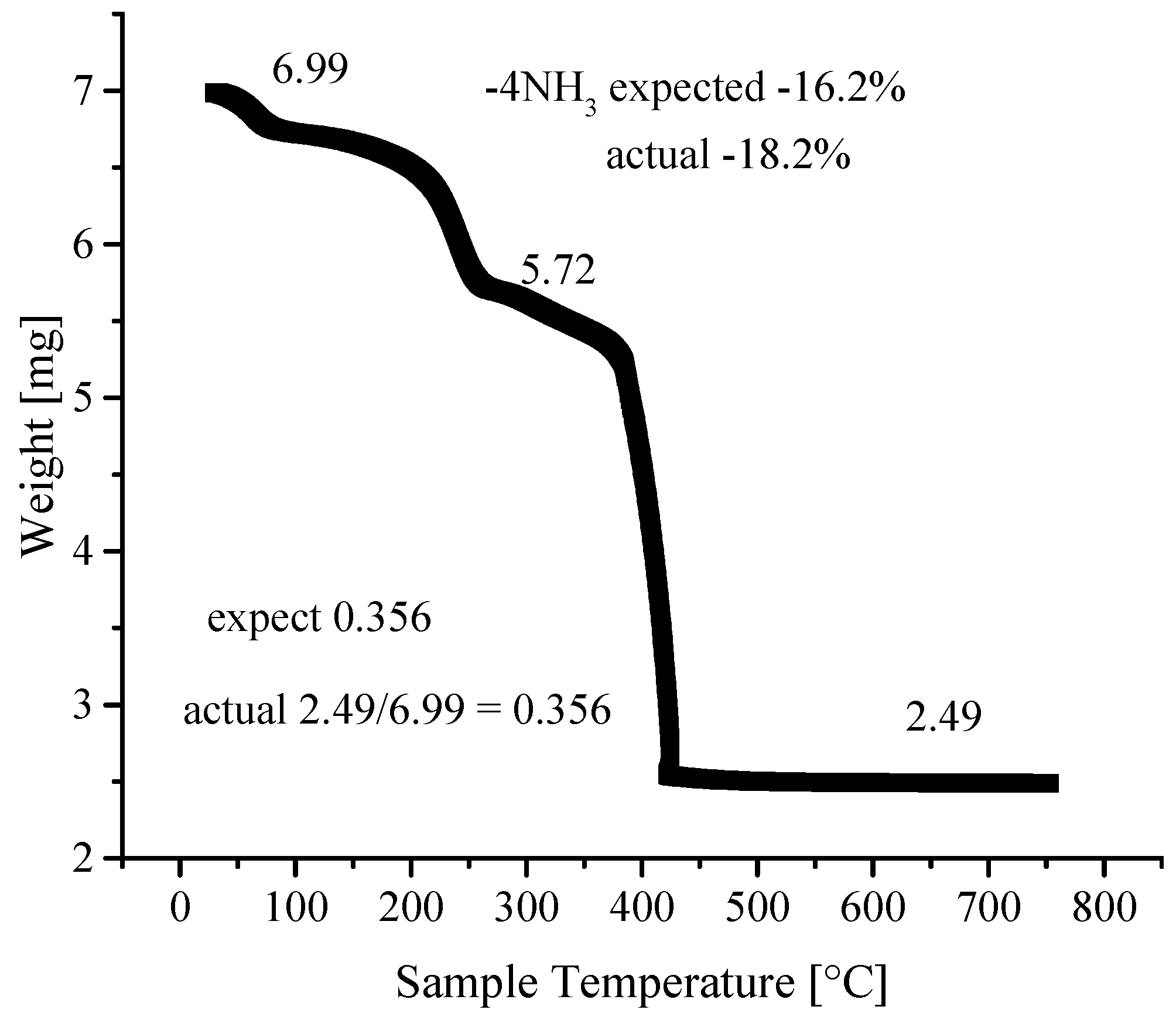
3.4. Thermogravimetric Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Etheridge, D.M.; Steele, L.P.; Langenfelds, R.L.; Francey, R.J.; Barnola, J.M.; Morgan, V.I. Natural and anthropogenic changes in atmospheric CO2 over the last 1000 years from air in Antarctic ice and firn. J. Geophys. Res. Atmos. 1996, 101, 4115–4128. [Google Scholar] [CrossRef]
- Gammon, R.H.; Sundquist, E.T.; Fraser, P.J. History of carbon dioxide in the atmosphere. In Atmospheric Carbon Dioxide and the Global Carbon Cycle; DOE/ER-239; Trabalka, J.R., Ed.; U.S. Department of Energy: Washington, DC, USA, 1985; pp. 25–62. [Google Scholar]
- Espinal, L.; Poster, D.L.; Wong-Ng, W.; Allen, A.J.; Green, M.L. Standards, Data, and Metrology Needs for CO2 Capture Materials—A Critical Review. Environ. Sci. Technol. 2009, 47, 11960–11975. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Drese, J.; Jones, C.W. Absorbent Materials for carbon Dioxide Capture from Large Anthropogenic Point Source. ChemSusChem 2009, 2, 796–854. [Google Scholar] [CrossRef]
- Wang, B.; Cote, A.P.; Furukawa, H.; O’Keeffe, M.J.; Yaghi, O.M. Colossal Cages in Zeolite Imidazolate Frameworks as Selective Carbon Dioxide Reservoirs. Nature 2008, 453, 207. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ng, W.; Kaduk, J.A.; Huang, Q.; Espinal, L.; Li, L.; Burress, J.W. Investigation of NaY Zeolite with Adsorbed CO2 by Neutron Powder Diffraction. Microporous Mesoporous Mater. 2013, 172, 95–104. [Google Scholar] [CrossRef]
- Siriwardane, R.V.; Shen, M.-S.; Fisher, E.P.; Poston, J.A. Adsorption of CO2 on Molecular Sieves and Activated Carbon. Energy Fules 2001, 15, 279–284. [Google Scholar] [CrossRef]
- Fripiat, J.J.; Cruz, M.M.; Bohor, B.F.; Thomas, J., Jr. Interlamellar Adsorption of Carbon Dioxide by Smectites. Clays Clay Miner 1974, 22, 23–30. [Google Scholar] [CrossRef]
- Sayyah, I.; Lu, Y.; Mall, R.I.; Suslick, K.S. Mechancial Activation of CaO-Based Adsorbates for CO2 Capture. ChemSusChem 2013, 6, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Fernández, E.; Rønning, M.; Grande, T.; Chen, D. Synthesis and CO2 Capture Properties of Nanocrystalline Lithium Zirconate. Chem. Mater. 2006, 18, 6037–6046. [Google Scholar] [CrossRef]
- Hutson, N.D. Structural Effects on the High Temperature Adsorption of CO2 on a Synthetic Hydrotalcite. Chem. Mater. 2004, 16, 4135–4143. [Google Scholar] [CrossRef]
- Zhou, H.C.; Kitagawa, S. Metal–Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. [Google Scholar] [CrossRef] [PubMed]
- Dey, C.; Kundu, T.; Biswal, B.P.; Mallick, A.; Banerjee, R. Crystalline metal-organic frameworks (MOFs): Synthesis, structure and function. Acta Cryst. 2014, B70, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Woerner, W.R.; Plonka, A.M.; Chen, X.; Banerjee, D.; Thallapally, P.K.; Parise, J.B. Simultaneous in situ X-ray Diffraction and Calorimetric Studies as a Tool to Evaluate Gas Adsorption in Microporous Materials. J. Phys. Chem. 2016, 120, 360–369. [Google Scholar]
- Liu, Y.; Wang, Z.U.; Zhou, H.-C. Recent Advances in Carbon Dioxide Capture with Metal-Organic Frameworks. Greenh. Gas Sci. Technol. 2012, 2, 239–259. [Google Scholar] [CrossRef]
- Yaghi, O.M.; Li, Q. Reticular Chemistry and Metal-Organic Frameworks for Clean Energy. MRS Bull. 2009, 34, 682–690. [Google Scholar] [CrossRef]
- Morris, R.E.; Wheatley, P.S. Gas Storage in Nanoporous Materials. Angew. Chem. Int. Ed. 2008, 47, 4966–4981. [Google Scholar] [CrossRef] [PubMed]
- Rowsell, J.L.C.; Yaghi, O.M. Metal-Organic Frameworks: A New Class of Porous Materials. Microporous Mesoporous Mater. 2004, 73, 3–14. [Google Scholar] [CrossRef]
- Chen, R.; Yao, J.; Gu, Q.; Smeets, S.; Barlocher, C.; Gu, H.; Zhu, D.; Morris, W.; Yaghi, O.M.; Wang, H.A. Two-Dimensional Zeolitic Imidazolate Framework with a Cushion-Shaped Cavity for CO2 Adsorption. Chem. Commun. 2013, 49, 9500–9502. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Shen, D.; Bülow, M.; Lau, M.L.; Deng, S.; Fitch, F.R.; Lemcoff, N.O.; Semanscin, J. Metallo-organic molecular sieve for gas separation and purification. Microporous Mesoporous Mater. 2002, 55, 217–230. [Google Scholar] [CrossRef]
- Britt, D.; Furukawa, H.; Wang, B.; Glover, T.G.; Yaghi, O.M. Highly efficient separation of carbon dioxide by a metal-organic framework replete with open metal sites. Proc. Natl. Acad. Sci. USA 2009, 106, 20637–20640. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.-Y.; Chrzanowski, M.; Ma, S. Metal-Metalloporphyrin Frameworks: Resurging Class of Functional Materials. Chem. Soc. Rev. 2014, 43, 5841–5866. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, Y.; Ma, D.; Ma, T.; Shi, Z.; Ma, S. Metal Cation Directed de novo Assembly of a Functionalized Guest Molecule into the Nanospace of a Metal-Organic Framework. J. Am. Chem. Soc. 2014, 136, 1202–1205. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Cheng, Q.; Kim, C.; Gao, W.-Y.; Wojtas, L.; Cheng, Y.-S.; Zaworotko, M.J.; Zhang, X.P.; Ma, S. Crystal engineering of a microporous, catalytically active fcu topology MOF using a custom-designed metalloporphyrin linker. Angew. Chem. Int. Ed. 2012, 51, 10082–10085. [Google Scholar] [CrossRef] [PubMed]
- Tranchemontagne, D.J.; Hunt, J.R.; Yaghi, O.M. Room Temperature Synthesis of Metal-Organic Framework: MOF-5, MOF-74, MOF-177, MOF-199, and IRMOF-0. Tetrahedron 2008, 64, 8553–8557. [Google Scholar] [CrossRef]
- Feng, D.; Gu, Z.-Y.; Chen, Y.-P.; Park, J.; Wei, Z.; Sun, Y.; Bosch, M.; Yuan, S.; Zhou, H.-C. A Highly Stable Porphyrinic Zirconium Metal-Organic Framework with shp-a Topology. J. Am. Chem. Soc. 2014, 136, 17714–17717. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Wang, K.; Su, J.; Liu, T.-F.; Park, J.; Wei, Z.; Bosch, M.; Yakovenko, A.; Zou, X.; Zhou, H.-C. A Highly Stable Zeotype Mesoporous Zirconium Metal-Organic Framework with Ultralarge Pores. Angew. Chem. Int. Ed. 2014, 54, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Queen, W.L.; Hudson, M.R.; Bloch, E.D.; Mason, J.A.; Gonzalez, M.L.; Lee, J.S.; Gygi, D.; Howe, J.D.; Lee, K.; Darwish, T.A.; et al. Comprehensive study of carbon dioxide adsorption in the metal-organic frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci. 2014, 5, 4569–4581. [Google Scholar] [CrossRef]
- Bloch, E.D.; Hudson, M.R.; Mason, J.A.; Queen, W.L.; Zadrozny, J.M.; Chavan, S.; Bordiga, S.; Brown, C.M.; Long, J.R. Reversible CO Binding Enables Tunable CO/H2 and CO/N2 Separations in Metal-Organic Frameworks with Exposed Divalent Metal Cations. J. Am. Chem. Soc. 2014, 136, 10752–10761. [Google Scholar] [CrossRef] [PubMed]
- Caskey, S.R.; Wong-Foy, A.G.; Matzger, A.J. Dramatic Tuning of Carbon Dioxide Uptake via Metal substitution in a Coordination Polymer with Cylindrical Pores. J. Am. Chem. Soc. 2008, 130, 10870–10871. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Simmons, J.M.; Srinivas, G.; Zhou, W.; Yildirim, T. Adsorption sites and binding nature of CO2 in prototypical metal-organic frameworks-A combined neutron diffraction and first-principles study. J. Phys. Chem. Lett. 2010, 1, 1946–1951. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ng, W.; Kaduk, J.A.; Siderius, D.L.; Allen, A.L.; Espinal, L.; Boyerinas, B.M.; Levin, I.; Suchomel, M.R.; Ilavsky, J.; Li, L. Reference Diffraction Patterns, Microstructure, and Pore Size Distribution for the Copper (II) benzene-1,3,5-tricarboxylate Metal Organic Framework (Cu-BTC) Compounds. Powder Diffr. 2015, 30, 2–13. [Google Scholar] [CrossRef]
- Bourrelly, S.; Serre, C.; Vimont, A.; Ramsahye, N.A.; Maurin, G.; Daturi, M.; Filinehuk, Y.; Férey, G.; Llewellyn, P.L. A Multidisciplary Approach to Understanding Sorption Induced Breathing in the Metal Organic Framework MIL53(Cr). In Zeolites to Porous Materials—The 40th Anniversary of International Zeolite Conference; Xu, R., Gao, Z., Chen, J., Yan, W., Eds.; Elsevier: Amsterdam, Netherlands, 2007; pp. 1008–1014. [Google Scholar]
- Espinal, L.; Wong-Ng, W.; Kaduk, J.A.; Allen, A.J.; Snyder, C.R.; Chiu, C.; Siderius, D.W.; Li, L.; Cockayne, E.; Espinal, A.E. Time dependent CO2 sorption hysteresis in a one-dimensional microporous octahedral molecular sieve. J. Am. Chem. Soc. 2012, 134, 7944–7951. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.J.; Espinal, L.; Wong-Ng, W.; Queen, W.L.; Brown, C.M.; Kline, S.R.; Kauffman, K.L.; Culp, J.T.; Matranga, C. In Situ Structural Dynamics of Flexible Metal-Organic Frameworks for Selective CO2 Adsorption. J. Alloys Compd. 2015, 647, 24–34. [Google Scholar]
- Skoulidas, A.I. Molecular Dynamics Simulations of Gas Diffusion in Metal-Organic Frameworks: Argon in CuBTC. J. Am. Chem. Soc. 2004, 126, 1356–1357. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ng, W.; Kaduk, J.A.; Espinal, L.; Suchomel, M.; Allen, A.J.; Wu, H. High-Resolution Synchrotron X-ray Diffraction Study of Bis(2-methylimidazolyl)-Zinc, C8H10N4Zn (ZIF-8). Powder Diffr. 2011, 26, 234–237. [Google Scholar] [CrossRef]
- Wong-Ng, W.; Kaduk, J.A.; Wu, H.; Suchomel, M. Synchrotron X-ray Studies of Metal-Organic Framework M2(2,5-dihydroxyterephthalte), M = (Mn,Co,Ni,Zn) (MOF74). Powder Diffr. 2012, 27, 256–262. [Google Scholar] [CrossRef]
- Wong-Ng, W.; Culp, J.T.; Chen, Y.-S.; Deschamps, J.; Marti, A. Flexible Metal Organic Framework {[Ni(DpBz)][Ni(CN)4]}n, DpBz=1,4-Bis(4-pyridyl)benzene) with an Unusual Ni-N Bond. Solid State Sci. 2016, 52, 1–9. [Google Scholar] [CrossRef]
- Zaman, M.B.; Udachin, K.; Ripmeester, J.A.; Smith, M.D.; Zur Loye, H.-C. Synthesis and Characterizaiton of Diverse Coordination Polymers. Linear and Zigzag Chains Involving Their Structural Transformation via Intermolecular Hydrogen-Bonded, Interpenetrating Ladders Polycatenane, and Noninterpenetrating Square Grid from Long, Rigid N,N’-Bidentate Ligands: 1,4-Bis[(x-pyridyl)ethynyl]benaene (x = 3,4). Inorg. Chem. 2005, 44, 5047–5059. [Google Scholar] [PubMed]
- Hu, J.S.; Shang, Y.-J.; Yao, X.-Q.; Qin, L.; Li, Y.Z.; Guo, Z.-J.; Zheng, H.-G.; Xue, Z.-L. Syntheses, Structures, and Photoluminescence of Five New Metal-Organic Frameworks Based on Flexible Tetrapyridines and Aromatic Polycarboxylate Acids. Cryst. Growth Des. 2010, 10, 2676–2684. [Google Scholar] [CrossRef]
- Serre, C. Role of Solvent-Host Interactions that Lead to Very Large Swelling of Hybrid Frameworks. Science 2007, 315, 1828–1831. [Google Scholar] [CrossRef] [PubMed]
- Cussen, E.J.; Claridge, J.B.; Rosseinsky, M.J.; Kepert, C.J. Flexible Sorption and Transformation Behavior in a Microporous Metal-Organic Framework. J. Am. Chem. Soc. 2002, 124, 9574–9581. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Kondo, M. Chemistry of Crystalline metal Complex-Assembled Compounds. Bull. Chem. Soc. Jpn. 1998, 71, 1739–1753. [Google Scholar] [CrossRef]
- Kitagawa, S.; Uemura, K. Dynamic Porous Properties of Coordination Polymers Inspired by Hydrogen Bonds. Chem. Soc. Rev. 2005, 34, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Uemura, K.; Matsuda, R.; Kitagawa, S. Flexible Microporous Coordination Polymers. J. Solid State Chem. 2005, 178, 2420–2429. [Google Scholar] [CrossRef]
- Uemura, K.; Kitagawa, S.; Kondo, M.; Fukui, K.; Kitaura, R.; Chang, H.-C.; Mizutani, T. Novel Flexible Frameworks of Porous Cobalt (II) Coordination Polymers That Show Selective Guest Adsorption Based on the Switching of Hydrogen-Bond Pairs of Amide Groups. Chem. Eur. J. 2002, 8, 3587–3590. [Google Scholar] [CrossRef]
- Flexible MOFs. Berkeley Lectures on Energy: Carbon Capture and Sequestration, UC Berkeley Main Site. Available online: http://www.cchem.berkeley.edu/molsim/teaching/spring2013/CCS/Group8/ (accessed on 12 April 2014).
- Liu, Y.-Y.; Couck, S.; Vandichel, M.; Grzywa, M.; Leus, K.; Biswas, S.; Volkmer, D.; Gascon, J.; Kapteijn, F.; Denayer, J.F.M. New VIV-Based Metal-Organic Framework Having Framework Flexibility and High CO2 Adsorption Capacity. Inorg. Chem. 2013, 52, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Reah, R.S.; Smith, D.A.; Trachtenberg, M.C.; Li, J. Highly Selective CO2 Capture by a Flexible Microporous Metal–Organic Framework (MMOF) Material. Chem. Eur. J. 2010, 16, 13951–13954. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-S.; Suh, M. Highly Selective CO2 Capture in Flexible 3-D Coordination Polymer Networks. Angew. Chem. Int. Ed. 2009, 48, 6865–6869. [Google Scholar] [CrossRef] [PubMed]
- Nijem, N.; Thissen, P.; Yao, Y.; Longo, R.; Roodenko, K.; Wu, H.; Zhao, Y.; Cho, K.; Li, J.; Langreth, D.C. Understanding the Preferential Adsorption of CO2 over N2 in a Flexible Metal–Organic Framework. J. Am. Chem. Soc. 2011, 133, 12849–12857. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, D.; Nakagawa, K.; Higuchi, M.; Horike, S.; Kubota, Y.; Kobayashi, T.C.; Takata, M.; Kitagawa, S. Kinetic Gate-Opening Process in a Flexible Porous Coordination Polymer. Angew. Chem. 2008, 120, 3978–3982. [Google Scholar] [CrossRef]
- Kauffman, K.L.; Culp, J.T.; Allen, A.J.; Espinal-Thielen, L.; Wong-Ng, W.; Brown, T.D.; Goodman, A.; Bernardo, M.P.; Pancoast, R.J.; Chirdon, D. Selective Adsorption of CO2 from Light Gas Mixture by Using a Structurally Dynamic Porous Coordination Polymer. Angew. Chem. Int. Ed. 2011, 50, 10888–10892. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, D.V.; Moudrakovski, I.L.; Ratcliffe, C.I.; Dutrisac, R.; Ripmeester, J.A. Sorption of Xenon, Methane, and Organic Solvents by a Flexible Microporous Polymer Catena-Bis(Dibenzoylmethanato)-(4,4′-bipyridyl)nickel(II). Chem. Mater. 2003, 15, 4810–4818. [Google Scholar] [CrossRef]
- Hofmann, K.A.; Küspert, F. Verbindungen von Kohlenwasserstoffen mit Metallsalzen. Z. Anorg. Allgem. Chem. 1897, 15, 204–207. [Google Scholar] [CrossRef]
- Niel, V.; Martinez-Agudo, J.M.; Muñoz, M.C.; Gaspar, A.B.; Real, J.A. Cooperative spin crossover behavior in cyanide-bridged Fe(II)-M(II) bimetallic 3-D Hofmann-like networks (M = Ni, Pd, and Pt). Inorg. Chem. 2001, 40, 3838–3839. [Google Scholar] [CrossRef] [PubMed]
- Agustí, G.; Cobo, S.; Gaspar, A.B.; Molná, G.; Moussa, N.O.; Szilagyi, P.Á.; Pálfi, V.; Vieu, C.; Muñoz, M.C.; Real, J.A. Thermal and Light-Induced Spin Crossover Phenomena in New 3-D Hofmann-Like Microporous Metalorganic Frameworks Produced As Bulk Materials and Nanopatterned Thin Films. Chem Mater. 2008, 20, 6721–6732. [Google Scholar] [CrossRef]
- Culp, J.T.; Madden, C.; Kauffman, K.; Shi, F.; Matranga, C. Screening Hofmann Compounds as CO2 Srobents: Nontraditional Synthetic Route to over 40 Different Pore-Functionalized and Flexible Pillard Cyanonickelates. Inorg. Chem. 2013, 52, 4205–4216. [Google Scholar] [CrossRef] [PubMed]
- Culp, J.T.; Smith, M.R.; Bittner, E.; Bockrath, B. Hysteresis in the Physisorption of CO2 and N2 in a Flexible Pillard layer Nickel Cyanide. J. Am. Chem. Soc. 2008, 130, 12427–12434. [Google Scholar] [CrossRef] [PubMed]
- Culp, J.T.; Natesakhawat, S.; Smith, M.R.; Bittner, E.; Matranga, C.S.; Bockrath, B. Hydrogen storage Properties of Rigid tThree-dimensional Hofmann Clathrate Derivatives: The effect of Pore Size. J. Phys. Chem. 2008, 112, 7079–7083. [Google Scholar]
- Kauffman, K.L.; Culp, J.T.; Goodman, A.; Matranga, C. FT-IR Study of CO2 Adsorption in a Dynamic Copper (II) Benzoate-Pyrazine Host with CO2-CO2 Interactions in the Adsorbed State. J. Phys. Chem. C 2011, 115, 1857–1866. [Google Scholar] [CrossRef]
- Culp, J.T.; Goodman, A.L.; Chirdon, D.; Sankar, S.G.; Matranga, C. Mechanism for the Dynamic Adsorption of CO2 and CH4 in a Flexible Linear Chain Coordination Polymer as Determined from in situ Infrared Spectroscopy. J. Phys. Chem. C 2010, 114, 2184–2191. [Google Scholar] [CrossRef]
- Bradshaw, S.; Claridge, J.B.; Cussen, E.J.; Prior, T.J.; Rosseinsky, M.J. Design, Chirality, and Flexibility in Nanoporous Molecule-Based Materials. Acc. Chem. Res. 2005, 38, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Bureekaew, S.; Shimonura, S.; Kitagawa, S. Chemistry and application of flexible porous coordination polymers. Sci. Technol. Adv. Mater. 2008, 9. [Google Scholar] [CrossRef]
- Ferey, G. Hybrid Porous Solids: Past, Present, Future. Chem. Soc. Rev. 2008, 37, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Thomas, K.M.; Rosseinsky, M.J. Flexibility in Metal-Organic Framework Materials: Impact on Sorption Properties. J. Solid State Chem. 2005, 178, 2491–2510. [Google Scholar] [CrossRef]
- Kawano, M.; Fujita, M. Direct observation of crystalline-state guest exchange in coordination networks. Coord. Chem. Rev. 2007, 251, 2592–2605. [Google Scholar]
- Kitagawa, S.; Kitaura, R.; Noro, S. Functional Porous Coordination Polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, S.; Masuda, R. Chemistry of coordination space of porous coordination polymers. Coord. Chem. Rev. 2007, 251, 2490–2509. [Google Scholar] [CrossRef]
- Tanaka, D.; Henke, A.; Albrecht, K.; Moeller, M.; Nakagawa, K.; Kitagawa, S.; Groll, J. Rapid preparation of flexible porous coordination polymer nanocrystals with accelerated guest adsorption kinetics. Nat. Chem. 2010, 2, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Maji, T.K.; Kitagawa, S. Chemistry of porous coordination polymers. Pure Appl. Chem. 2007, 79, 2155–2177. [Google Scholar] [CrossRef]
- Soldatov, D.V.; Ukraintseva, E.A.; Logvinenko, A. Structure and stability of a clathrate of bis (dibenzoylmethanato)-dipyridine-nickel (II) with acetone (1:2). J. Struct. Chem. 2007, 48, 938–948. [Google Scholar] [CrossRef]
- Choi, H.J.; Dinca, M.; Long, J.R. Broadly Hysteretic H2 Adsorption in the Microporous Metal−Organic Framework Co(1,4-benzenedipyrazolate). J. Am. Chem. Soc. 2008, 130, 7848–7850. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Her, J.H.; Dailly, A.; Ramirez-Cuesta, A.J.; Neumann, D.A.; Brown, C.M. Reversible structural transition in MIL-53 with large temperature hysteresis. J. Am. Chem. Soc. 2008, 130, 11813–11818. [Google Scholar] [CrossRef] [PubMed]
- Chandler, B.D.; Enright, G.D.; Udachin, K.A.; Pawsey, S.; Ripmeester, J.A.; Cramb, D.T.; Shimizu, G.K.H. Mechanical gas capture and release in a network solid via multiple single-crystalline transformations. Nat. Mater. 2008, 7, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Maji, T.K.; Matsuda, R.; Kitagawa, S. A flexible interpenetrating coordination framework with a bimodal porous functionality. Nat. Mater. 2007, 6, 142–148. [Google Scholar]
- Shimomura, S.; Horike, S.; Matsuda, R.; Kitagawa, S. Guest-specific function of a flexible undulating channel in a 7,7,8,8-tetracyano-p-quinodimethane dimer-based porous coordination polymer. J. Am. Chem. Soc. 2007, 129, 10990–10991. [Google Scholar] [CrossRef]
- Snurr, R.Q.; Hupp, J.T.; Nguyen, S.T. Prospects for nanoporous metal-organic materials in advanced separations processes. AIChE J. 2004, 50, 1090–1095. [Google Scholar] [CrossRef]
- Llewellyn, P.L.; Maurin, G.; Devic, T.; Lorea-Serna, S.; Rosenbach, N.; Serre, C.; Bourrelly, S.; Horeajada, P.; Filinchuk, Y.; Ferey, G. Prediction of the conditions for breathing of metal organic framework materials using a combination of X-ray powder diffraction, microcalorimetry, and molecular simulation. J. Am. Chem. Soc. 2008, 130, 12808–12814. [Google Scholar] [CrossRef] [PubMed]
- Hagrman, D.; Hagr, P.J.; Zubieta, J. Solid-State Coordination Chemistry: The Self-Assembly of Microporous Organic–Inorganic Hybrid Frameworks Constructed from Tetrapyridylporphyrin and Bimetallic Oxide Chains or Oxide Clusters. Angew. Chem. Int. Ed. 1999, 38, 3165–3168. [Google Scholar] [PubMed]
- Wong-Ng, W.; Culp, J.T.; Chen, Y.S.; Zavalij, P.; Espinal, L.; Siderius, D.W.; Allen, A.J.; Scheins, S.; Matranga, C. Improved Synthesis and Crystal Structure of the Flexible Pillared Layer Porous Coordination Polymer: Ni(1,2-bis(4-pyridyl)ethylene)[Ni(CN)4]. CryEngComm. 2013, 15, 4684–4693. [Google Scholar] [CrossRef]
- Nishikiori, S.; Hasegawa, T.; Iwamoto, T. The crystal structures of α, ω- diaminoalkanecadmium(II) tetracyanonickelate(II)-Aromatic molecule inclusion compounds. V. Toluidine clathrates of the hosts built of the diamines, 1,4-diaminobutane, 1.5-diaminonentane, and 1,8-diaminooctane. J. Incl. Phenom. 1991, 11, 137–152. [Google Scholar] [CrossRef]
- Akitsu, T.; Einaga, Y. Extremely long axial Cu-N bonds in chiral One-dimensional zigzag cyanide-bricged CuII-NiII and CuII-PtII Bimetallic assemblies. Inorg. Chem. 2006, 45, 9826–9833. [Google Scholar]
- Kürkçüoğlu, G.S.; Yeşilel, O.Z.; Kavlak, I.; Büyükgüngör, O. Syntheses, spectral and thermal analyses of heteronuclear aqua(2-methylpyrazine)metal(II) complexes with tetracyanonickelate ion and crystal structure of supramolecular [Cd(H2O)(2mpz)Ni(μ-CN)4]n complex. Struct. Chem. 2008, 19, 879–888. [Google Scholar] [CrossRef]
- Hibble, S.J.; Chippindale, A.M.; Pohl, A.H.; Hannon, A.C. Surprises from a simple metal-The structure and properties of nickel cyanide. Angew. Chem. Int. Ed. 2007, 46, 7116–7118. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Kung, M.C.; Kung, H. IR Studies of NH3, Pyridine, CO, and NO Adsorbed on Transition Metal Oxides. Catal. Rev. Sci. Eng 1985, 27, 425–460. [Google Scholar] [CrossRef]
- Klauber, C.; Alvey, M.D.; Yates, T. Evidence for chemisorption site selection based on an electron-donor mechanism. Chem. Phy. Lett. 1984, 106, 477–481. [Google Scholar] [CrossRef]
- Madey, T.E.; Houston, J.E.; Seabury, C.W.; Rhodin, T.N. The Structure of NH3 on Ni(111). J. Vac. Sci. Technol. 1981, 1892, 476–479. [Google Scholar] [CrossRef]
- Netzer, F.P.; Madey, T.E. Coadsorption-Induced Azimuthal ordering in Molecular Adsorbate Layers: H2O and NH3 on Oxgyen-Precovered Ni (111). Phy. Rev. Lett. 1981, 47, 928–931. [Google Scholar] [CrossRef]
- Calderón-Casado, A.; Barandika, G.; Bazán, B.; Urtiaga, M.-K.; Vallcorba, O.; Rius, J.; Miravittles, C.; Arriortua, M.-I. Solid-state transformation of the MOF [Ni2(bipy)1.5(PDC)2(H2O)2]·3.5H2O. CrystEngComm 2011, 13, 6831–6838. [Google Scholar] [CrossRef]
- Kizzie, A.C.; Wong-Foy, A.G.; Matzger, A.J. Effect of Humidity on the Performance of Microporous Coordination Polymers as Adsorbents for CO2 Capture. Langmuir 2011, 27, 6368–6373. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.; Vitillo, J.G.; Groppo, E.; Bonino, F.; Lamberti, C.; Dietzel, P.D.C.; Bordiga, S. Polymer: Spectroscopic Features and Interaction Energy. J. Phys. Chem. C 2009, 113, 3292–3299. [Google Scholar]
- Černák, J.; Abboud, K.A. Ni(bipy)2Ni(CN)4, a new type of one-dimensional square tetracyano complex. Acta Crystallogr. 2000, C56, 783–785. [Google Scholar] [CrossRef]
- APEX2 (2009.11-0). Program for Bruker CCD X-ray Diffractometer Control; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]
- Sheldrick, G.M. SHELXTL, Version 6.14. Program for Solution and Refinement of Crystal Structures; Universität Göttingen: Göttingen, Germany, 2000. [Google Scholar]
- Lemmon, E.W.; Huber, M.L. A Computer Program, REFPROP (Reference Fluid Thermodynamic and Transport Properties, ver. 9.1); National Institute of Standards and Technology: Gaithersburg, MD, USA.
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Woodward, P.A. Mercury, CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
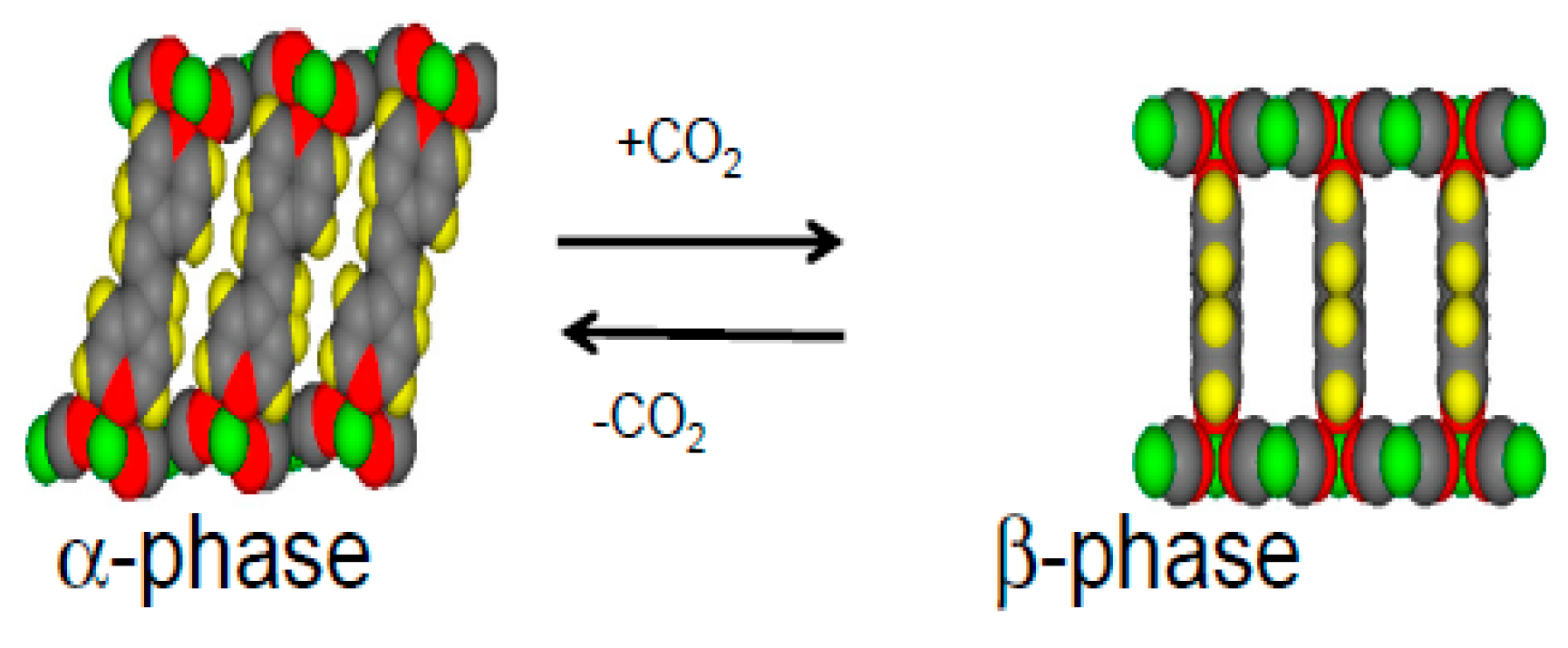



{kind=link}
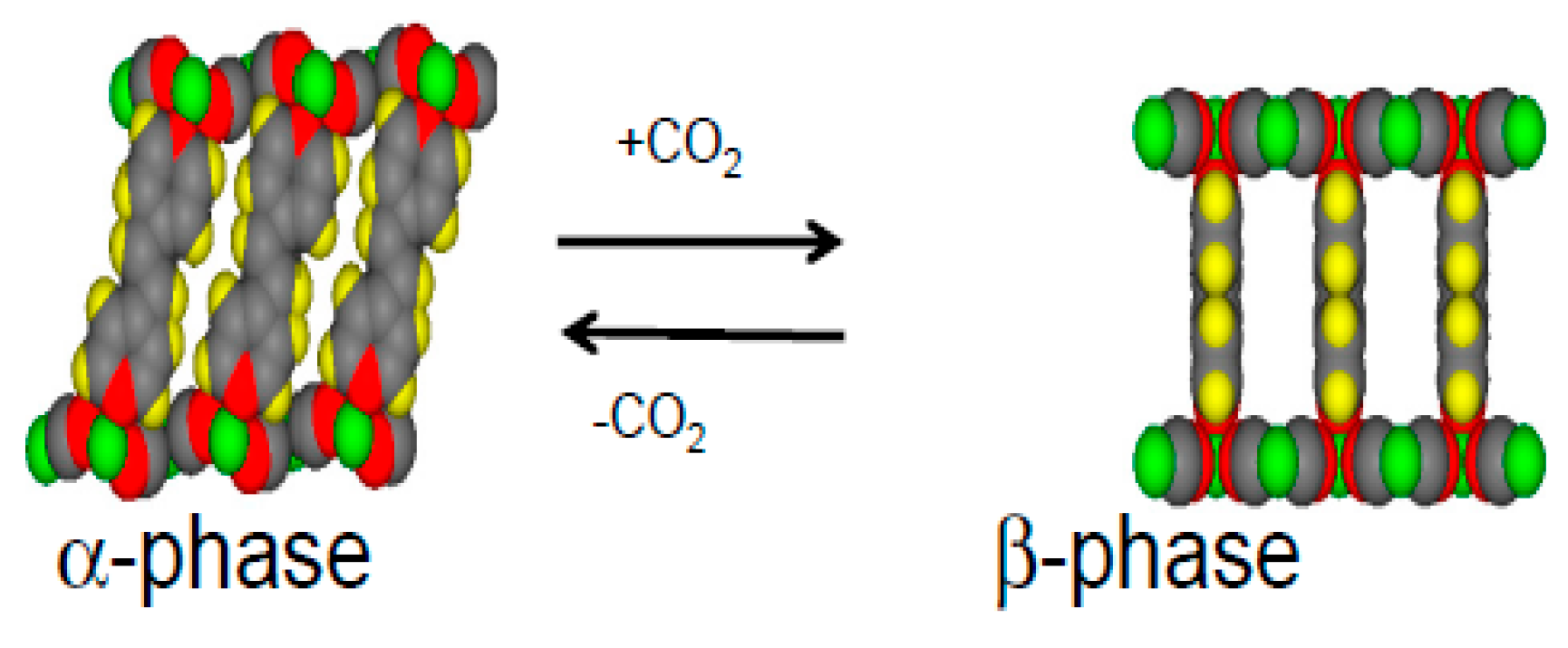{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ni-BPyMe | Ni-BPyNH2 | Ni-BPy | Ni-Naph | |
|---|---|---|---|---|
| Formula (Expected) | C15H10N6Ni2 | C14H9N9Ni2 | C14H8N6Ni2 | C12H6N6Ni2 |
| Formula (Determined) | C19H22N6Ni2O2S2 | C18H21N7Ni2O2S2 | C7H7N4Ni | C12H18N10Ni2 |
| Formula weight | 546.96 | 543.92 | 205.88 | 419.78 |
| Temperature (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| Wavelength (Å) | 0.41328 | 0.4428 | 0.41328 | 0.41328 |
| Crystal system | Monoclinic | Orthorhombic | Orthorhombic | Triclinic |
| Space group | P2/n | Cmca | Pnnm | P-1 |
| Unit cell dimension | ||||
| a (Å) | 13.3483(14) | 14.7000(5) | 7.2641(9) | 7.0315(3) |
| b (Å) | 7.1002(7) | 22.6879(7) | 9.9285(12) | 7.9219(3) |
| c (Å) | 13.5625(14) | 13.8028(4) | 11.4008(14) | 8.6103(3) |
| α (°) | 67.8160(10) | |||
| β (°) | 114.834(2) | 72.2820(10) | ||
| γ (°) | 73.6980(10) | |||
| V (Å3) | 1166.5(2) | 4603.4(2) | 822.2(2) | 415.51(3) |
| Z | 2 | 8 | 4 | 1 |
| Dcal, mg/m3 | 1.557 | 1.570 | 1.663 | 1.678 |
| µ (mm−1) | 0.412 | 0.462 | 0.475 | 0.517 |
| F(000) | 562 | 2216 | 420 | 216 |
| θ range (°) | 1.044 to 14.349 | 1.379 to 23.456 | 1.581 to 14.351 | 1.524 to 23.837 |
| Index ranges | −16 ≤ h ≤ 1 | −24 ≤ h ≤ 20 | −8 ≤ h ≤ 8 | −13 ≤ h ≤ 13 |
| −8 ≤ k ≤ 8 | −40 ≤ k ≤ 39 | −11 ≤ k ≤ 9 | −15 ≤ k ≤ 14 | |
| −16 ≤ l ≤ 16 | −17 ≤ l ≤ 22 | −13 ≤ l ≤ 12 | −10 ≤ l ≤ 15 | |
| No. reflections | 14101 | 33326 | 6410 | 12740 |
| R(intensity) | 0.0866 | 0.0862 | 0.112 | 0.0435 |
| Completeness, θ(%) | 14.357(99.9) | 15.407(97.1) | 14.357(94.3) | 14.357(96.9) |
| Refinement method | Full matrix LSQ on F2 | Full matrix LSQ on F2 | Full matrix LSQ on F2 | Full matrix LSQ on F2 |
| Data/restraints/parameters | 2116/6/169 | 5839/48/159 | 740/0/81 | 5162/0/148 |
| Goodness-of-fit on F2 | 1.152 | 1.054 | 1.086 | 1.030 |
| R indices [I>2sigma(I)] | ||||
| R1 | 0.0586 | 0.0923 | 0.0393 | 0.0300 |
| wR2 | 0.1463 | 0.2582 | 0.1072 | 0.0750 |
| R indices (all data) | ||||
| R1 | 0.0991 | 0.1587 | 0.0411 | 0.0424 |
| wR2 | 0.1754 | 0.3022 | 0.1103 | 0.0801 |
| Extinction coefficient | 0.18(3) | |||
| Largest diff. peak (e.Å−3) | 0.902 | 2.952 | 1.008 | 0.751 |
| Largest diff. hole (e.Å−3) | −0.523 | −2.676 | −0.65 | −0.729 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong-Ng, W.; Culp, J.T.; Chen, Y.-S. Crystallography of Representative MOFs Based on Pillared Cyanonickelate (PICNIC) Architecture. Crystals 2016, 6, 108. https://doi.org/10.3390/cryst6090108
Wong-Ng W, Culp JT, Chen Y-S. Crystallography of Representative MOFs Based on Pillared Cyanonickelate (PICNIC) Architecture. Crystals. 2016; 6(9):108. https://doi.org/10.3390/cryst6090108
Chicago/Turabian StyleWong-Ng, Winnie, Jeffrey T. Culp, and Yu-Sheng Chen. 2016. "Crystallography of Representative MOFs Based on Pillared Cyanonickelate (PICNIC) Architecture" Crystals 6, no. 9: 108. https://doi.org/10.3390/cryst6090108
APA StyleWong-Ng, W., Culp, J. T., & Chen, Y.-S. (2016). Crystallography of Representative MOFs Based on Pillared Cyanonickelate (PICNIC) Architecture. Crystals, 6(9), 108. https://doi.org/10.3390/cryst6090108