
N,O-Type Carborane-Based Materials



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Carboranyl Compounds with N,O-Donor Functionalities and Properties
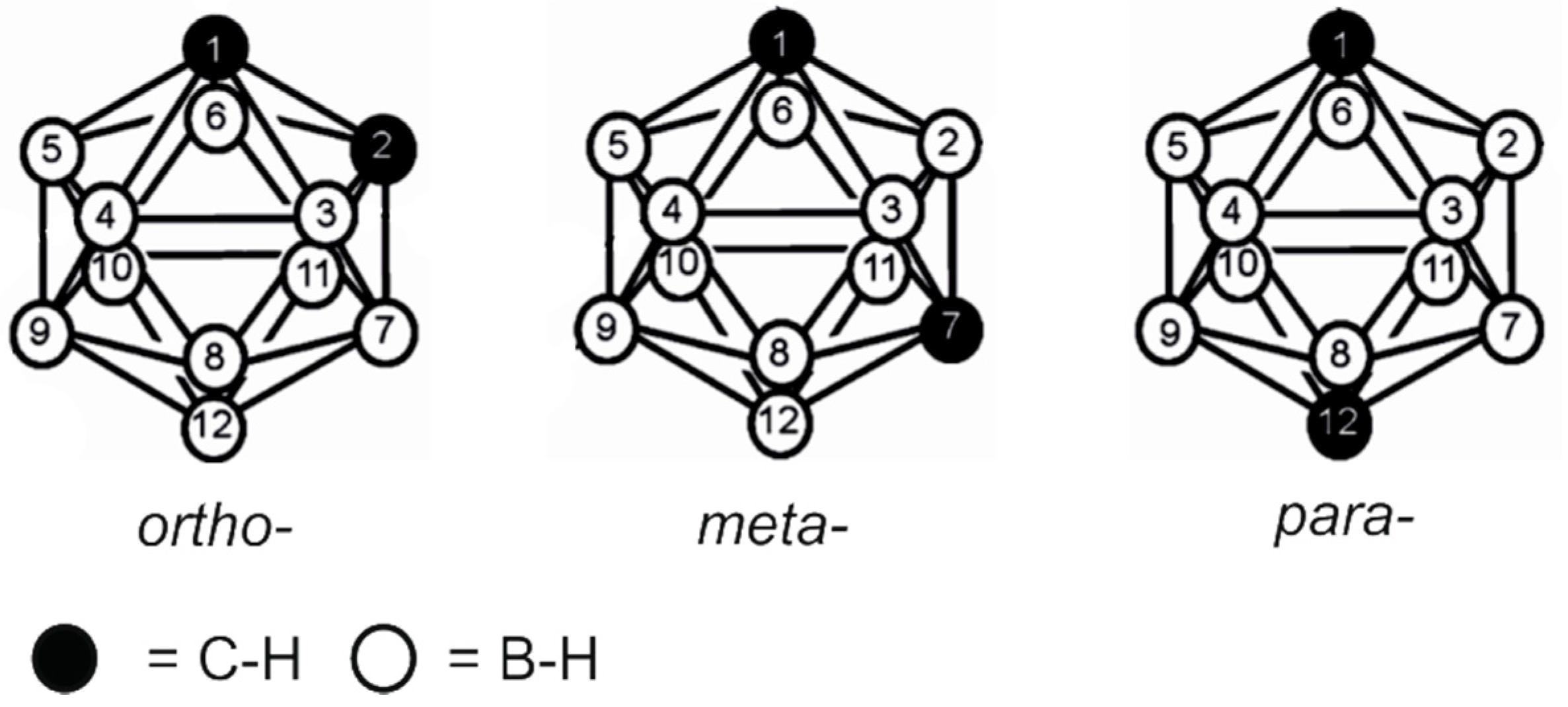
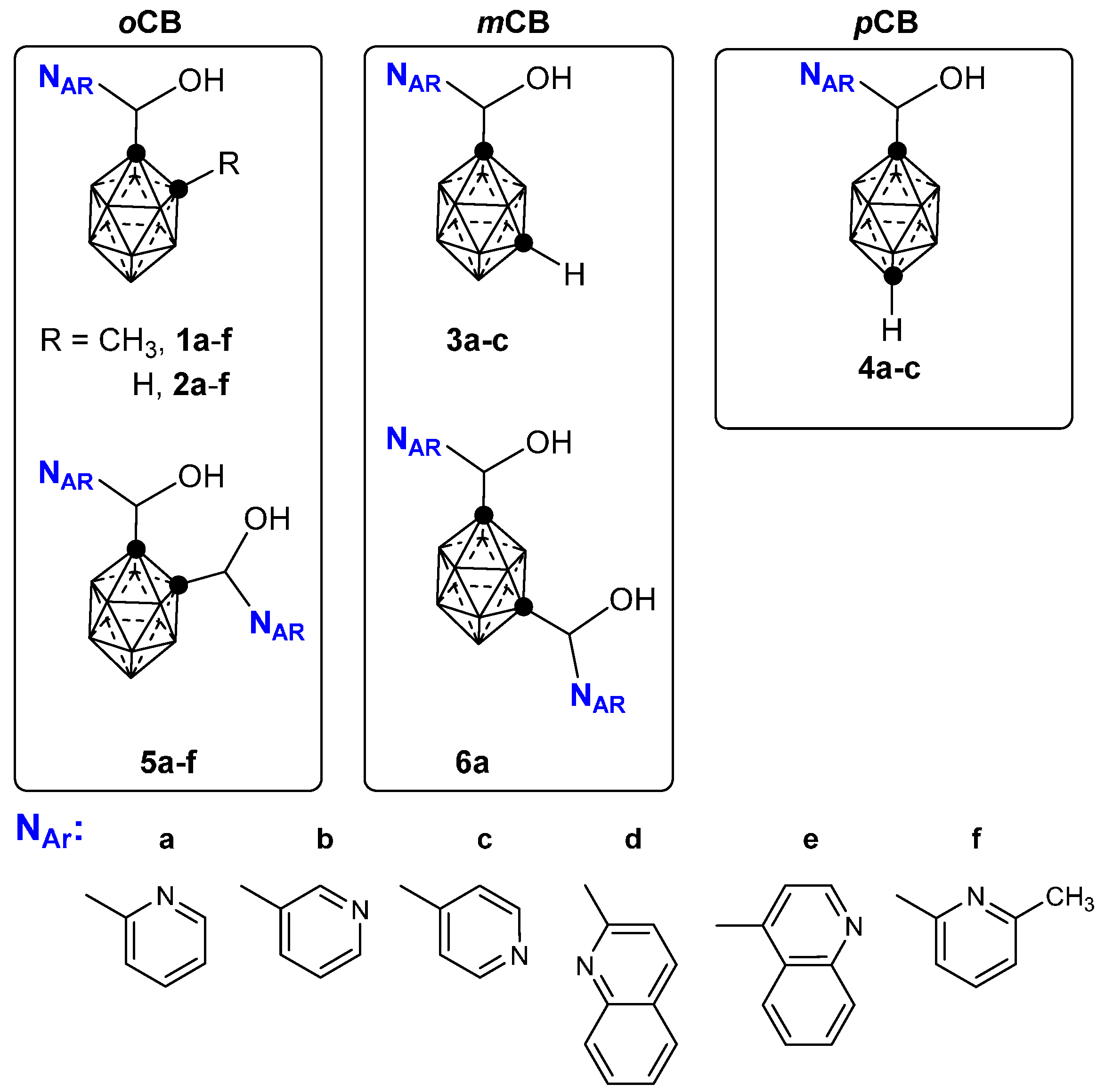
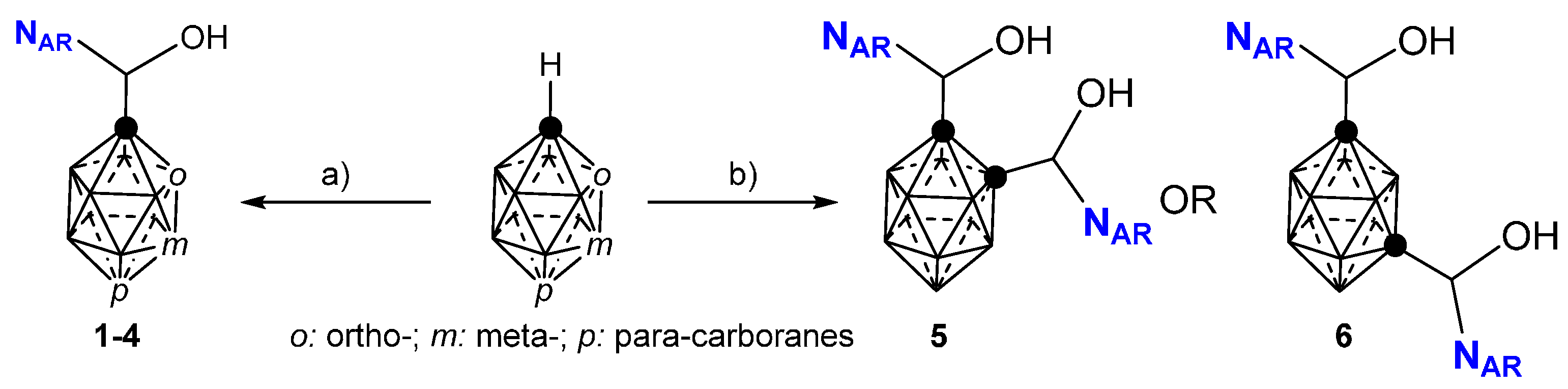
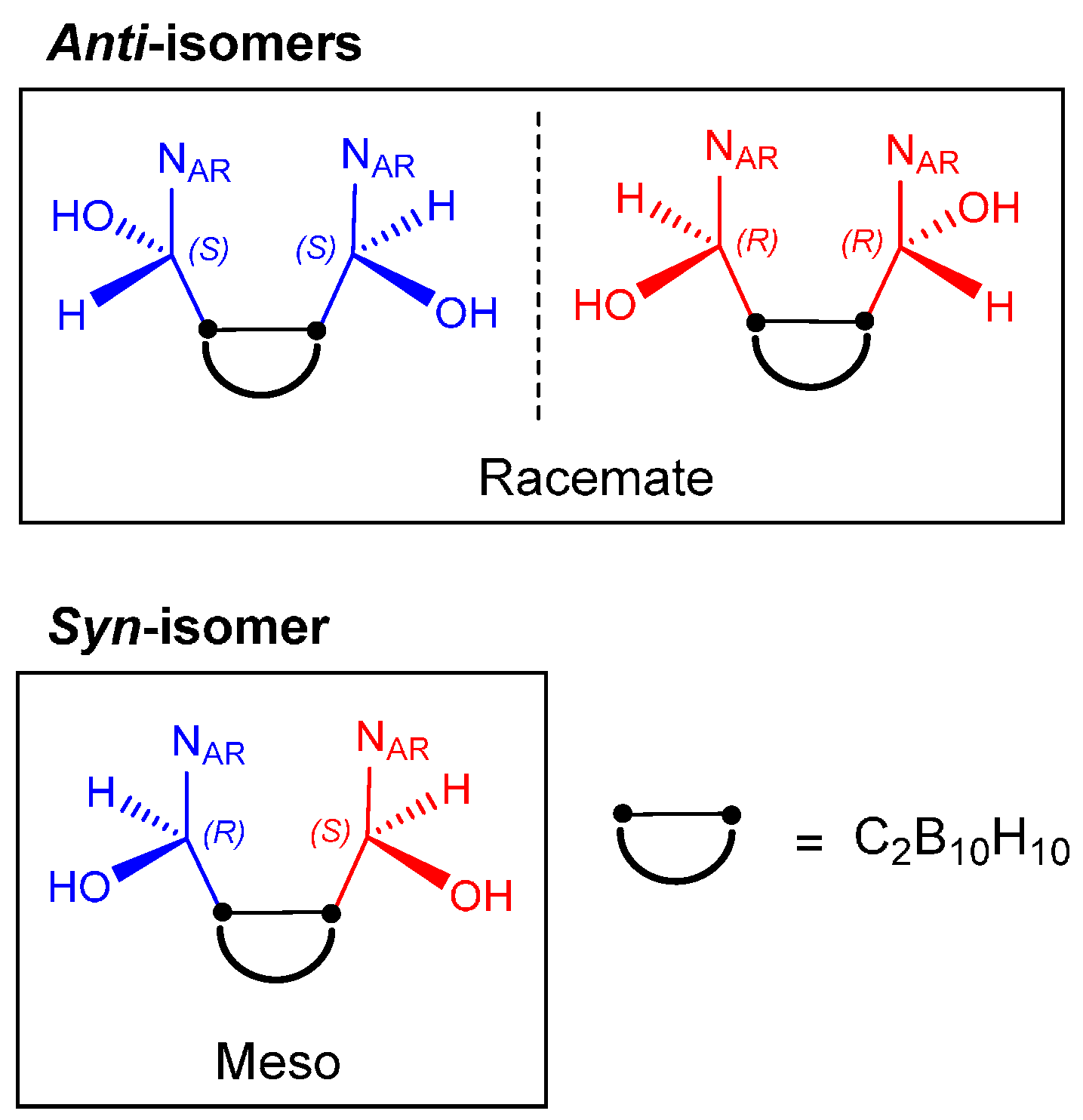
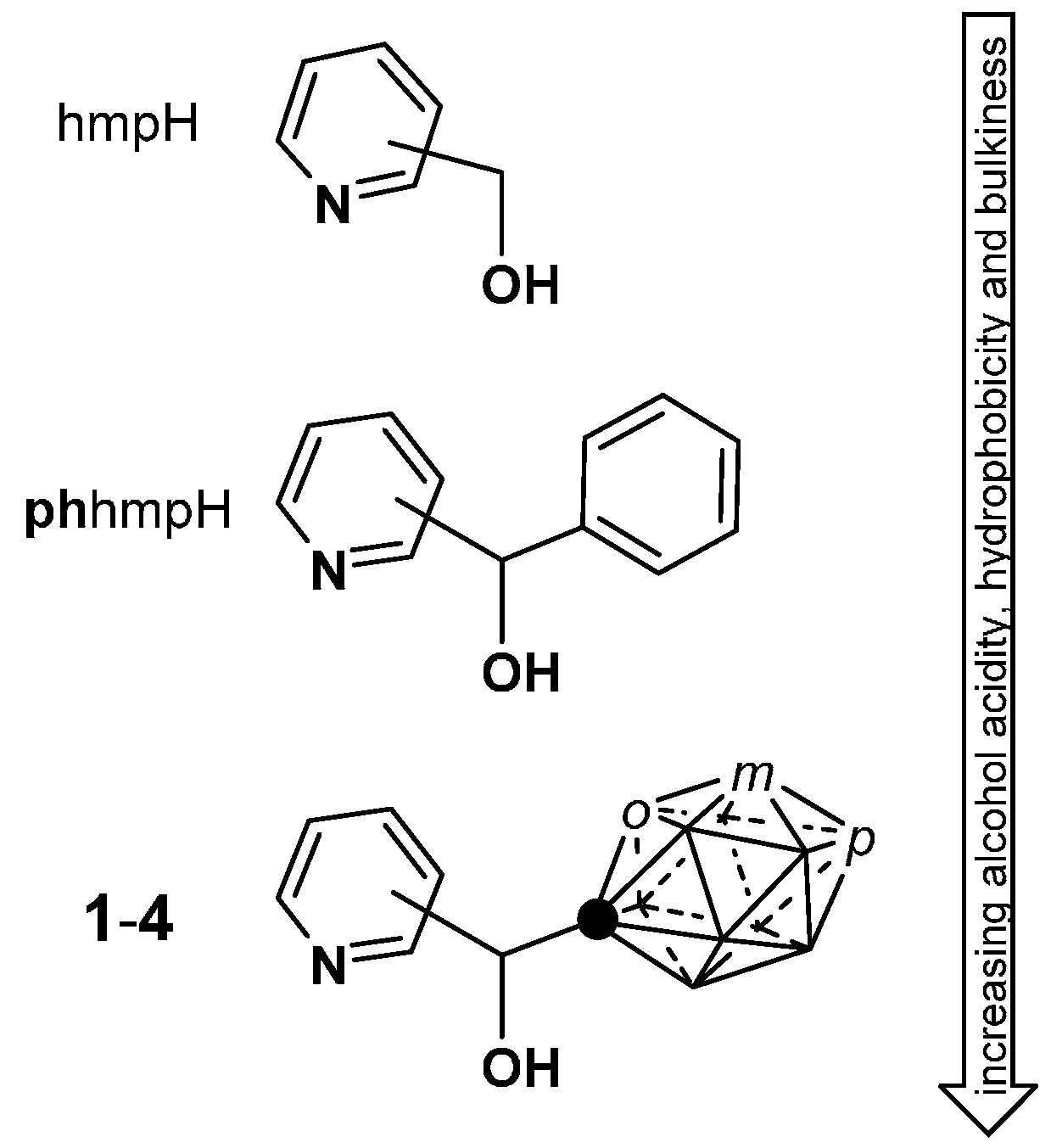
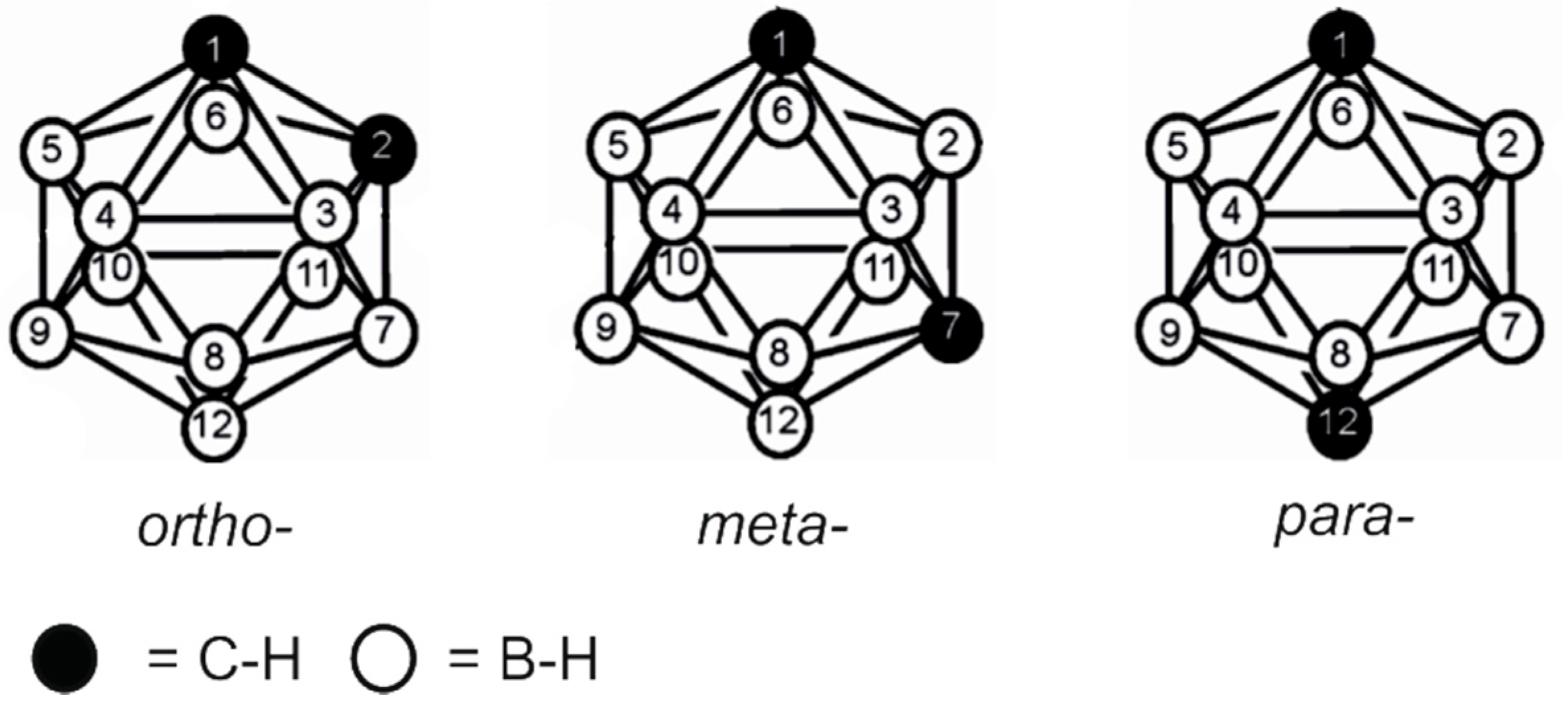
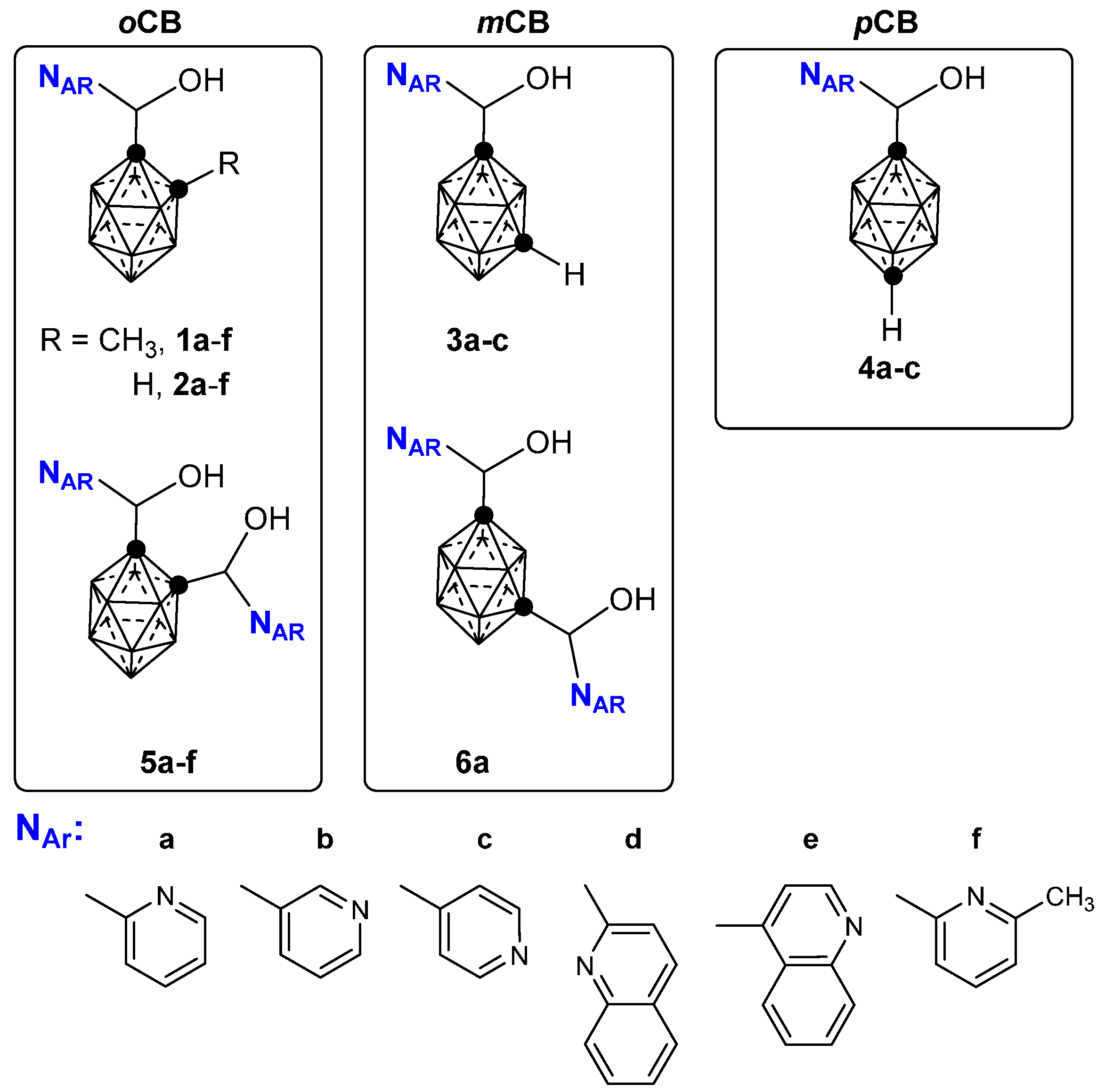
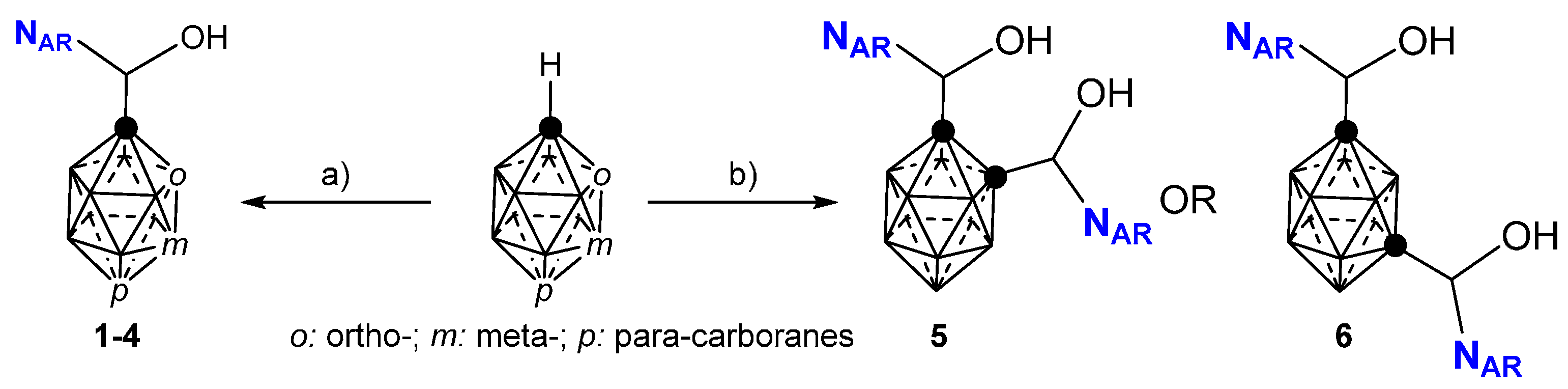
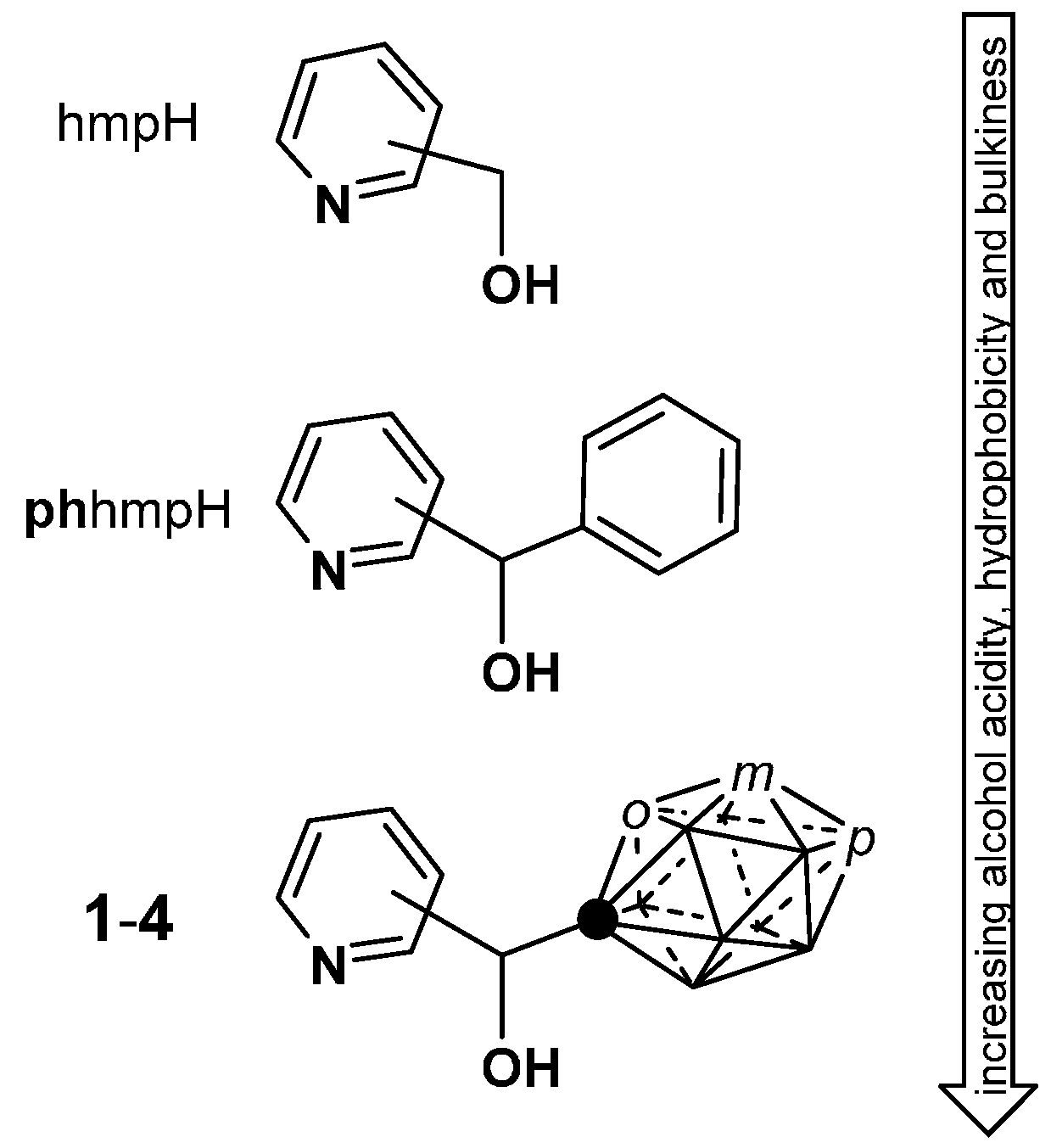
2.1. Closo-Carboranylmethylalcohols with Nitrogenated Aromatic Rings
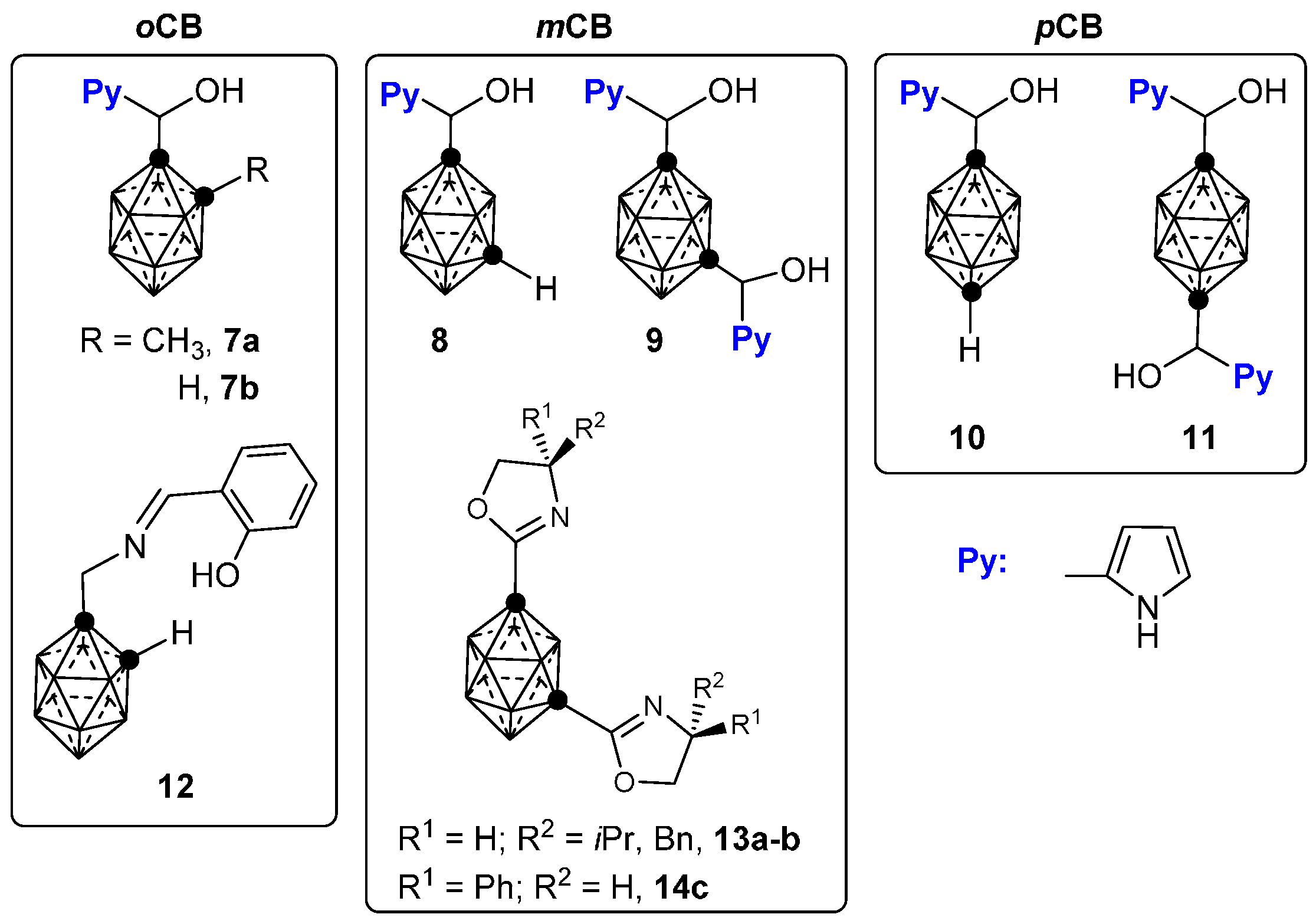
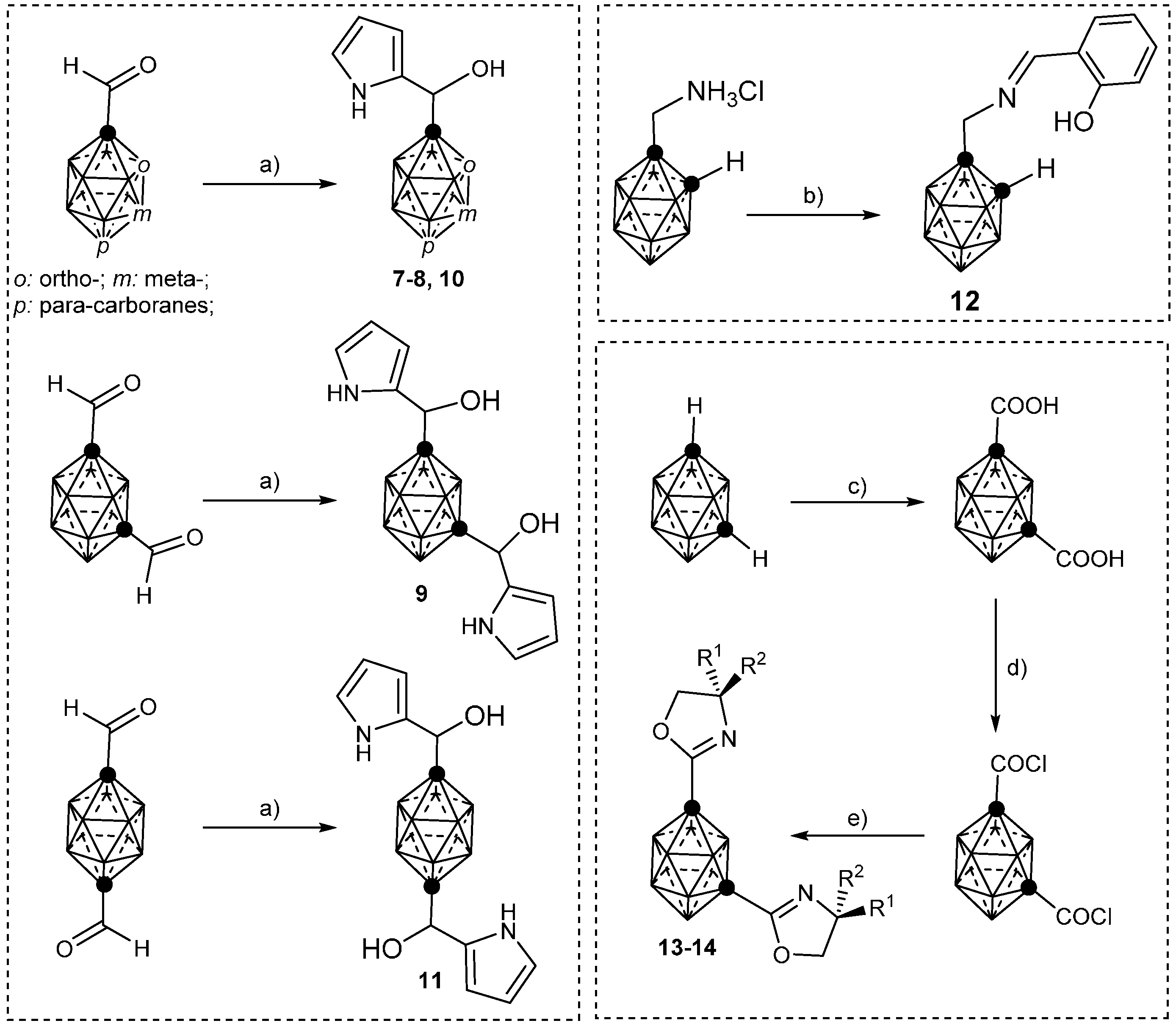
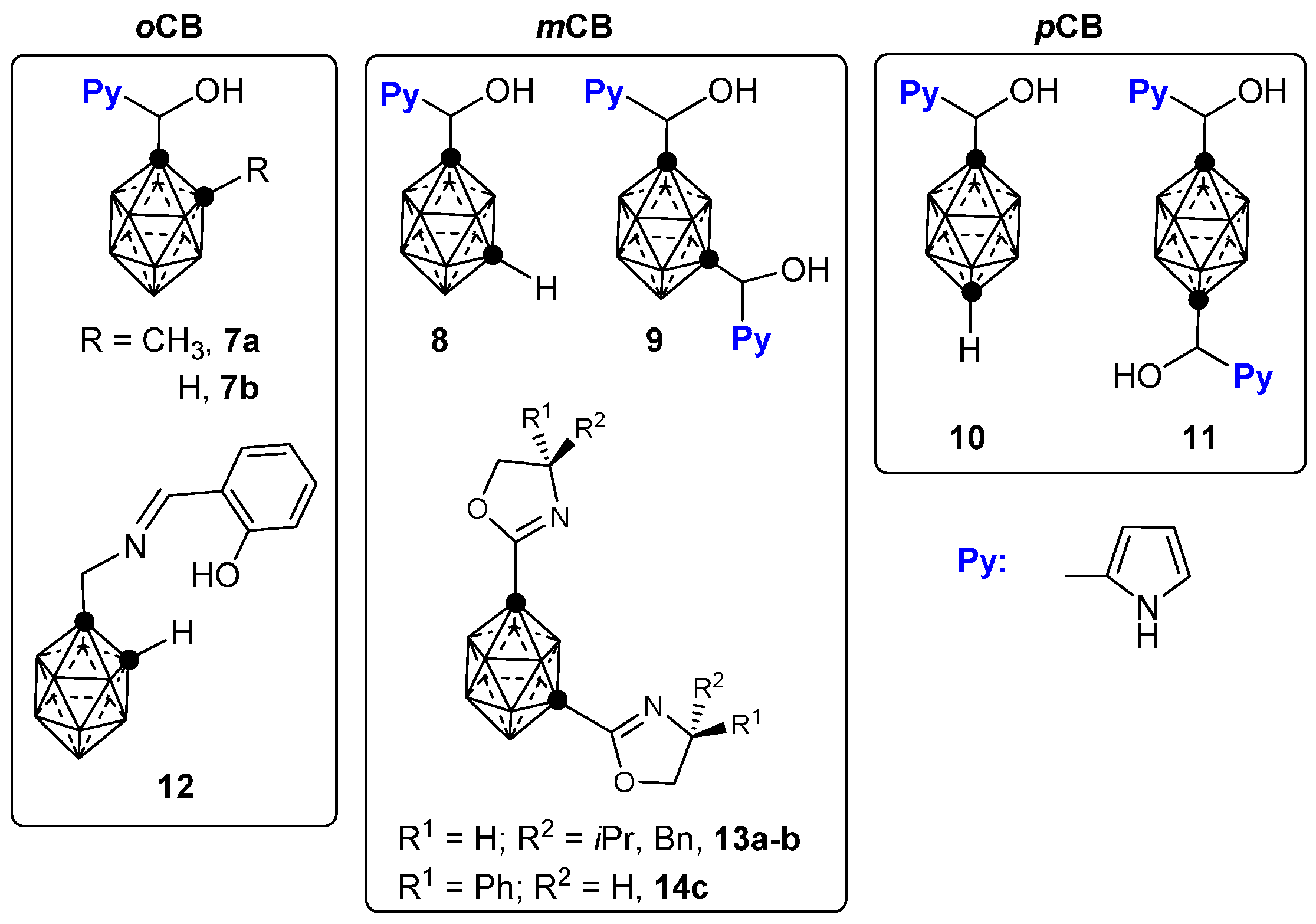
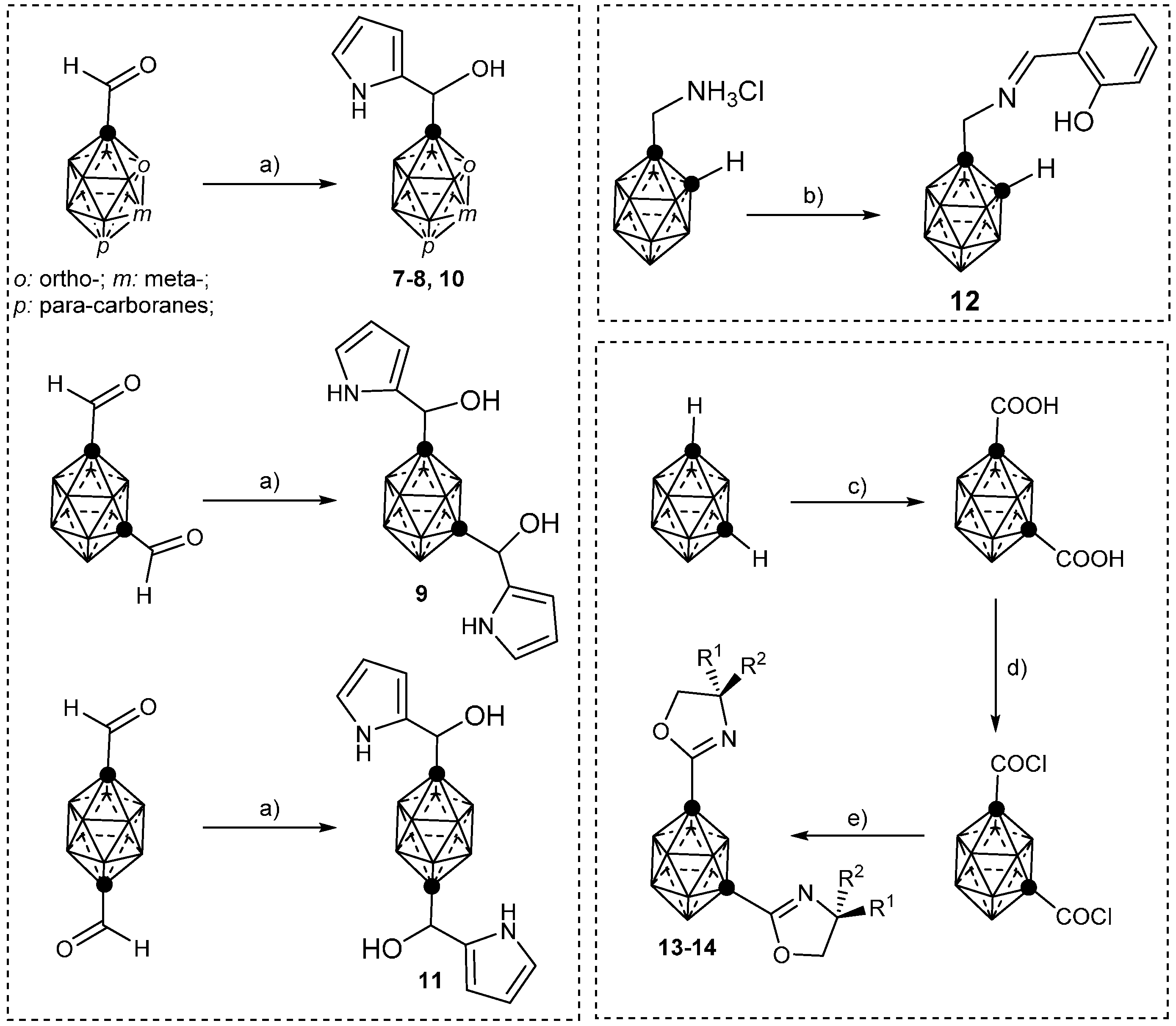
2.2. Other Closo-Carboranes Incorporating N and O Functionalities
3. Synthesis of Coordination Complexes and Properties
3.1. Complexes of Monosubstituted 1–4 and 12
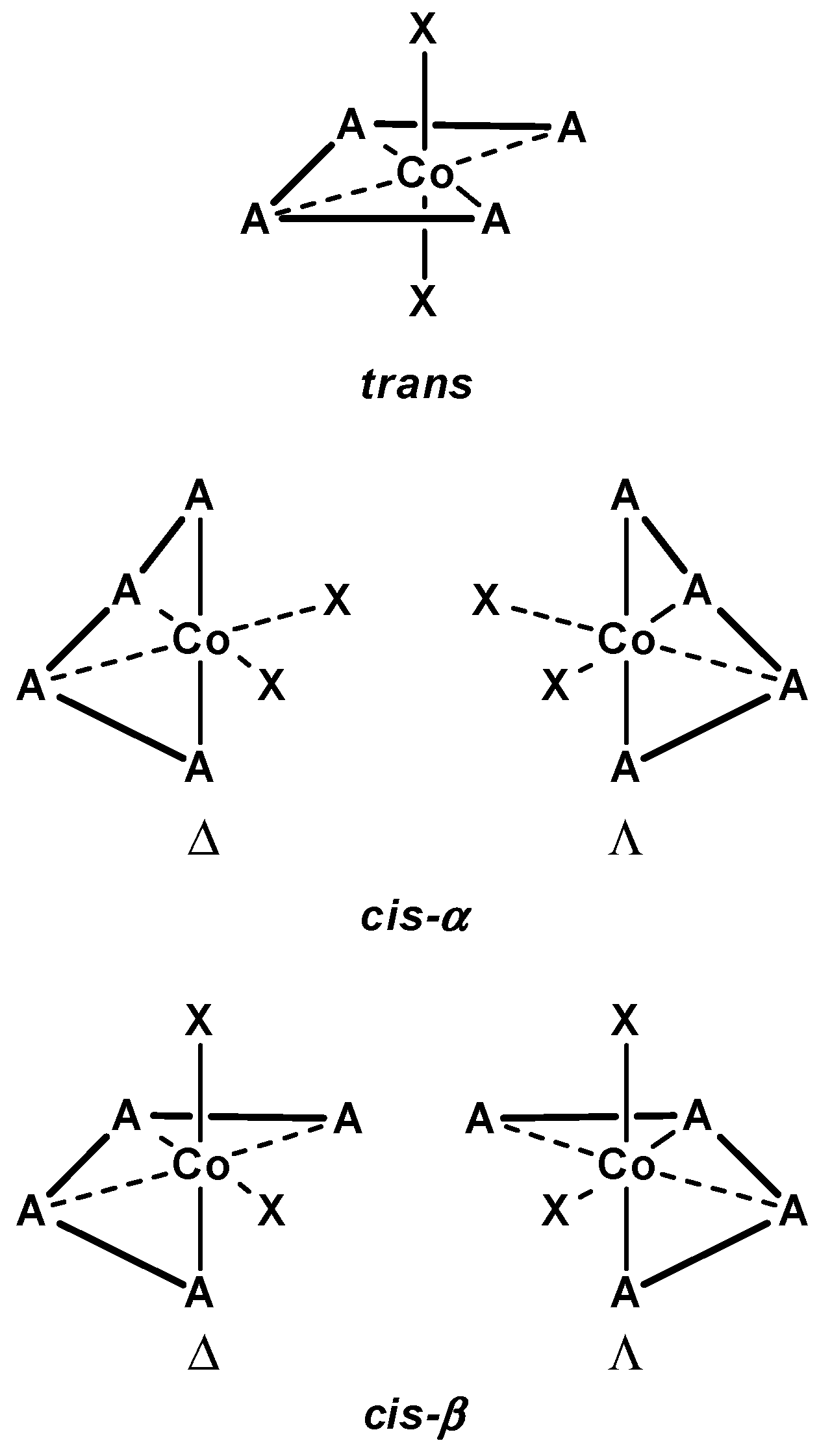
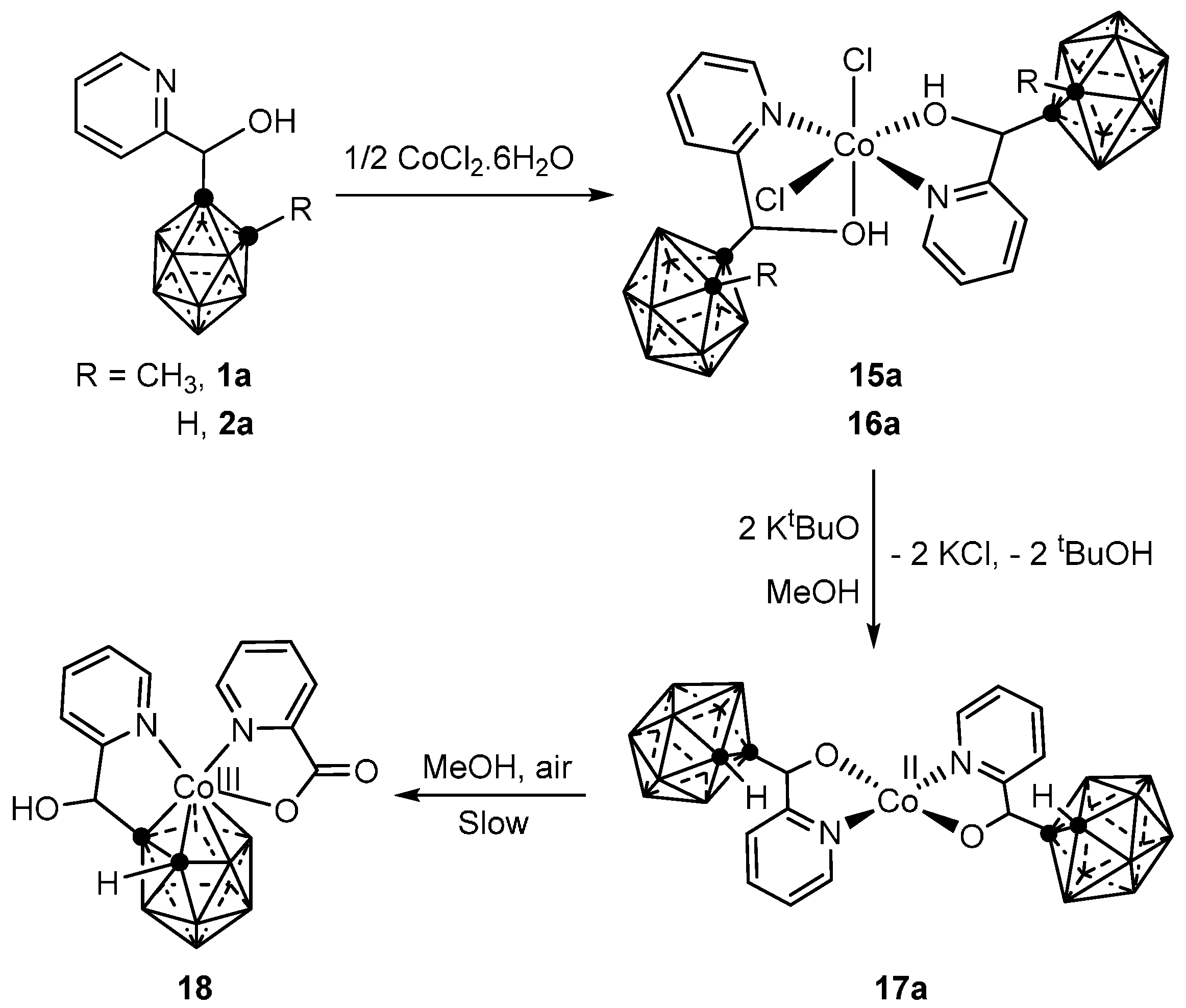
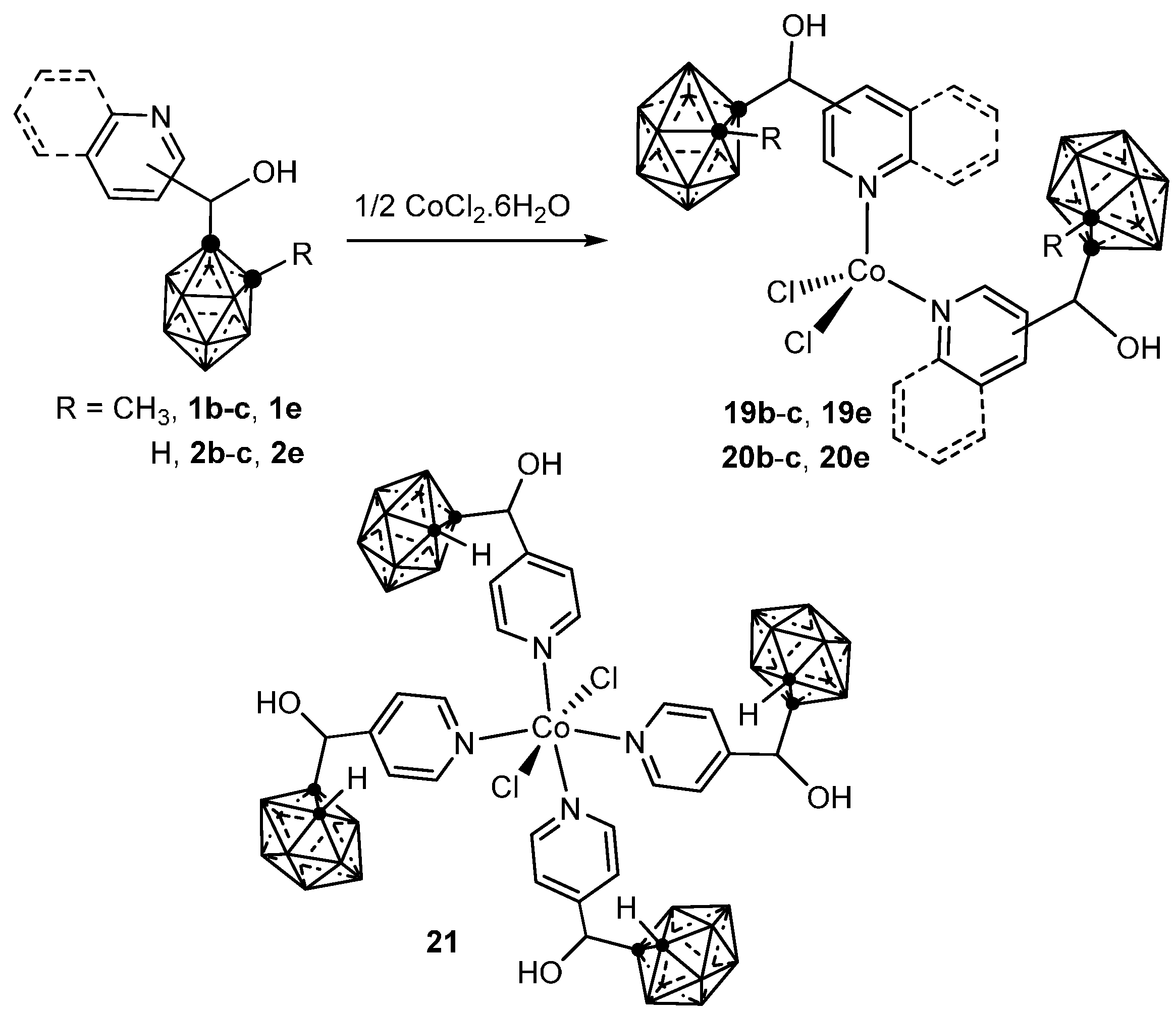
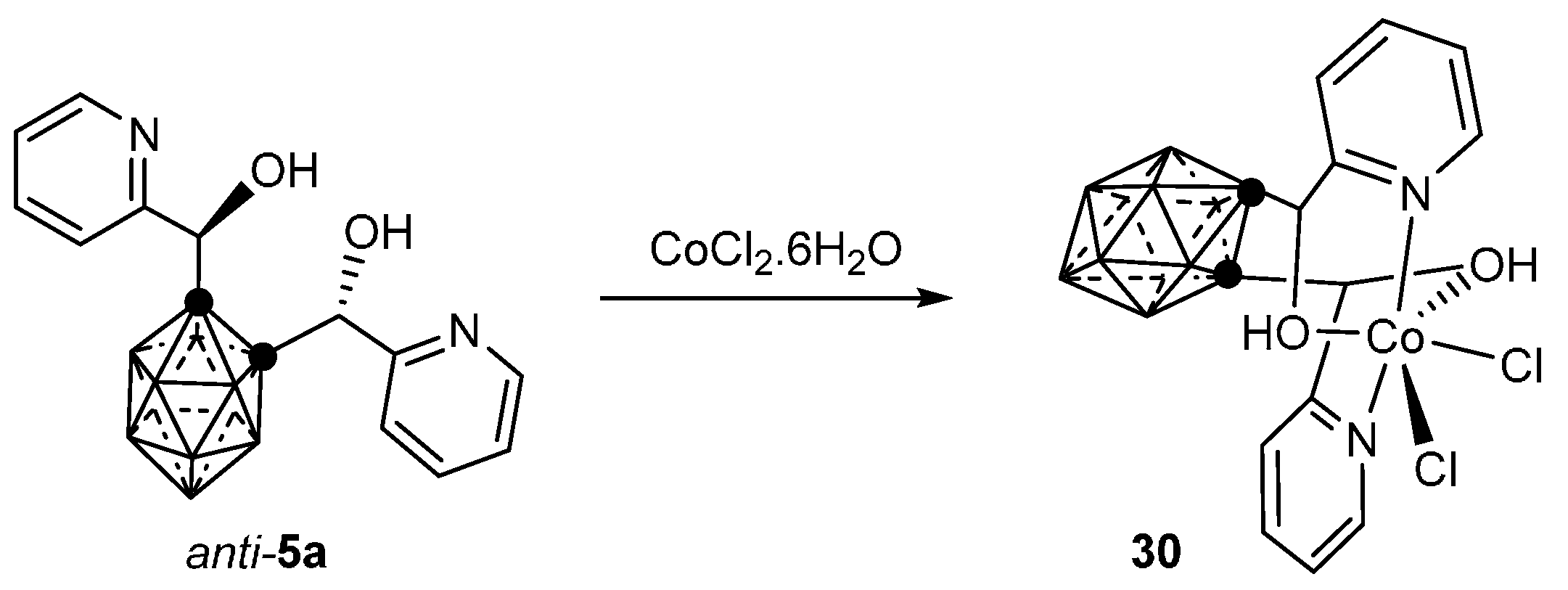
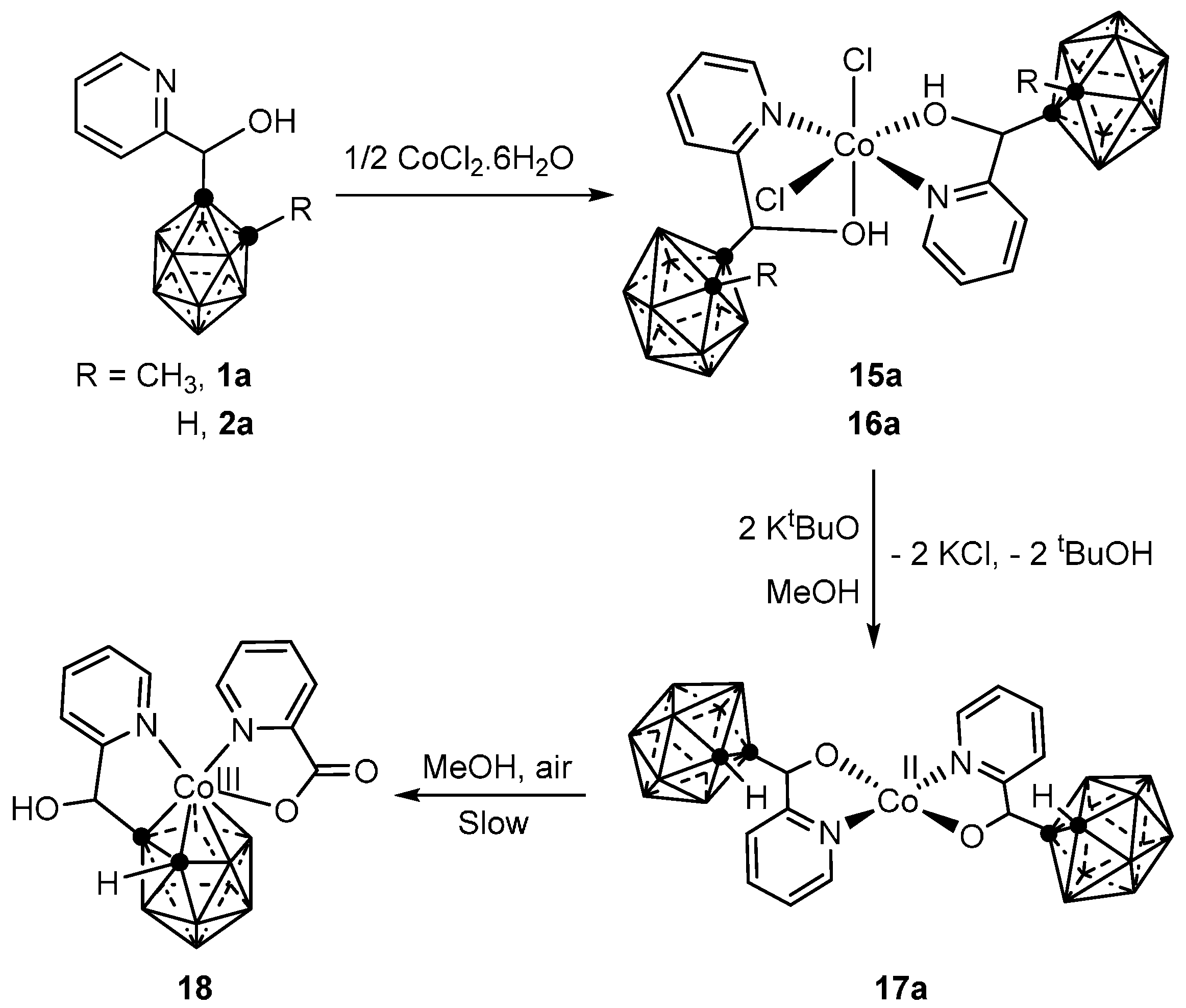
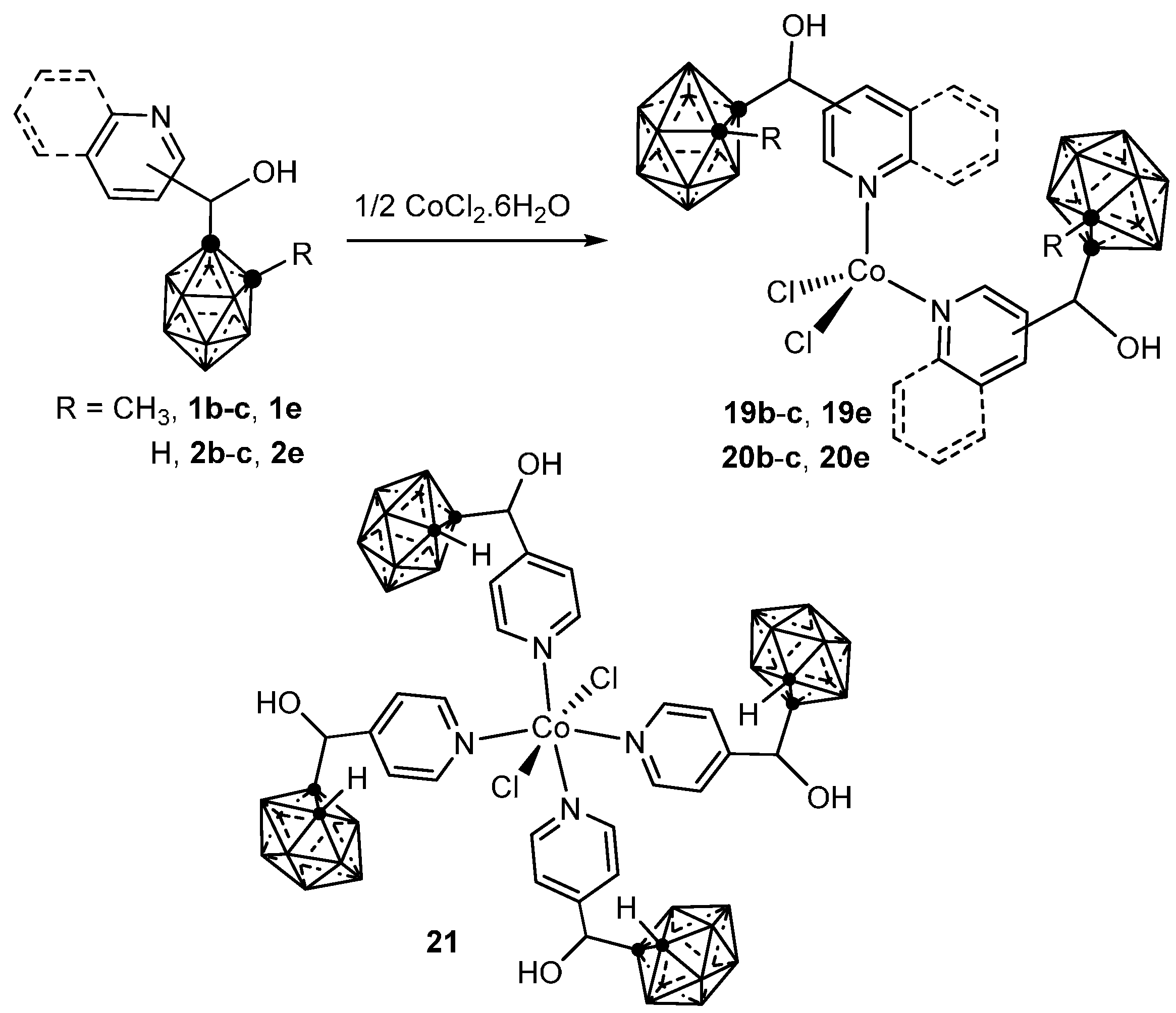
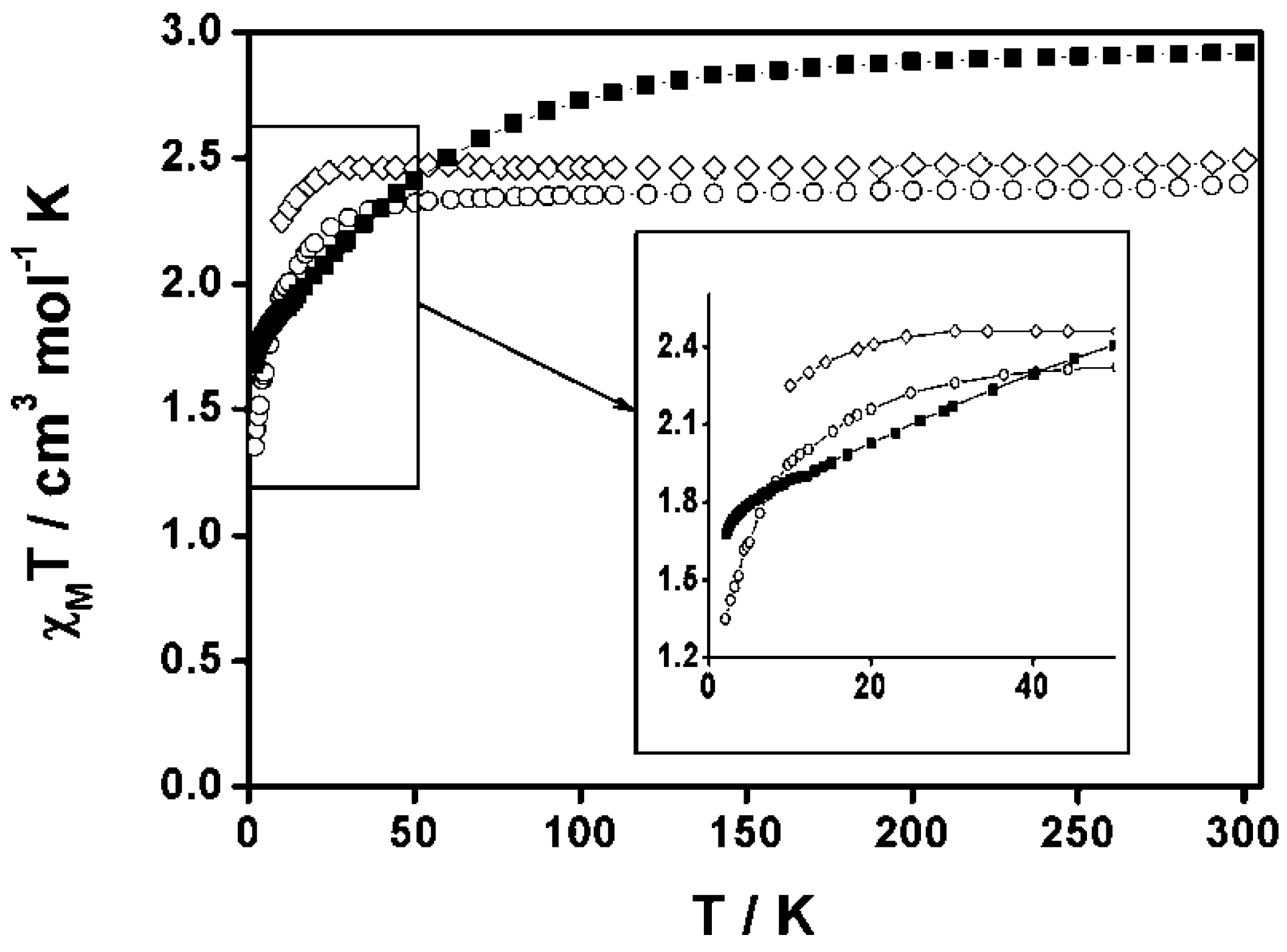
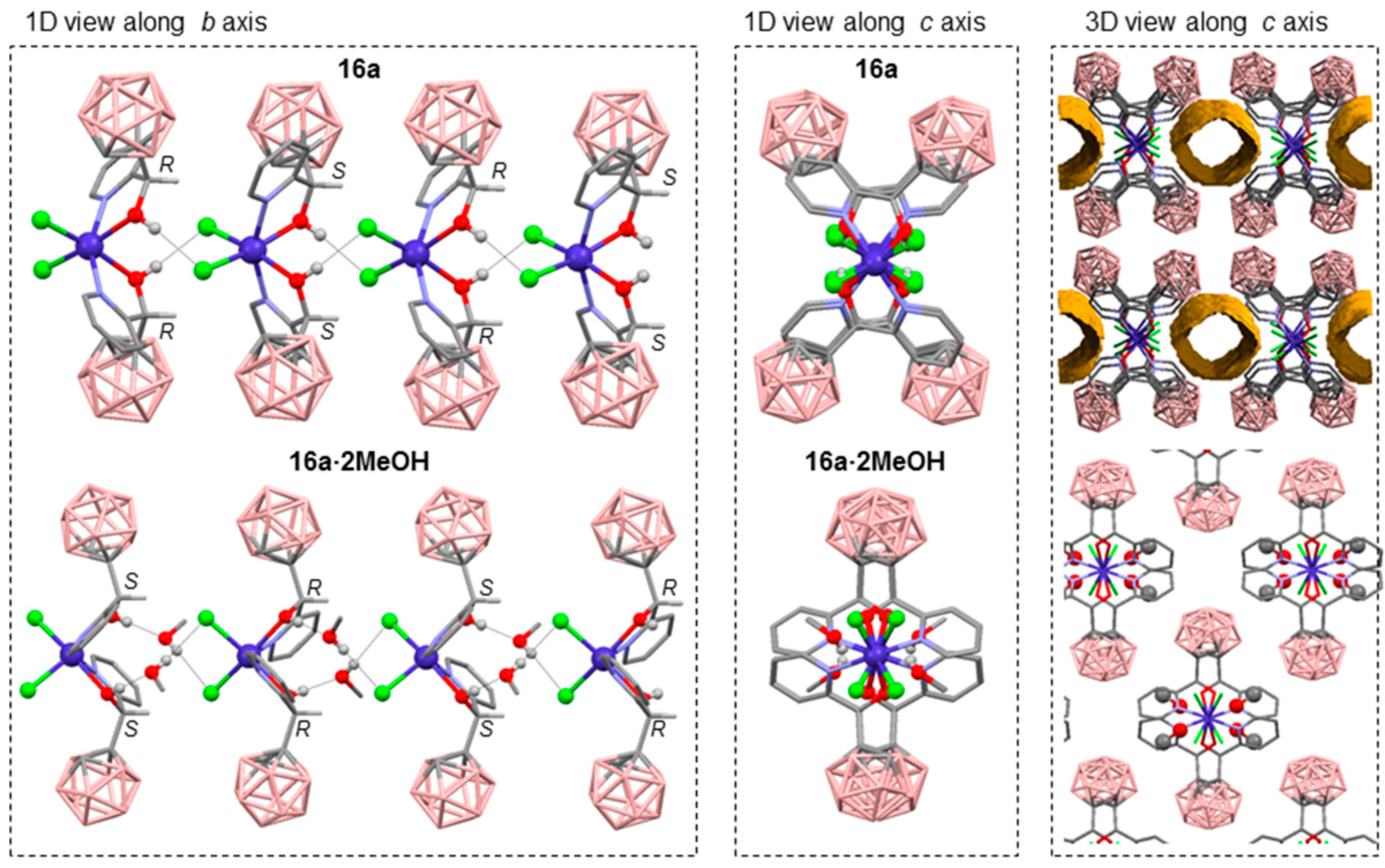
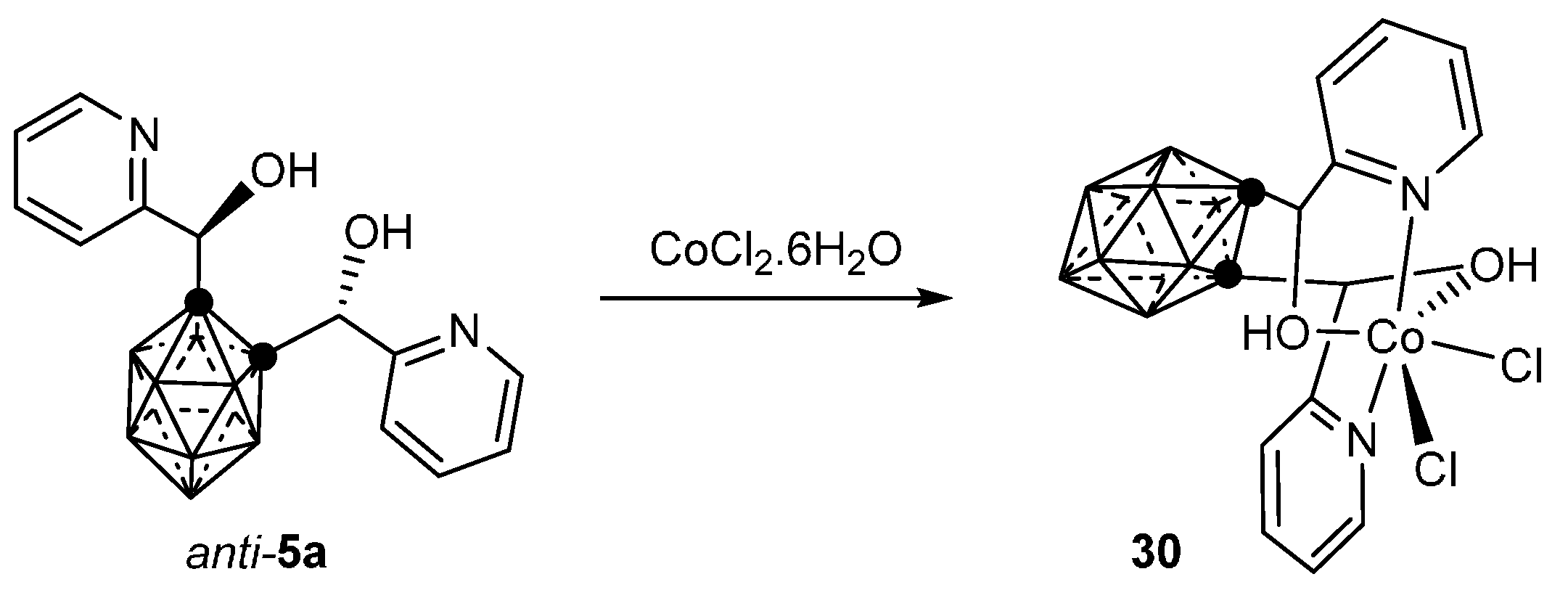
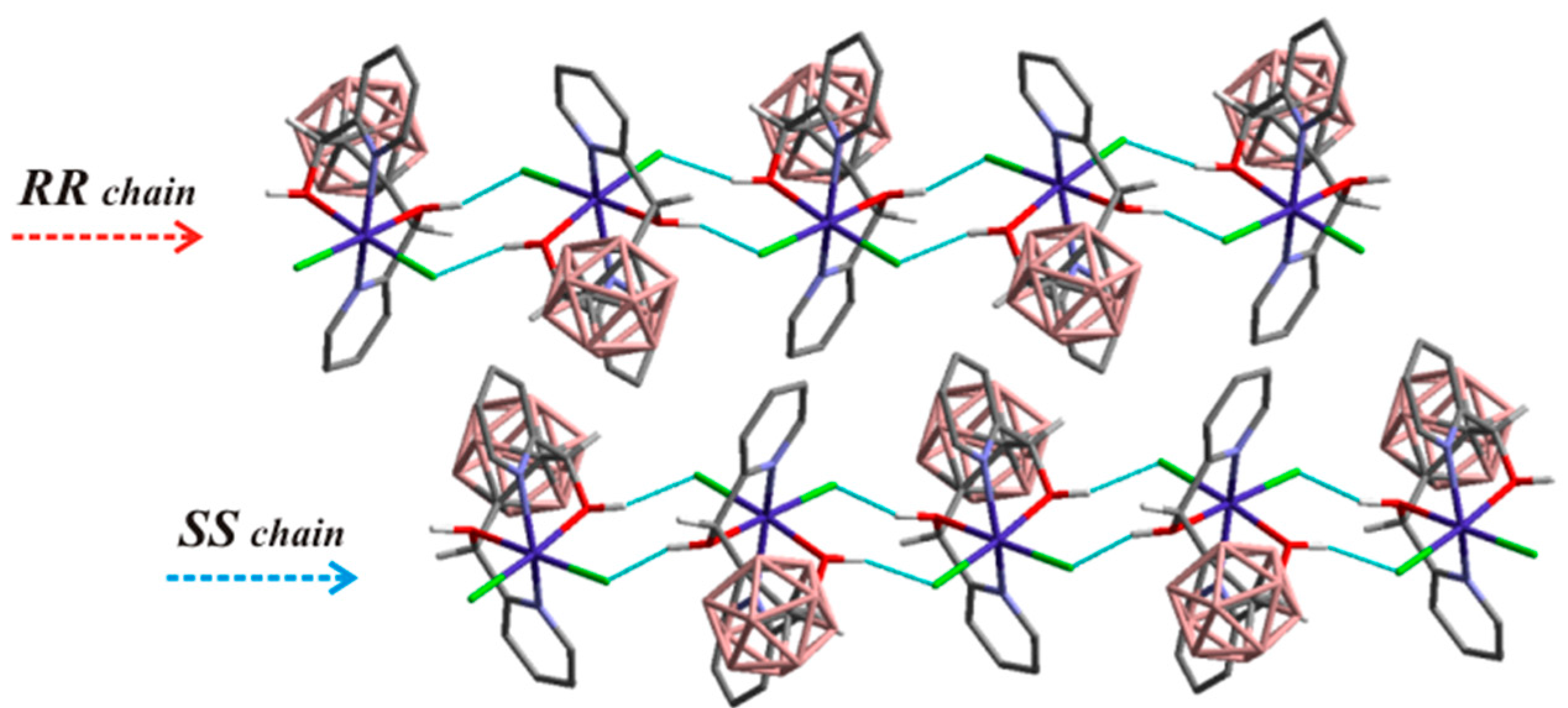
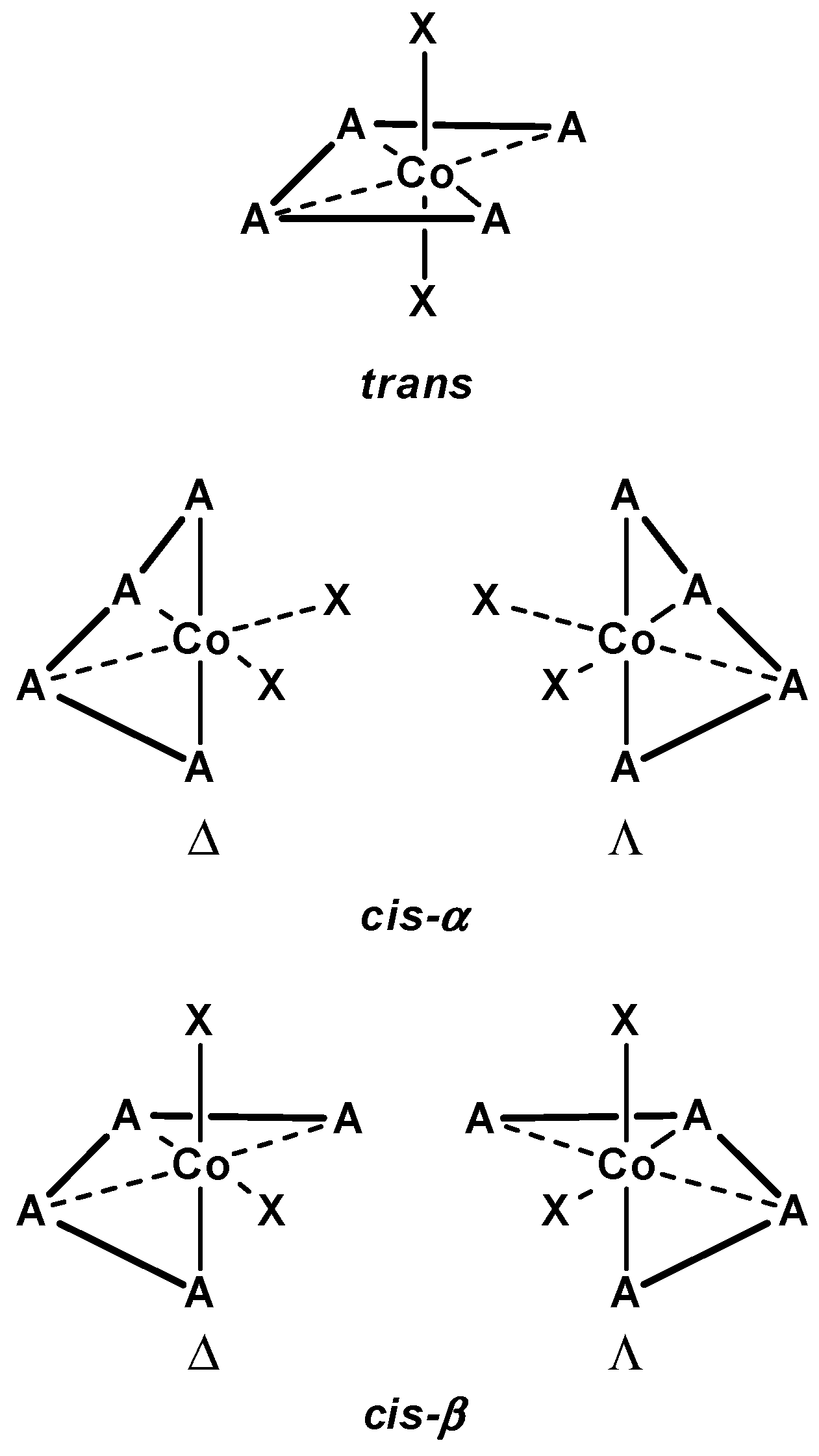
3.1.1. Cobalt
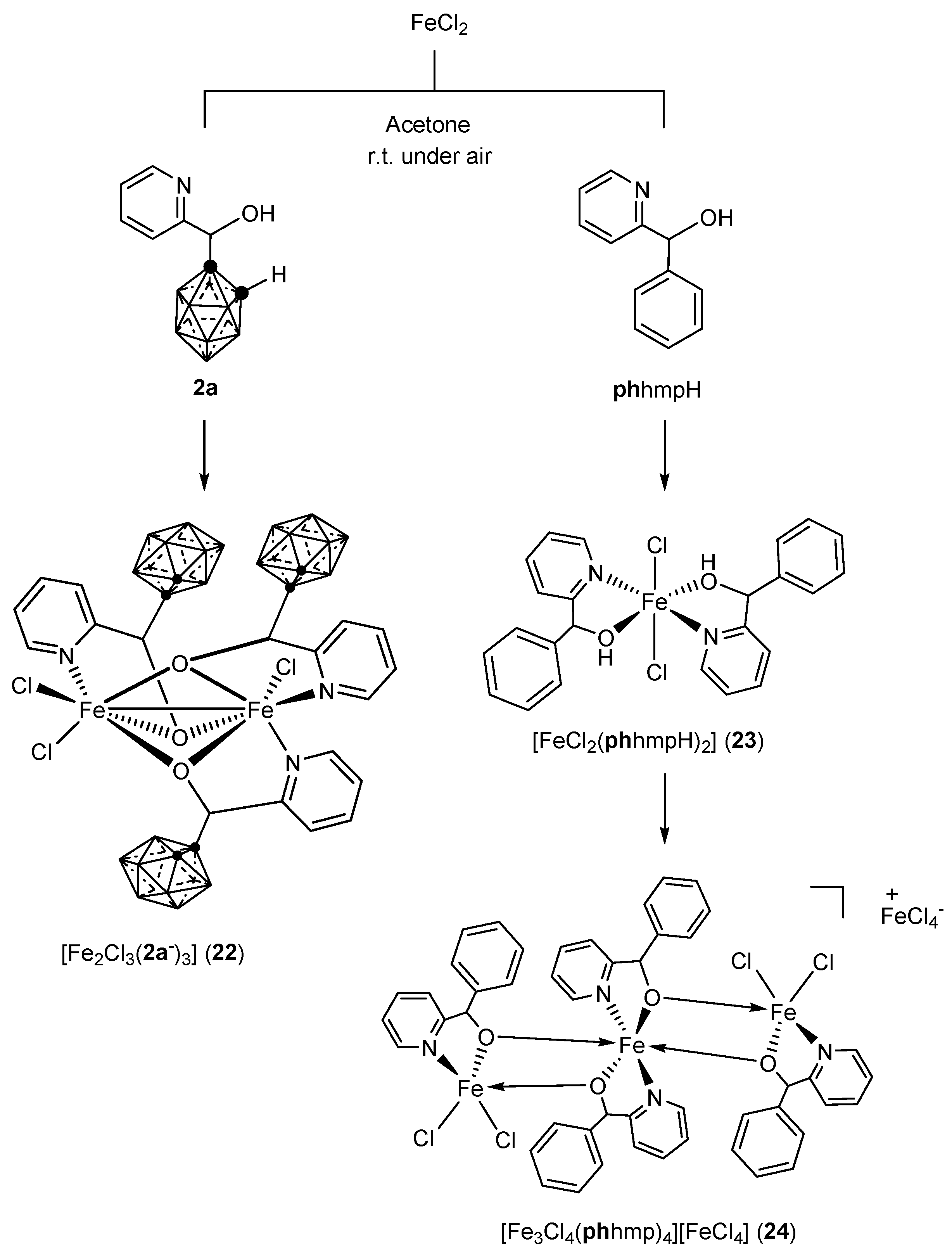
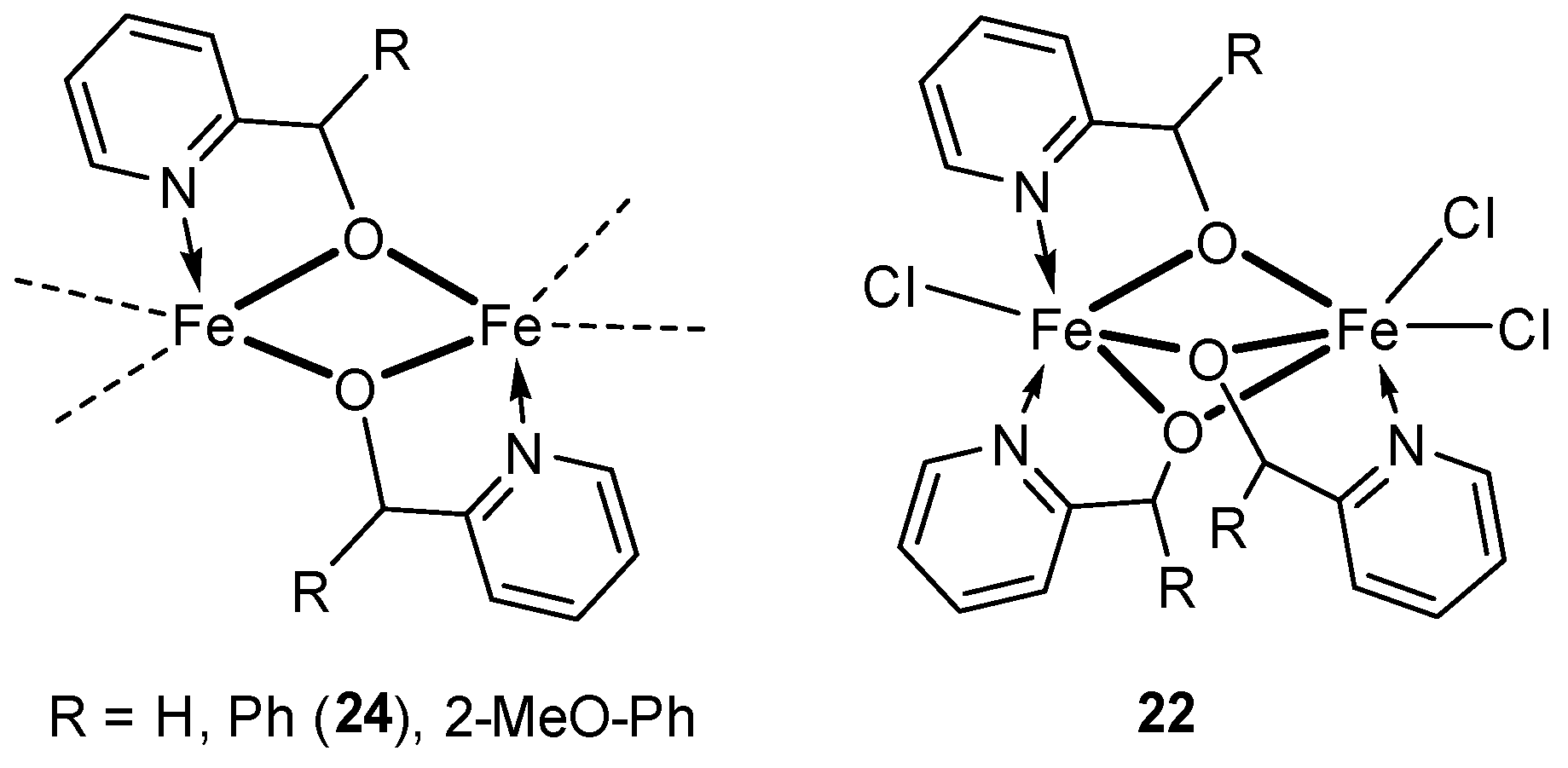
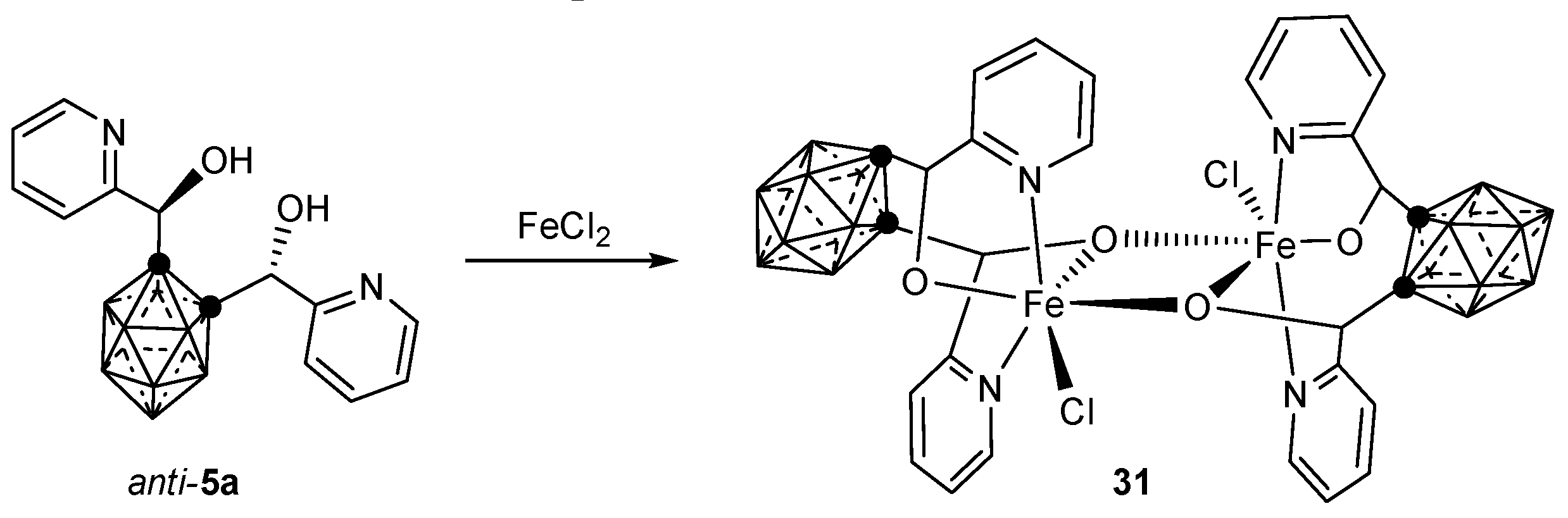
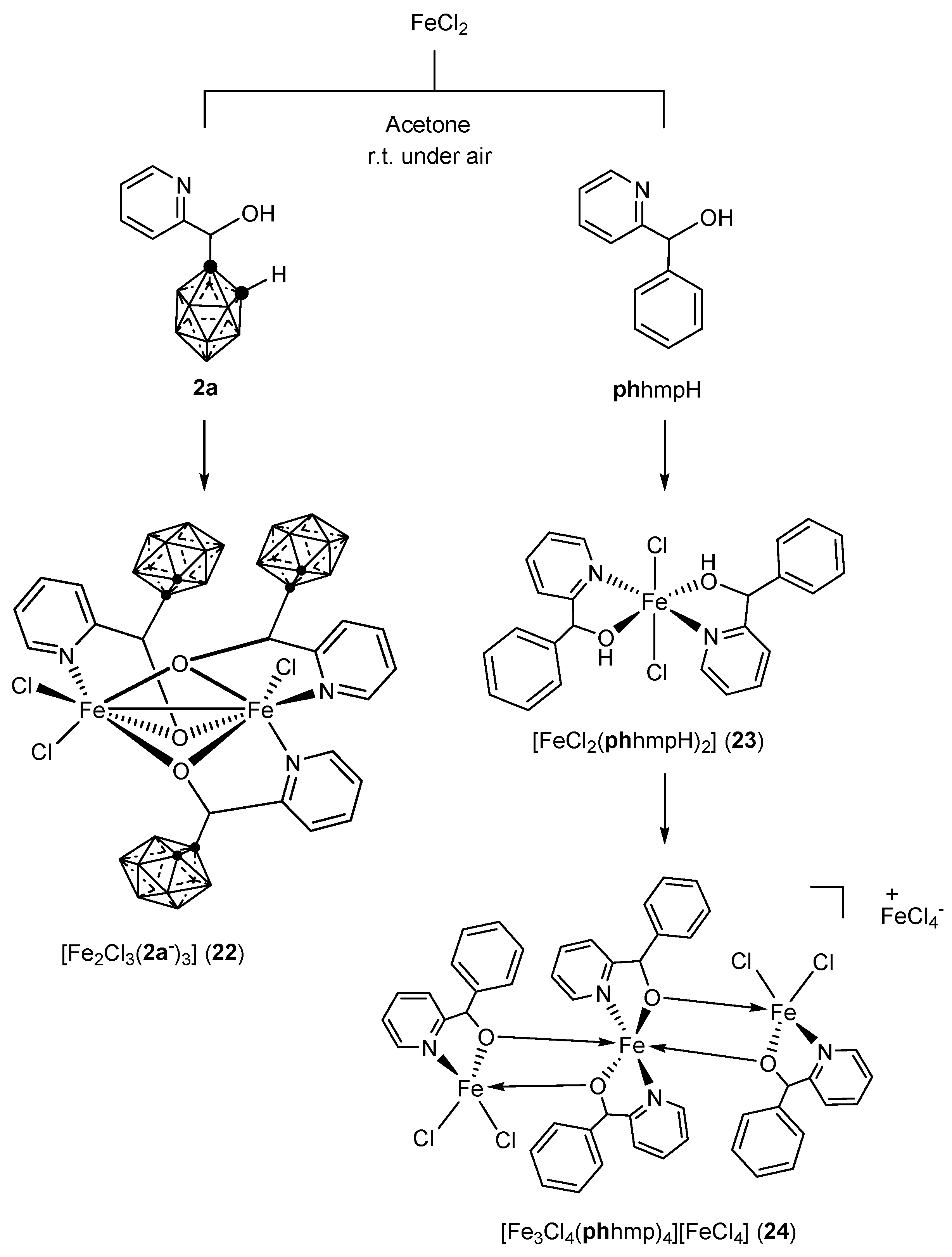
3.1.2. Iron
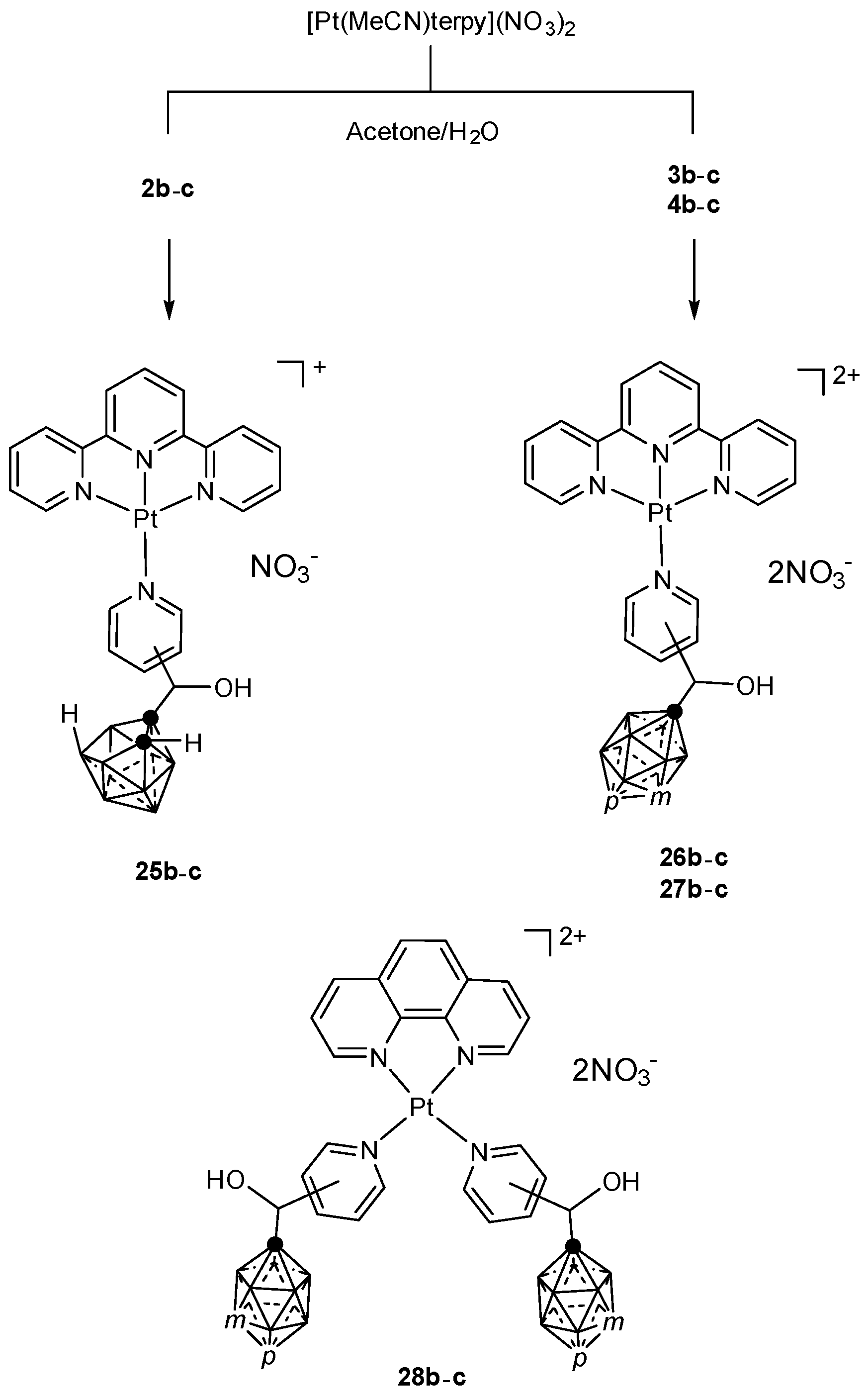
3.1.3. Platinum
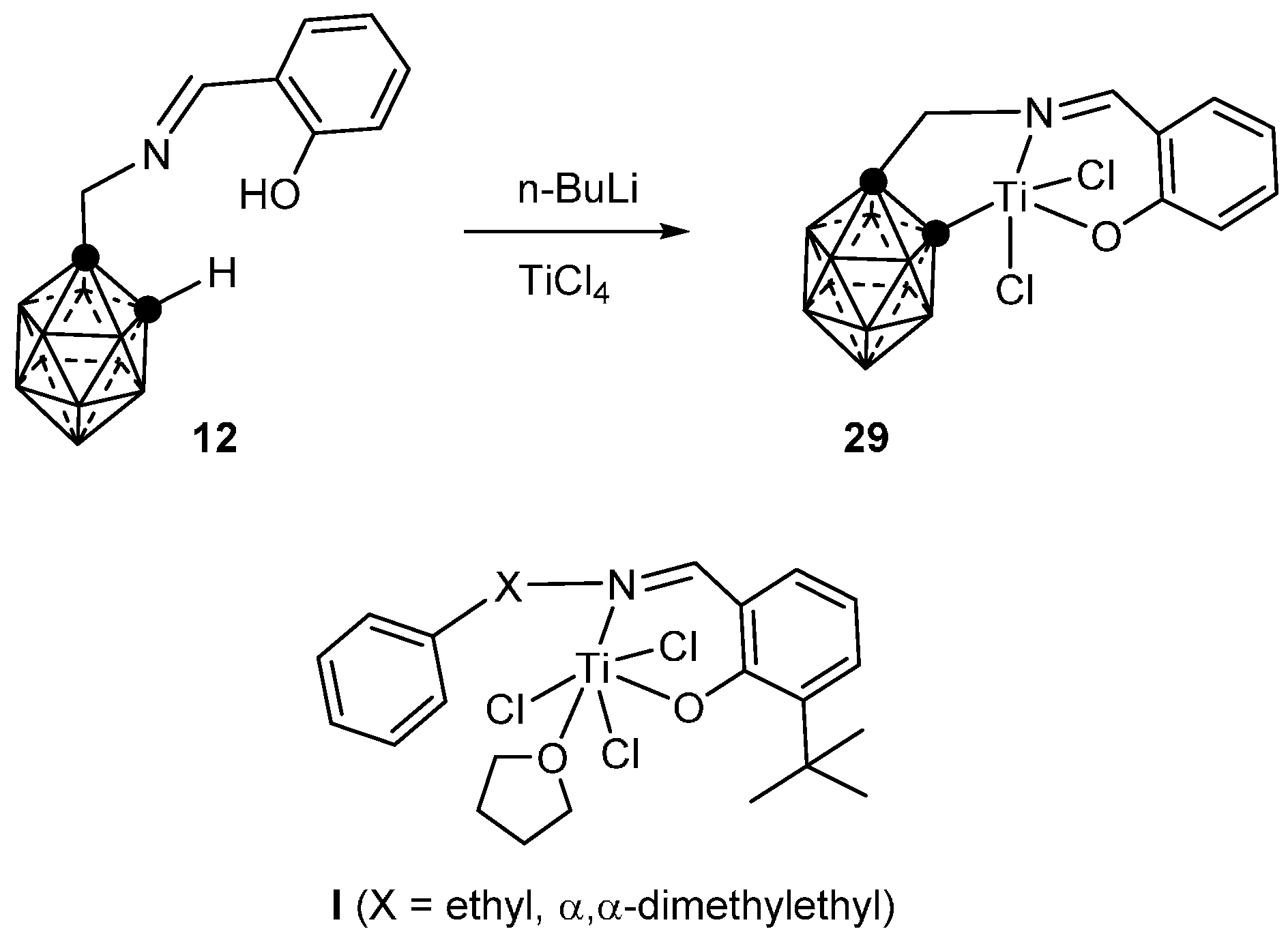
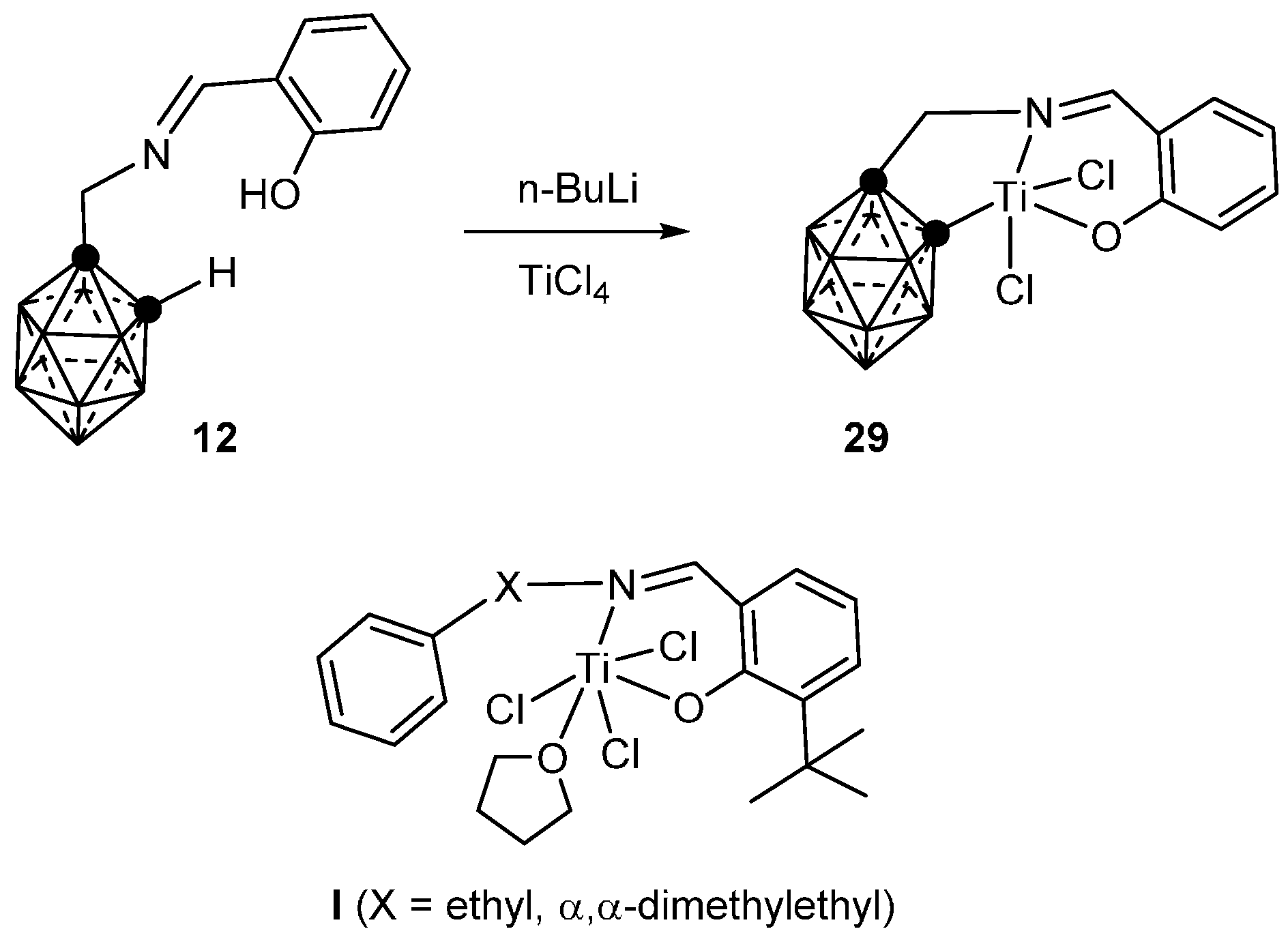
3.1.4. Titanium
3.2. Complexes of Disubstituted 5–6 and 13–14
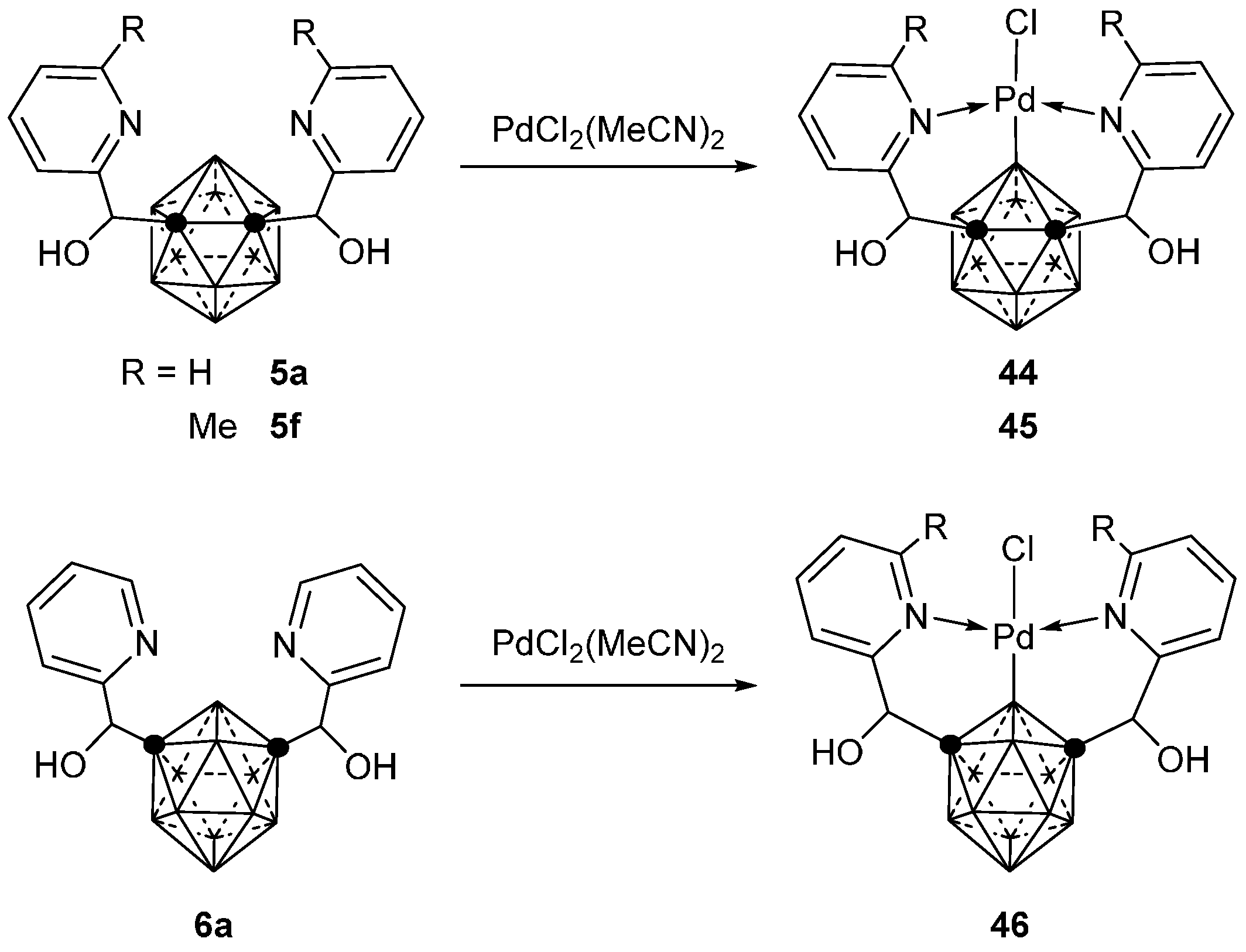
3.2.1. Cobalt
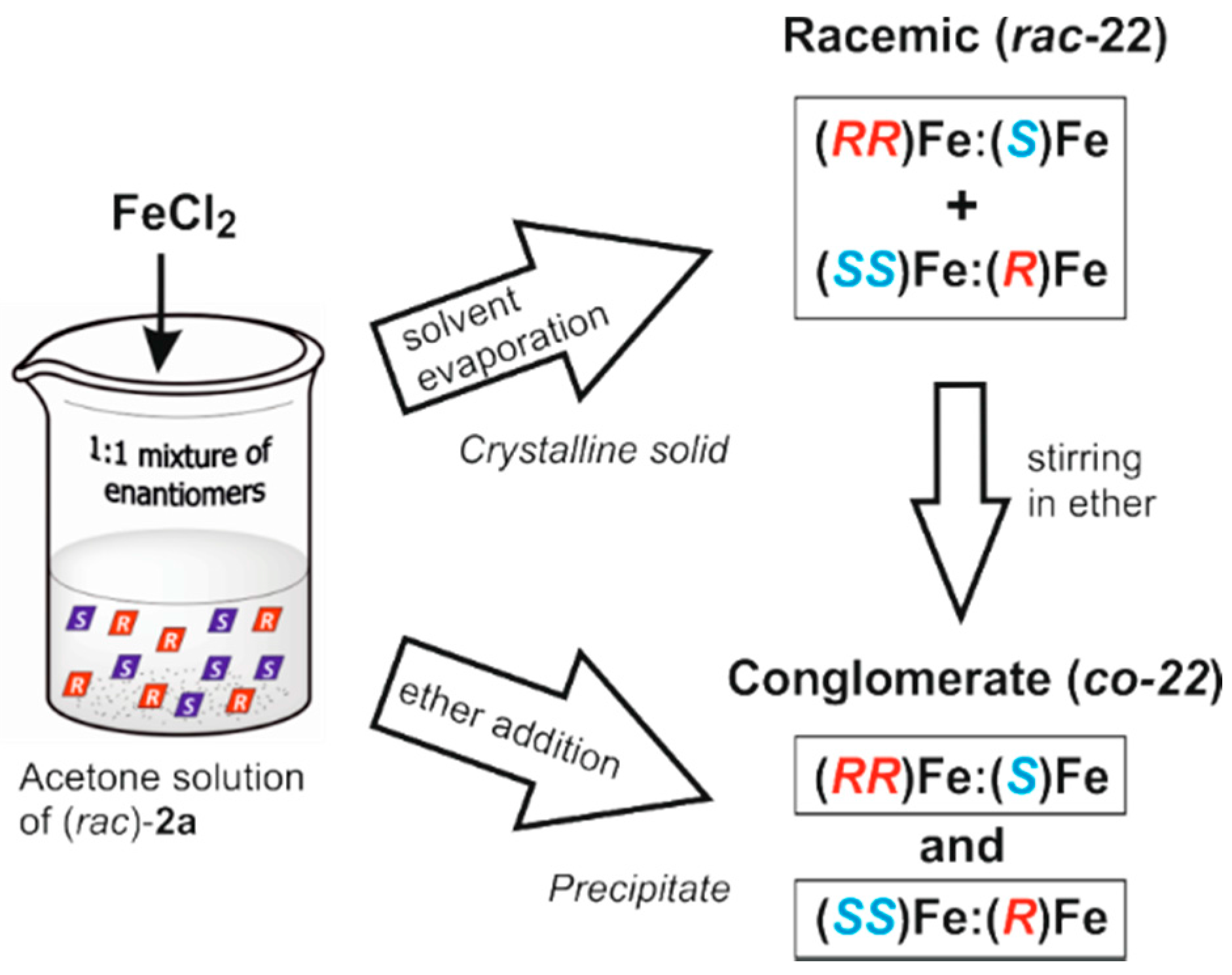
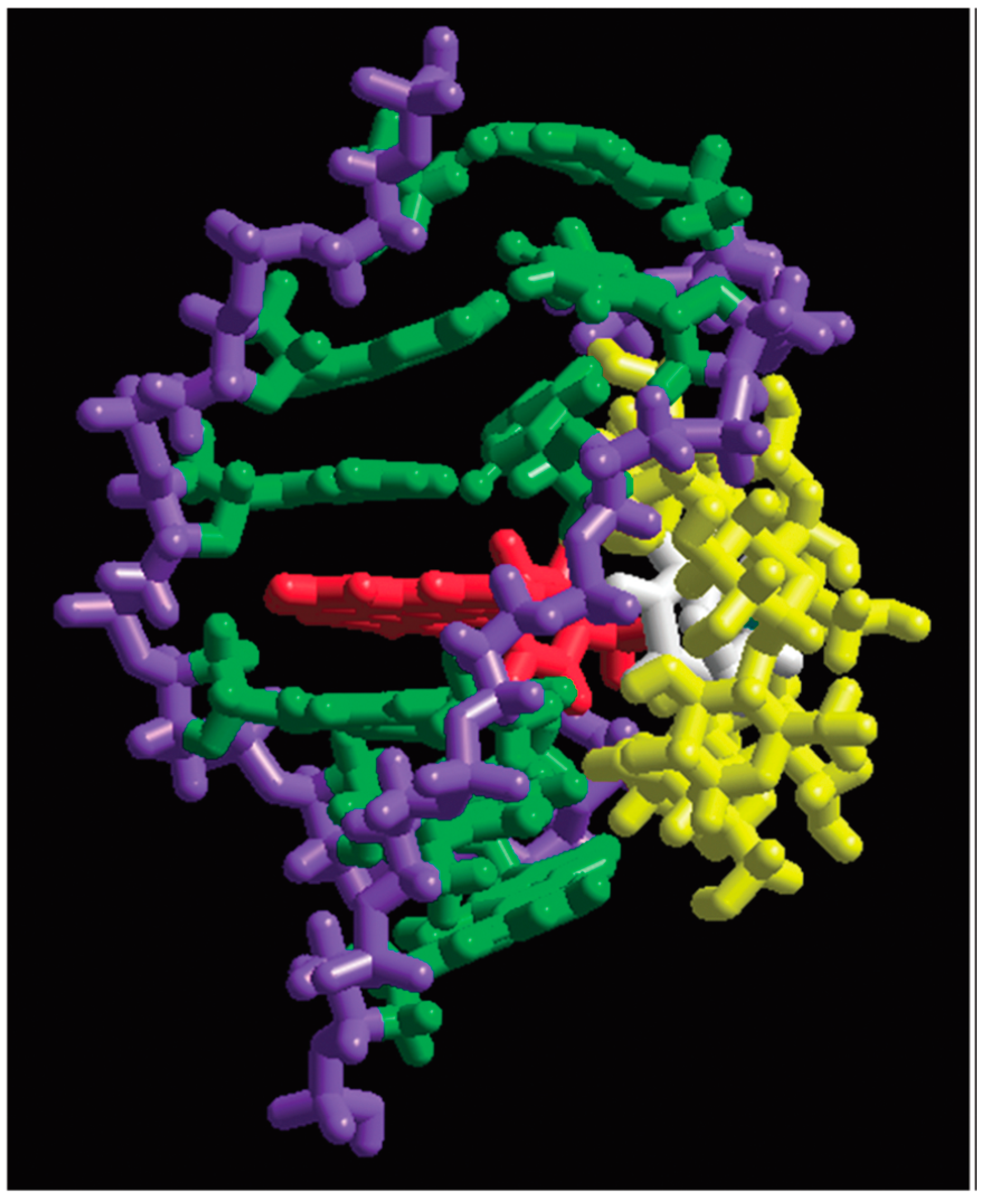
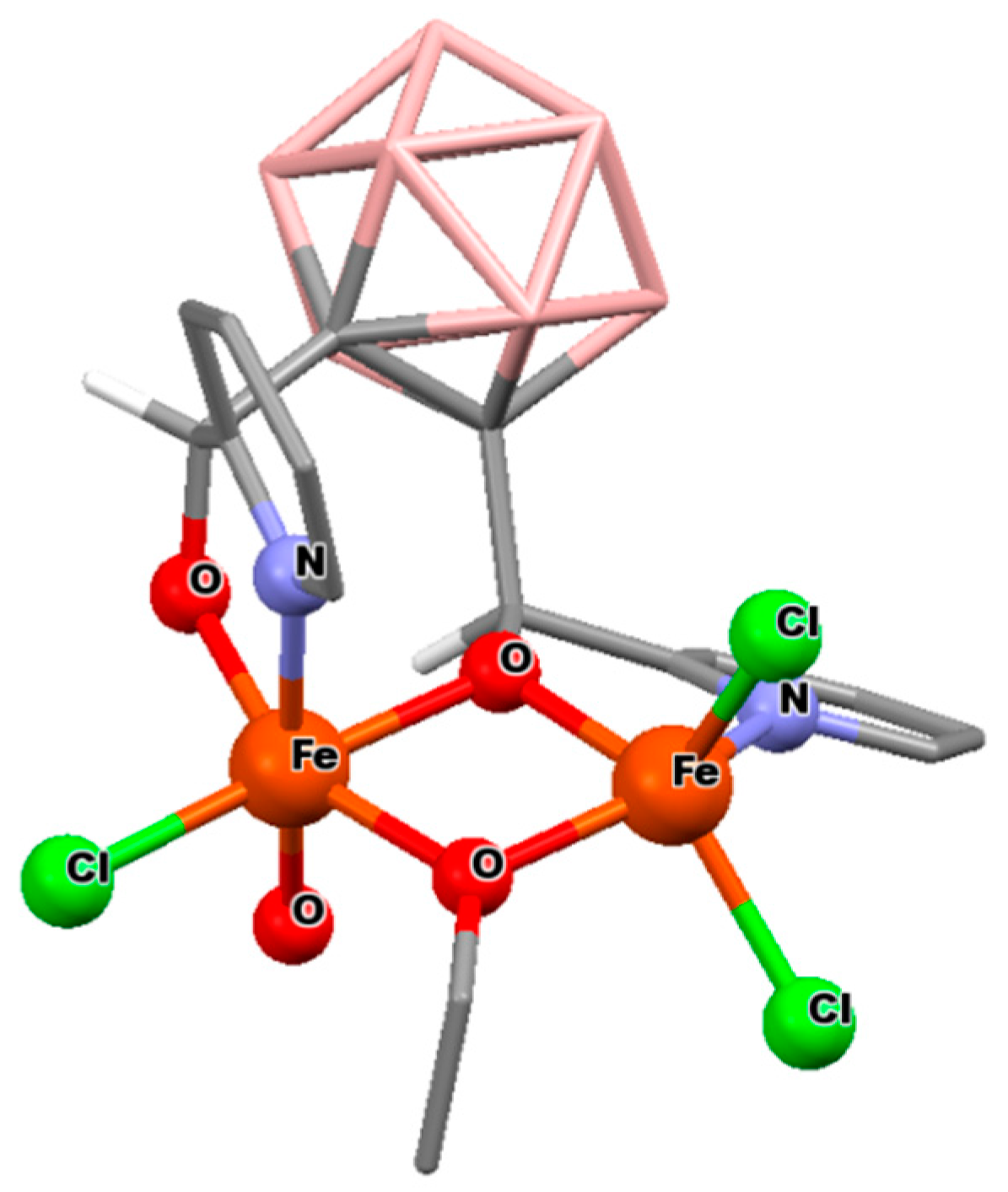
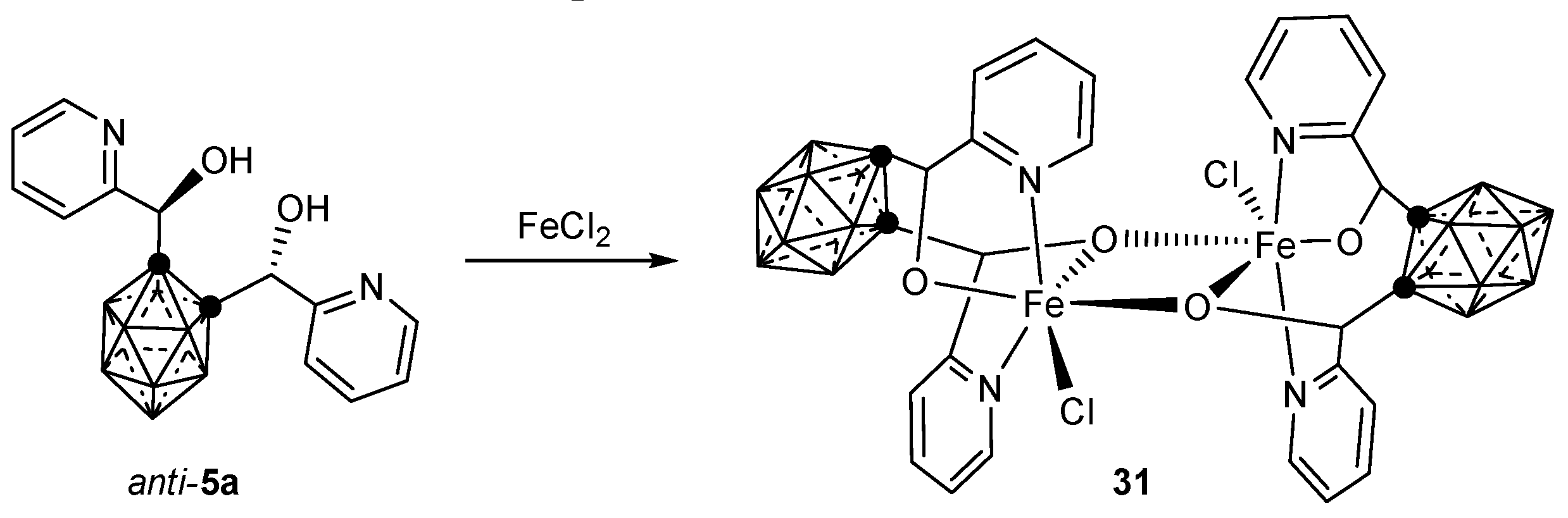
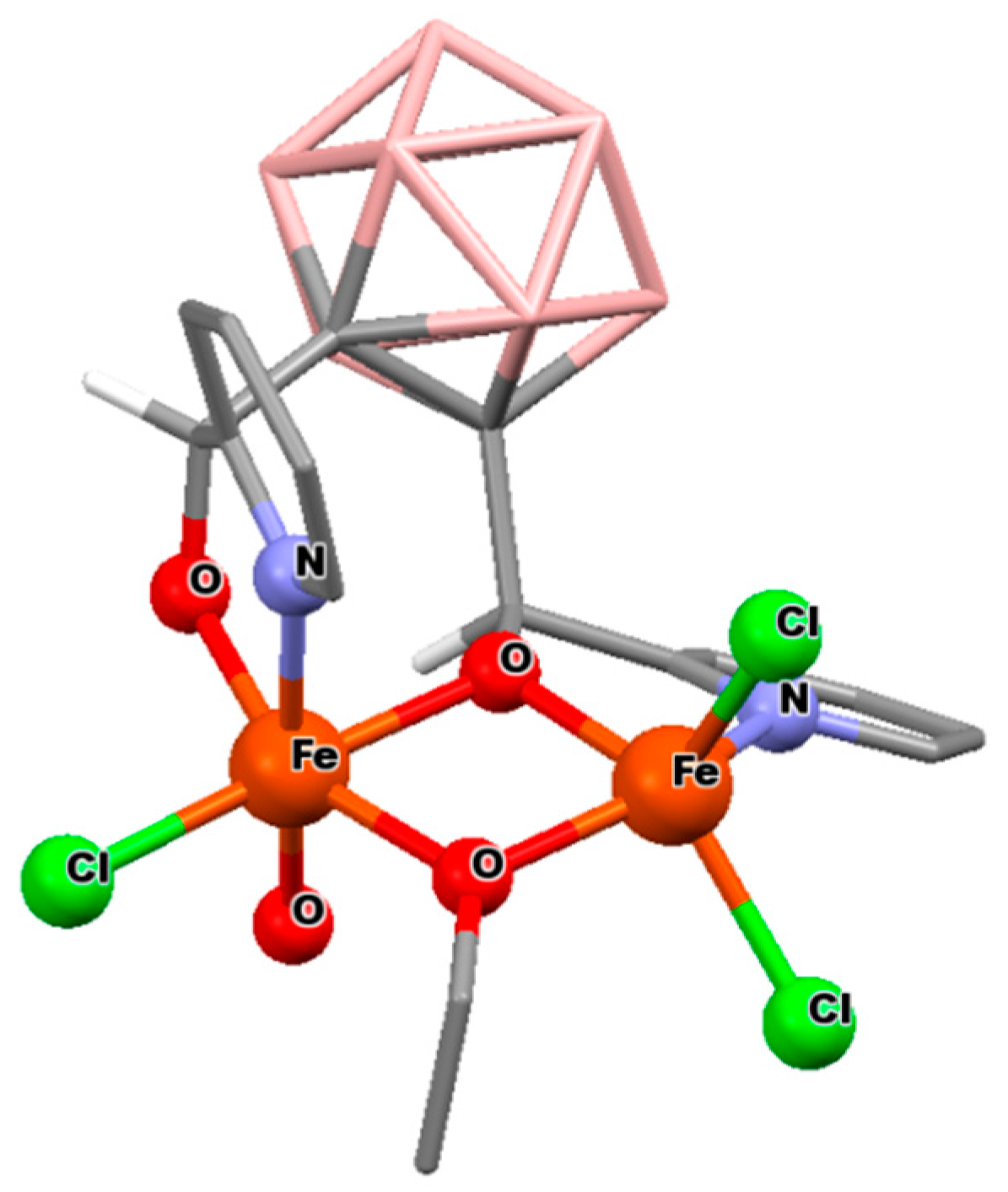
3.2.2. Iron
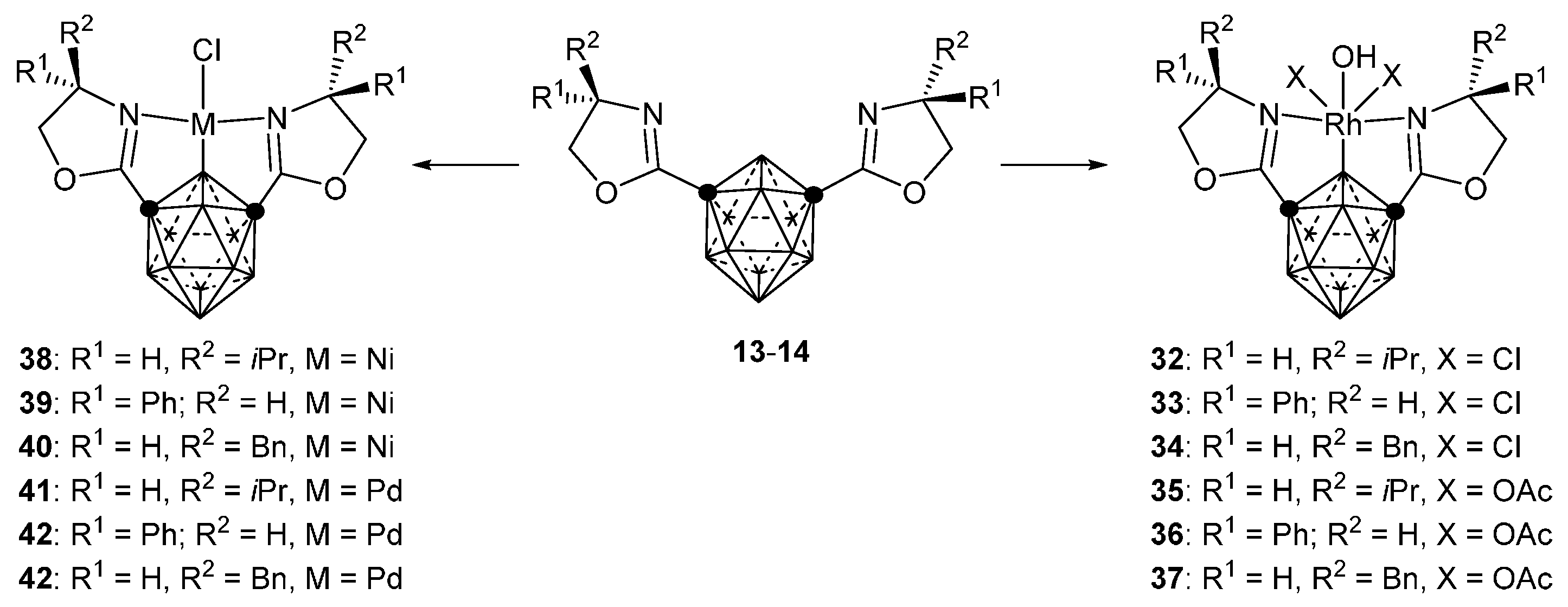
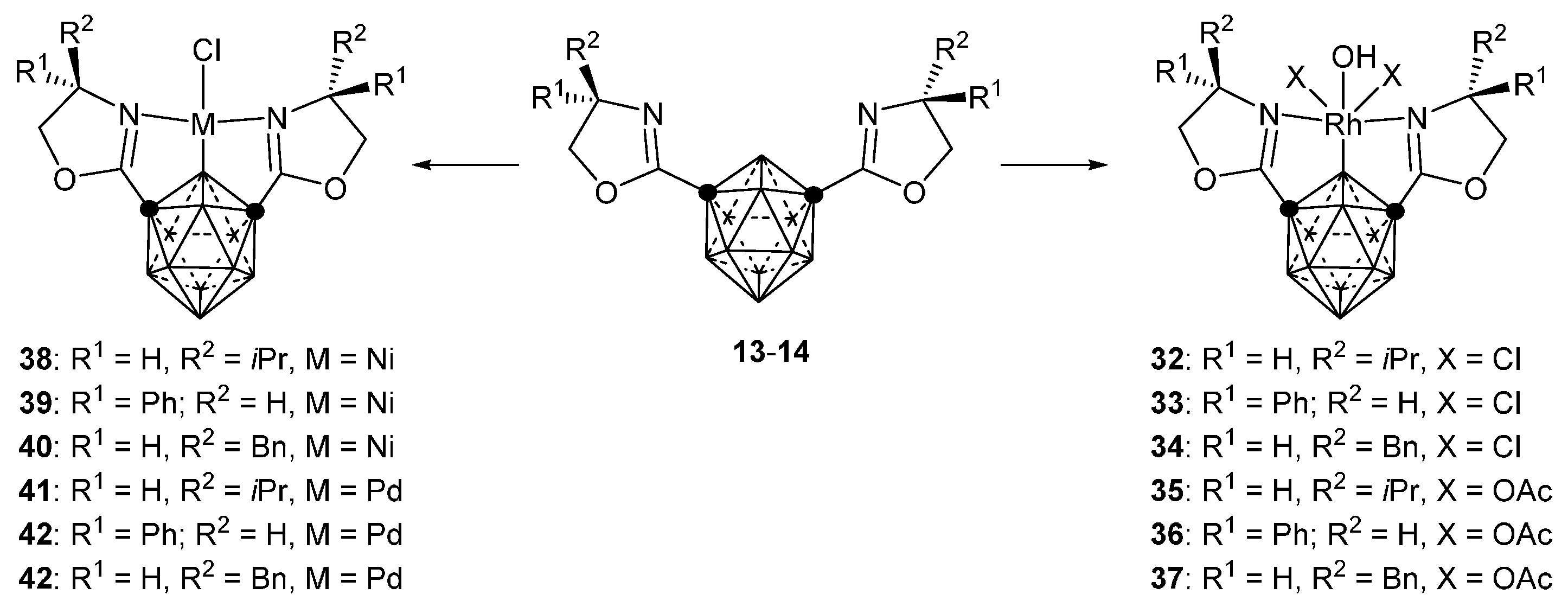
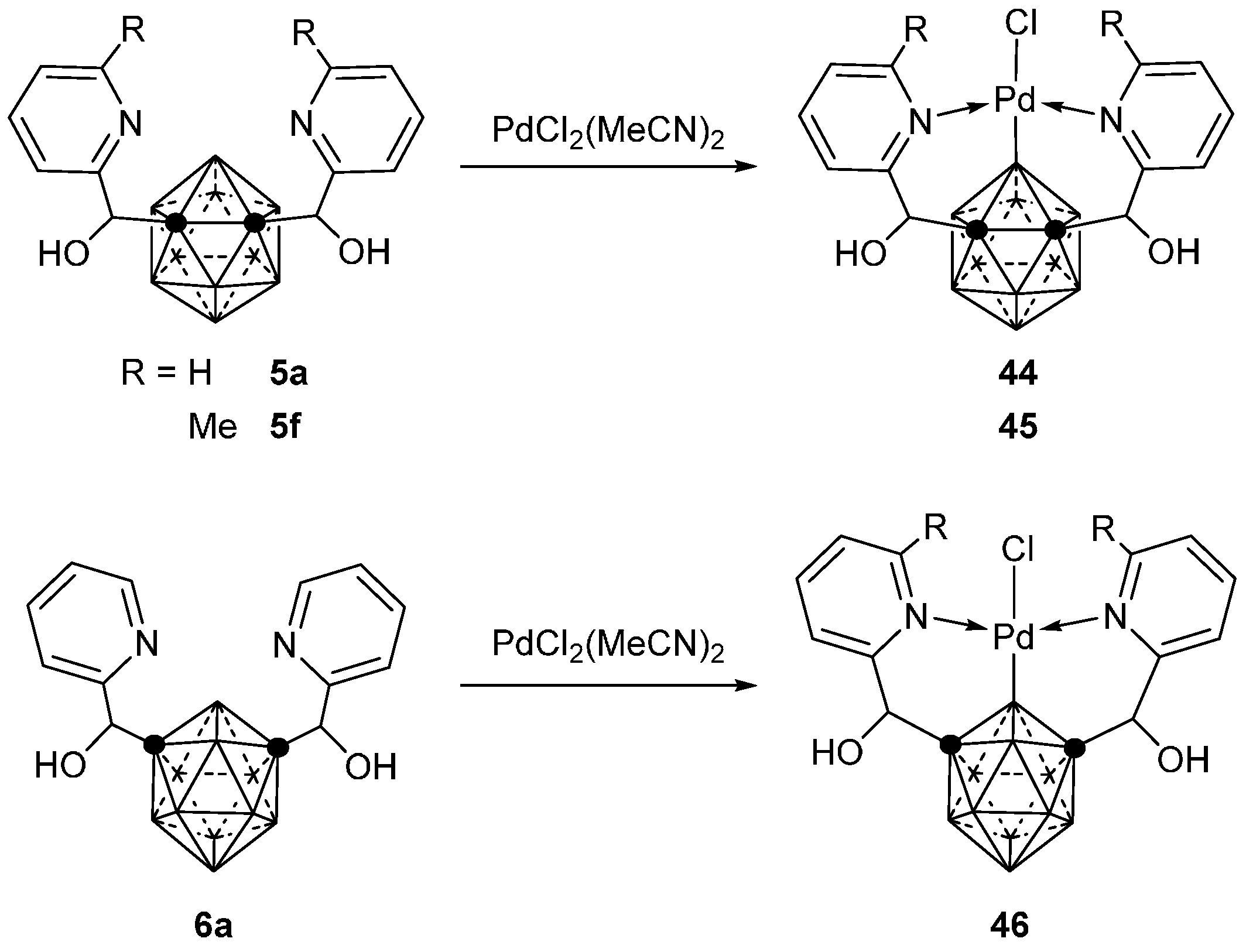
3.2.3. Nickel, Palladium and Rhodium Complexes
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grimes, R.N. Carboranes, 2nd ed.; Academic Press: Oxford, UK, 2011. [Google Scholar]
- Teixidor, F.; Viñas, C. Science of Synthesis; Thieme: Stuttgart, Germany, 2005; Volume 6, p. 1325. [Google Scholar]
- Hermansson, K.; Wójcik, M.; Sjöberg, S. o-, m-, and p-Carboranes and Their Anions: Ab Initio Calculations of Structures, Electron Affinities, and Acidities. Inorg. Chem. 1999, 38, 6039–6048. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.N. Carboranes in the chemist’s toolbox. Dalton Trans. 2015, 44, 5939–5956. [Google Scholar] [CrossRef] [PubMed]
- Olid, D.; Núñez, R.; Viñas, C.; Teixidor, F. Methods to produce B–C, B–P, B–N and B–S bonds in boron clusters. Chem. Soc. Rev. 2013, 42, 3318–3336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.H.; Hosmane, N.S. Carborane-based transition metal complexes and their catalytic applications for olefin polymerization: Current and future perspectives. J. Organomet. Chem. 2013, 747, 25–29. [Google Scholar]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in Drug Discovery: Carboranes as Unique Pharmacophores in Biologically Active Compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as Pharmacophores: Properties, Synthesis, and Application Strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef] [PubMed]
- Dash, B.P.; Satapathy, R.; Maguire, J.A.; Hosmane, N.S. Polyhedral boron clusters in materials science. New J. Chem. 2011, 35, 1955–1972. [Google Scholar] [CrossRef]
- Hardie, M.J. The use of carborane anions in coordination polymers and extended solids. J. Chem. Crystallogr. 2007, 37, 69–80. [Google Scholar] [CrossRef]
- Grimes, R.N. Metallacarboranes in the new millennium. Coord. Chem. Rev. 2000, 200–202, 773–811. [Google Scholar] [CrossRef]
- Adams, G.B.; O’Keeffe, M.; Ruoff, R.S. Van der Waals surface areas and volumes of fullerenes. J. Phys. Chem. 1994, 98, 9465–9469. [Google Scholar] [CrossRef]
- Fauchère, J.L.; Do, K.Q.; Jow, P.Y.C.; Hansch, C. Unusually strong lipophilicity of “fat” or “super” amino-acids, including a new reference value for glycine. Experientia 1980, 36, 1203–1204. [Google Scholar] [CrossRef] [PubMed]
- Zakharkin, L.I.; Grebennikov, A.V.; Kazantsev, A.V. Alkylation of carborane grignard reagents. Bull. Acad. Sci. USSR Div. Chem. Sci. 1968, 16, 1991–1993. [Google Scholar] [CrossRef]
- Viñas, C.; Benakki, R.; Teixidor, F.; Casabo, J. Dimethoxyethane as a Solvent for the Synthesis of C-Monosubstituted o-Carborane Derivatives. Inorg. Chem. 1995, 34, 3844–3845. [Google Scholar] [CrossRef]
- Gomez, F.A.; Hawthorne, M.F. A simple route to C-monosubstituted carborane derivatives. J. Org. Chem. 1992, 57, 1384–1390. [Google Scholar] [CrossRef]
- Popescu, A.R.; Musteti, A.D.; Ferrer-Ugalde, A.; Viñas, C.; Núñez, R.; Teixidor, F. Influential role of ethereal solvent on organolithium compounds: The case of carboranyllithium. Chem. Eur. J. 2012, 18, 3174–3184. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.-J.; Deng, W. Half-sandwich late transition metal complexes based on functionalized carborane ligands. Coord. Chem. Rev. 2016, 309, 21–35. [Google Scholar] [CrossRef]
- Popescu, A.R.; Teixidor, F.; Viñas, C. Metal promoted charge and hapticities of phosphines: The uniqueness of carboranylphosphines. Coord. Chem. Rev. 2014, 269, 54–84. [Google Scholar] [CrossRef]
- Yao, Z.J.; Jin, G.X. Transition metal complexes based on carboranyl ligands containing N, P, and S donors: Synthesis, reactivity and applications. Coord. Chem. Rev. 2013, 257, 2522–2535. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Machan, C.W.; Clingerman, D.J.; Rosen, M.S.; Wiester, M.J.; Kennedy, R.D.; Stern, C.L.; Sarjeant, A.A.; Mirkin, C.A. A coordination chemistry dichotomy for icosahedral carborane-based ligands. Nat. Chem. 2011, 3, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Crespo, O.; Gimeno, M.C.; Laguna, A. Carboranyl C-σ-bonded and C-functionalized carboranes as ligands in gold and silver chemistry. J. Organomet. Chem. 2009, 694, 1588–1598. [Google Scholar] [CrossRef]
- Kang, S.O.; Ko, J. Chemistry of o-carboranyl derivatives. In Advances in Organometallic Chemistry; Academic Press: Cambridge, MA, USA, 2001; Volume 47, pp. 61–99. [Google Scholar]
- Crespo, O.; Gimeno, M.C.; Laguna, A. Bis(diphenylphosphino)-ferrocene or -dicarba-closo-dodecaborane as ligands in gold and silver chemistry. Appl. Organomet. Chem. 2000, 14, 644–652. [Google Scholar] [CrossRef]
- Teixidor, F.; Viñas, C.; Demonceau, A.; Nuñez, R. Boron clusters: Do they receive the deserved interest? Pure Appl. Chem. 2003, 75, 1305–1313. [Google Scholar] [CrossRef]
- Teixidor, F.; Núñez, R.; Flores, M.A.; Demonceau, A.; Viñas, C. Forced exo-nido rhoda and ruthenacarboranes as catalyst precursors: A review. J. Organomet. Chem. 2000, 614–615, 48–56. [Google Scholar] [CrossRef]
- Teixidor, F.; Flores, M.A.; Viñas, C.; Sillanpää, R.; Kivekäs, R. Exo-nido-Cyclooctadienerhodacarboranes: Synthesis, reactivity, and catalytic properties in alkene hydrogenation. J. Am. Chem. Soc. 2000, 122, 1963–1973. [Google Scholar] [CrossRef]
- Teixidor, F.; Flores, M.A.; Viñas, C.; Kivekäs, R.; Sillanpää, R. [Rh(7-SPh-8-Me-7,8-C2B9H10)(PPh 3)2]: A new rhodacarborane with enhanced activity in the hydrogenation of 1-alkenes. Angew. Chem. Int. Ed. 1996, 35, 2251–2253. [Google Scholar] [CrossRef]
- Mitani, M.; Saito, J.; Ishii, S.I.; Nakayama, Y.; Makio, H.; Matsukawa, N.; Matsui, S.; Mohri, J.I.; Furuyama, R.; Terao, H.; et al. FI catalysts: New olefin polymerization catalysts for the creation of value-added polymers. Chem. Rec. 2004, 4, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Hemmert, C.; Meunier, B. A macrocyclic chiral manganese(III) Schiff base complex as an efficient catalyst for the asymmetric epoxidation of olefins. J. Catal. 2005, 234, 250–255. [Google Scholar] [CrossRef]
- Kruppa, M.; König, B. Reversible coordinative bonds in molecular recognition. Chem. Rev. 2006, 106, 3520–3560. [Google Scholar] [CrossRef] [PubMed]
- Burzlaff, N. Tripodal N,N,O-ligands for metalloenzyme models and organometallics. Adv. Inorg. Chem. 2008, 60, 101–165. [Google Scholar]
- Weber, B. Spin crossover complexes with N4O2 coordination sphere—The influence of covalent linkers on cooperative interactions. Coord. Chem. Rev. 2009, 253, 2432–2449. [Google Scholar] [CrossRef]
- Kuźnik, N.; Wyskocka, M. Iron(III) Contrast Agent Candidates for MRI: A Survey of the Structure-Effect Relationship in the Last 15 Years of Studies. Eur. J. Inorg. Chem. 2016, 2016, 445–458. [Google Scholar] [CrossRef]
- Spokoyny, A.M. New Ligand platforms featuring boron-rich clusters as organomimetic substituents. Pure Appl. Chem. 2013, 85, 903–919. [Google Scholar] [CrossRef] [PubMed]
- Viñas, C.; Teixidor, F.; Núñez, R. Boron clusters-based metallodendrimers. Inorg. Chim. Acta 2014, 409, 12–25. [Google Scholar] [CrossRef]
- Timofeev, S.V.; Sivaev, I.B.; Prikaznova, E.A.; Bregadze, V.I. Transition metal complexes with charge-compensated dicarbollide ligands. J. Organomet. Chem. 2014, 751, 221–250. [Google Scholar] [CrossRef]
- Farràs, P.; Juárez-Pérez, E.J.; Lepšík, M.; Luque, R.; Núñez, R.; Teixidor, F. Metallacarboranes and their interactions: Theoretical insights and their applicability. Chem. Soc. Rev. 2012, 41, 3445–3463. [Google Scholar] [CrossRef] [PubMed]
- Štíbr, B. Reactions associated with arene replacement in bis-(arene) iron complexes. J. Organomet. Chem. 2012, 716, 1–5. [Google Scholar] [CrossRef]
- Deng, L.; Xie, Z. Advances in the chemistry of carboranes and metallacarboranes with more than 12 vertices. Coord. Chem. Rev. 2007, 251, 2452–2476. [Google Scholar] [CrossRef]
- Corsini, M.; de Biani, F.F.; Zanello, P. Mononuclear metallacarboranes of groups 6–10 metals: Analogues of metallocenes. Electrochemical and X-ray structural aspects. Coord. Chem. Rev. 2006, 250, 1351–1372. [Google Scholar] [CrossRef]
- Nakamura, H.; Aoyagi, K.; Yamamoto, Y. Synthetic utility of o-carborane: Novel protective group for aldehydes and ketones. J. Organomet. Chem. 1999, 574, 107–115. [Google Scholar] [CrossRef]
- Nakamura, H.; Aoyagi, K.; Yamamoto, Y. o-Carborane as a Novel Protective Group for Aldehydes and Ketones. J. Org. Chem. 1997, 62, 780–781. [Google Scholar] [CrossRef]
- Terrasson, V.; Planas, J.G.; Prim, D.; Viñas, C.; Teixidor, F.; Light, M.E.; Hursthouse, M.B. Cooperative Effect of Carborane and Pyridine in the Reaction of Carboranyl Alcohols with Thionyl Chloride: Halogenation versus Oxidation. J. Org. Chem. 2008, 73, 9140–9143. [Google Scholar] [CrossRef] [PubMed]
- Terrasson, V.; Giner Planas, J.; Prim, D.; Teixidor, F.; Viñas, C.; Light, M.E.; Hursthouse, M.B. General Access to Aminobenzyl-o-carboranes as a New Class of Carborane Derivatives: Entry to Enantiopure Carborane-Amine Combinations. Chem. Eur. J. 2009, 15, 12030–12042. [Google Scholar] [CrossRef] [PubMed]
- Terrasson, V.; Garcia, Y.; Farras, P.; Teixidor, F.; Viñas, C.; Giner Planas, J.; Prim, D.; Light, M.E.; Hursthouse, M.B. Crystal engineering of o-carboranyl alcohols: Syntheses, crystal structures and thermal properties. CrystEngComm 2010, 12, 4109–4123. [Google Scholar] [CrossRef]
- Terrasson, V.; Planas, J.G.; Viñas, C.; Teixidor, F.; Prim, D.; Light, M.E.; Hursthous, M.B. Closo-o-Carboranylmethylamine-Pyridine Associations: Synthesis, Characterization, and First Complexation Studies. Organometallics 2010, 29, 4130–4134. [Google Scholar] [CrossRef]
- Di Salvo, F.; Camargo, B.; Garcia, Y.; Teixidor, F.; Viñas, C.; Giner Planas, J.; Light, M.E.; Hursthouse, M.B. Supramolecular architectures in o-carboranyl alcohols bearing N-aromatic rings: Syntheses, crystal structures and melting points correlation. Crystengcomm 2011, 13, 5788–5806. [Google Scholar] [CrossRef]
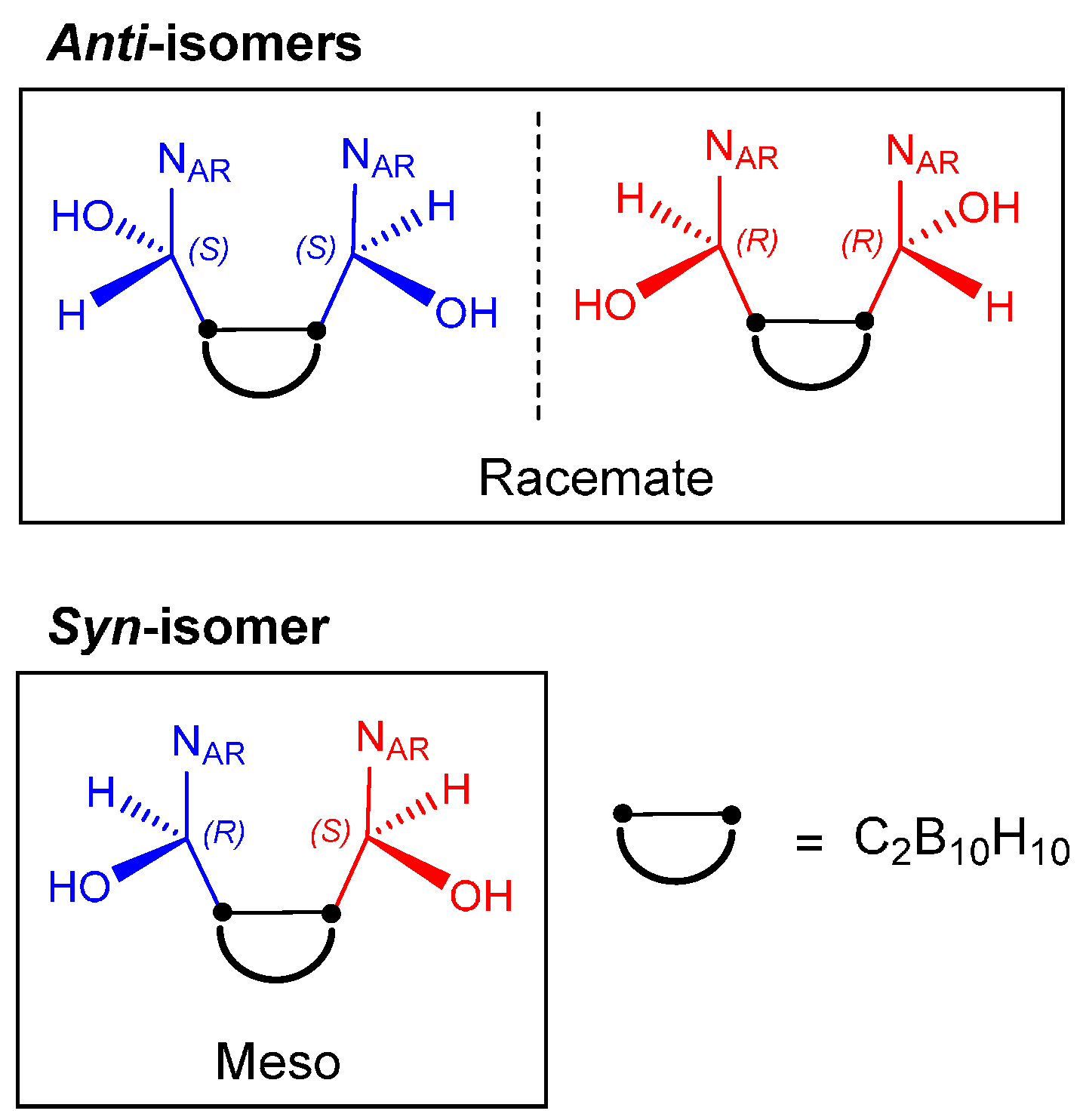
- Tsang, M.Y.; Di Salvo, F.; Teixidor, F.; Viñas, C.; Giner Planas, J.; Choquesillo-Lazarte, D.; Vanthuyne, N. Is Molecular Chirality Connected to Supramolecular Chirality? The Particular Case of Chiral 2-Pyridyl Alcohols. Cryst. Growth Des. 2015, 15, 935–945. [Google Scholar] [CrossRef]
- Ching, H.Y.V.; Buck, D.P.; Bhadbhade, M.; Collins, J.G.; Rendina, L.M. A ternary supramolecular system containing a boronated DNA-metallointercalator, β-cyclodextrin and the hexanucleotide d(GTCGAC) 2. Chem. Commun. 2012, 48, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Ching, H.Y.V.; Clifford, S.; Bhadbhade, M.; Clarke, R.J.; Rendina, L.M. Synthesis and supramolecular studies of chiral boronated platinum(ii) complexes: Insights into the molecular recognition of carboranes by β-cyclodextrin. Chem. Eur. J. 2012, 18, 14413–14425. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.Y.; Viñas, C.; Teixidor, F.; Planas, J.G.; Conde, N.; SanMartin, R.; Herrero, M.T.; Dominguez, E.; Lledos, A.; Vidossich, P.; et al. Synthesis, Structure, and Catalytic Applications for ortho- and meta-Carboranyl Based NBN Pincer-Pd Complexes. Inorg. Chem. 2014, 53, 9284–9295. [Google Scholar] [CrossRef] [PubMed]
- Di Salvo, F.; Paterakis, C.; Tsang, M.Y.; Garcia, Y.; Viñas, C.; Teixidor, F.; Giner Planas, J.; Light, M.E.; Hursthouse, M.B.; Choquesillo-Lazarte, D. Synthesis and Crystallographic Studies of Disubstituted Carboranyl Alcohol Derivatives: Prevailing Chiral Recognition? Cryst. Growth Des. 2013, 13, 1473–1484. [Google Scholar]
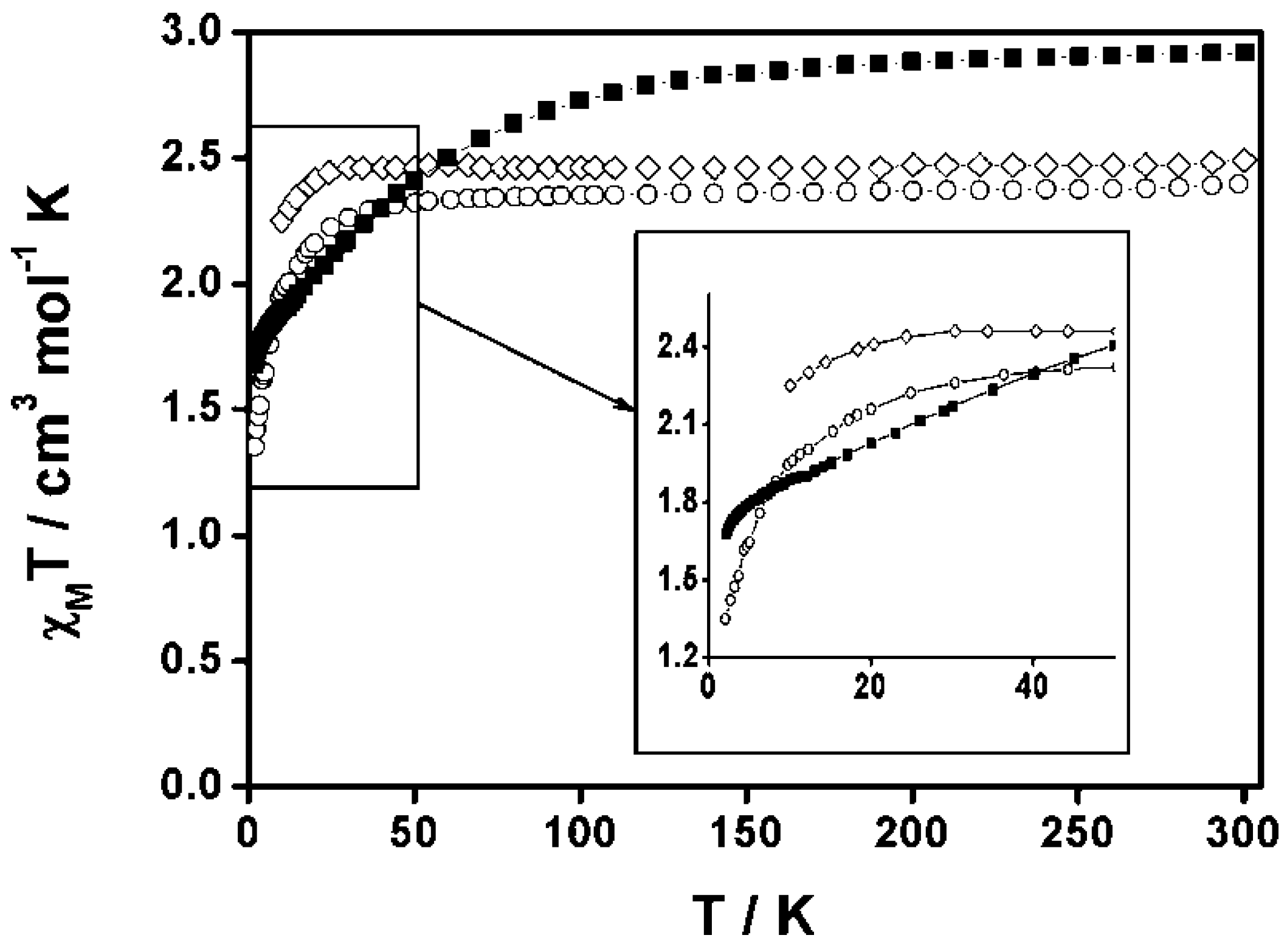
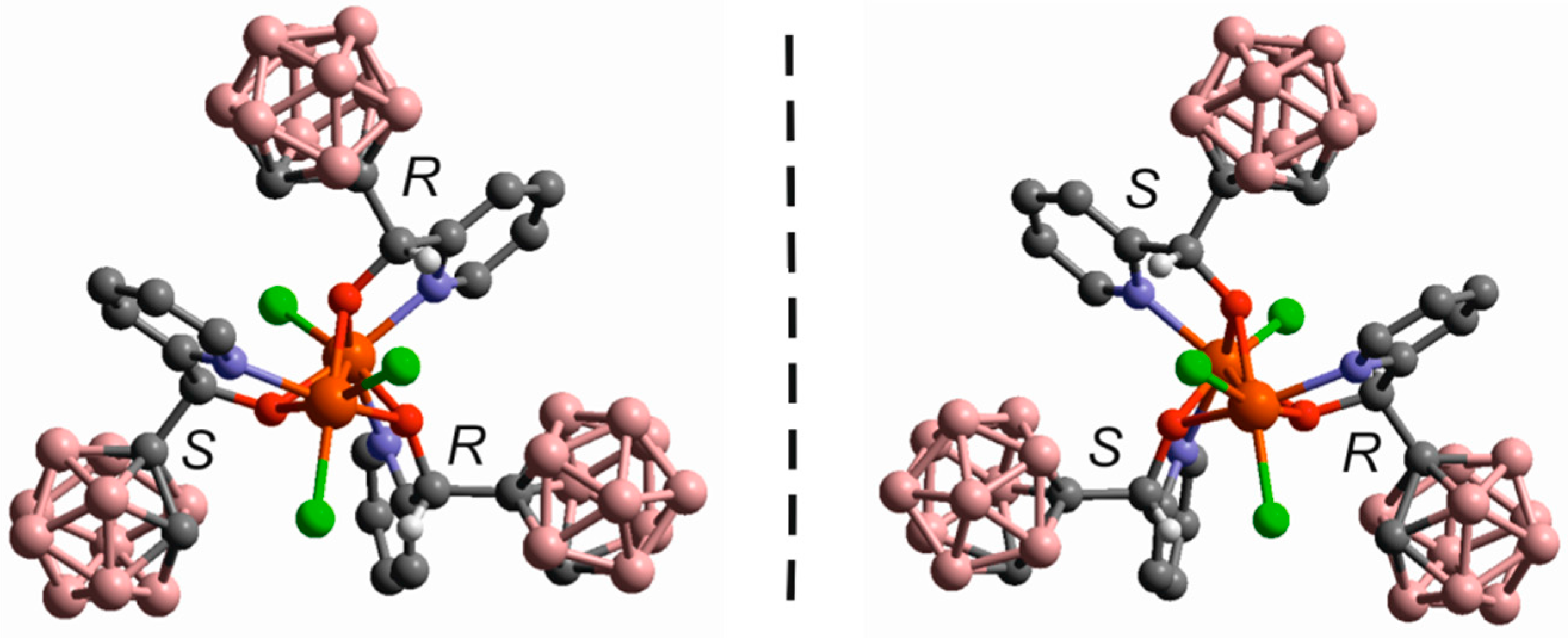
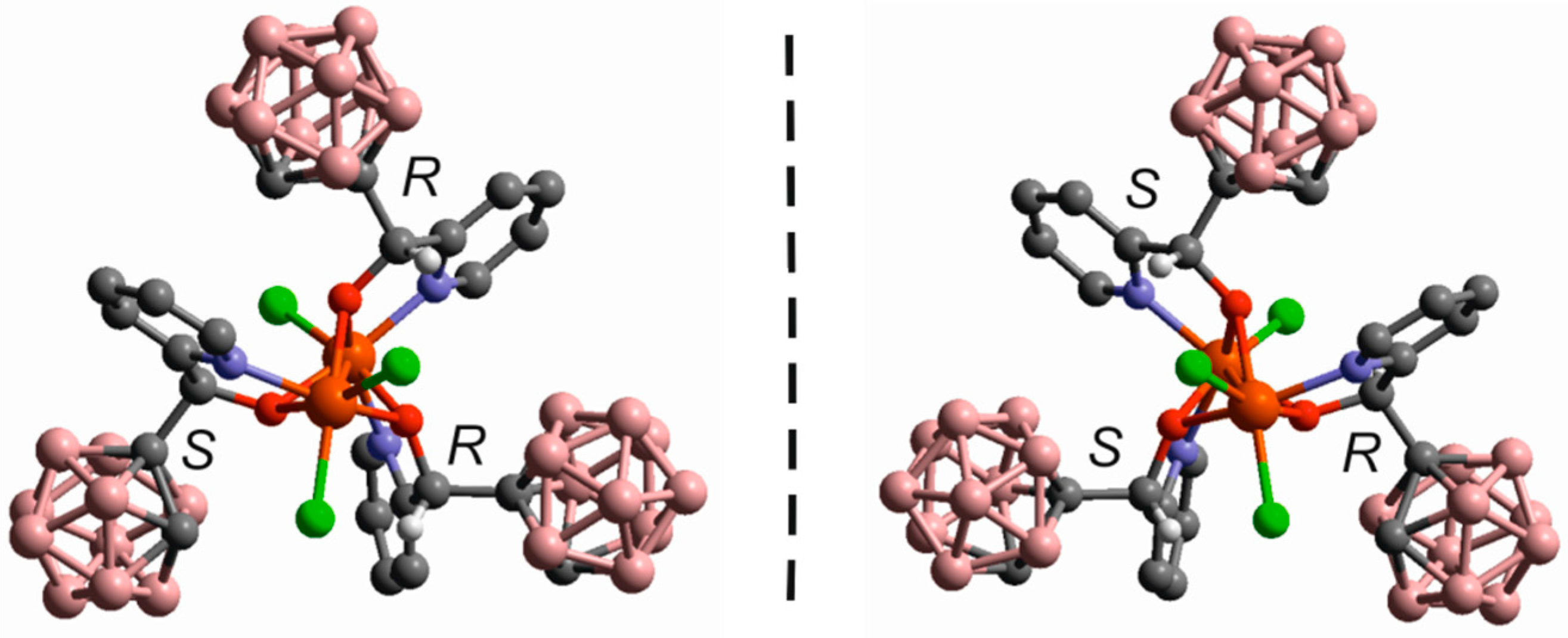
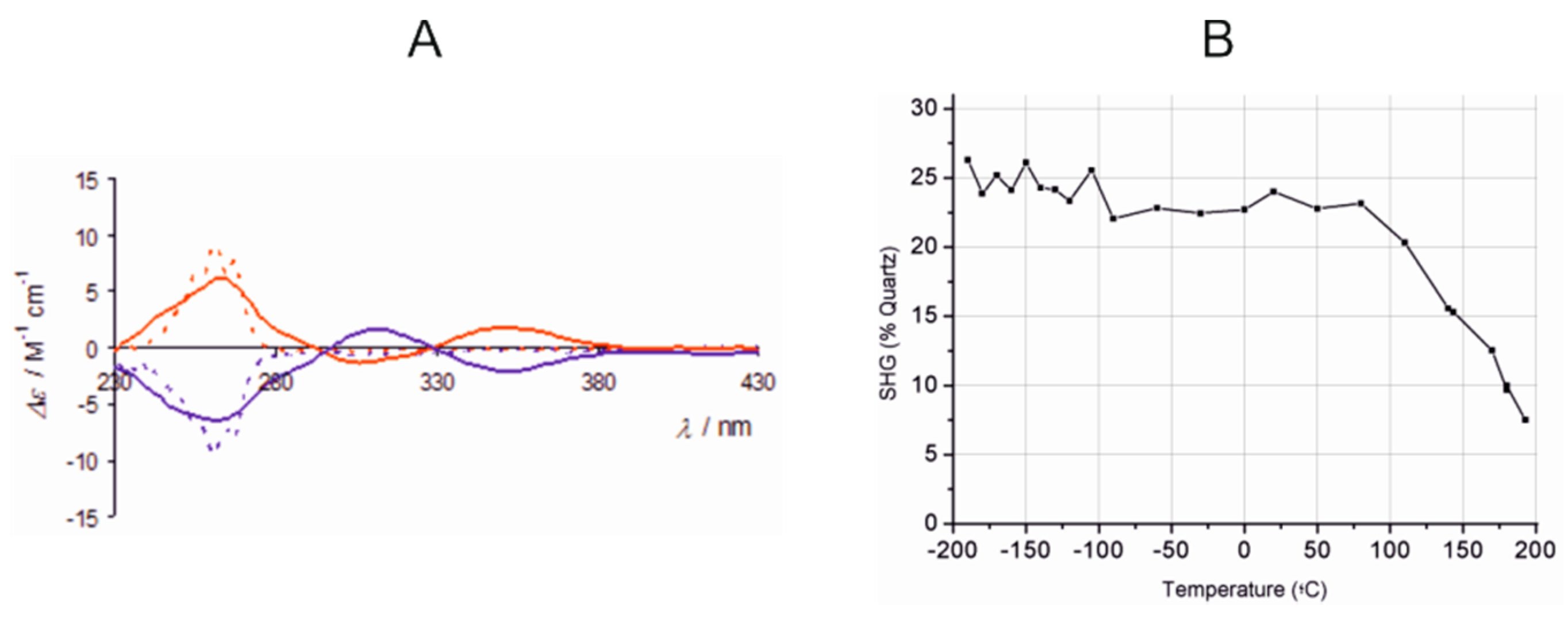
- Di Salvo, F.; Tsang, M.Y.; Teixidor, F.; Viñas, C.; Planas, J.G.; Crassous, J.; Vanthuyne, N.; Aliaga-Alcalde, N.; Ruiz, E.; Coquerel, G.; et al. A Racemic and Enantiopure Unsymmetric Diiron(III) Complex with a Chiral o-Carborane-Based Pyridylalcohol Ligand: Combined Chiroptical, Magnetic, and Nonlinear Optical Properties. Chem. Eur. J. 2014, 20, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
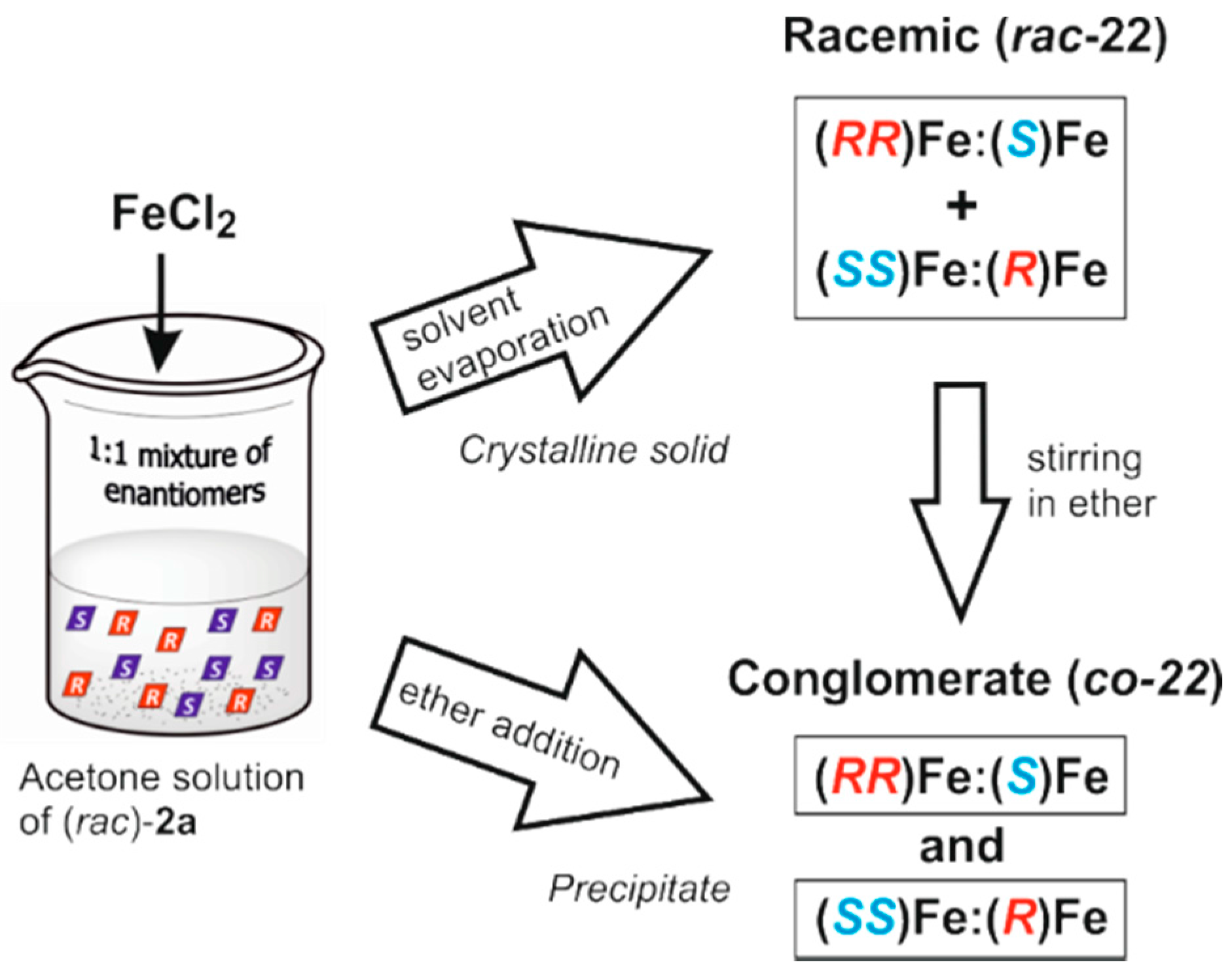
- Tsang, M.Y.; Teixidor, F.; Viñas, C.; Choquesillo-Lazarte, D.; Aliaga-Alcalde, N.; Planas, J.G. Synthesis, Structures and Properties of iron(III) complexes with (o-carboranyl)bis-(2-hydroxymethyl)pyridine: Racemic versus meso. Inorg. Chim. Acta 2016. [Google Scholar] [CrossRef]
- Satapathy, R.; Dash, B.P.; Zheng, C.; Maguire, J.A.; Hosmane, N.S. Carboranylpyrroles: A Synthetic Investigation. J. Org. Chem. 2011, 76, 3562–3565. [Google Scholar] [CrossRef] [PubMed]
- Yinghuai, Z.; Shiwei, X.; Vangala, V.R.; Chia, S.C.; Cheong, A.; Qin, O.N.; Hosmane, N.S. Carboranylimine-complexed titanium (IV) organometallics: An investigation of synthesis, structure and catalytic polymerization. J. Organomet. Chem. 2012, 721–722, 119–123. [Google Scholar] [CrossRef]
- El-Zaria, M.E.; Arii, H.; Nakamura, H. m-Carborane-Based Chiral NBN Pincer-Metal Complexes: Synthesis, Structure, and Application in Asymmetric Catalysis. Inorg. Chem. 2011, 50, 4149–4161. [Google Scholar] [CrossRef] [PubMed]
- Crassous, J. Chiral transfer in coordination complexes: Towards molecular materials. Chem. Soc. Rev. 2009, 38, 830–845. [Google Scholar] [CrossRef] [PubMed]
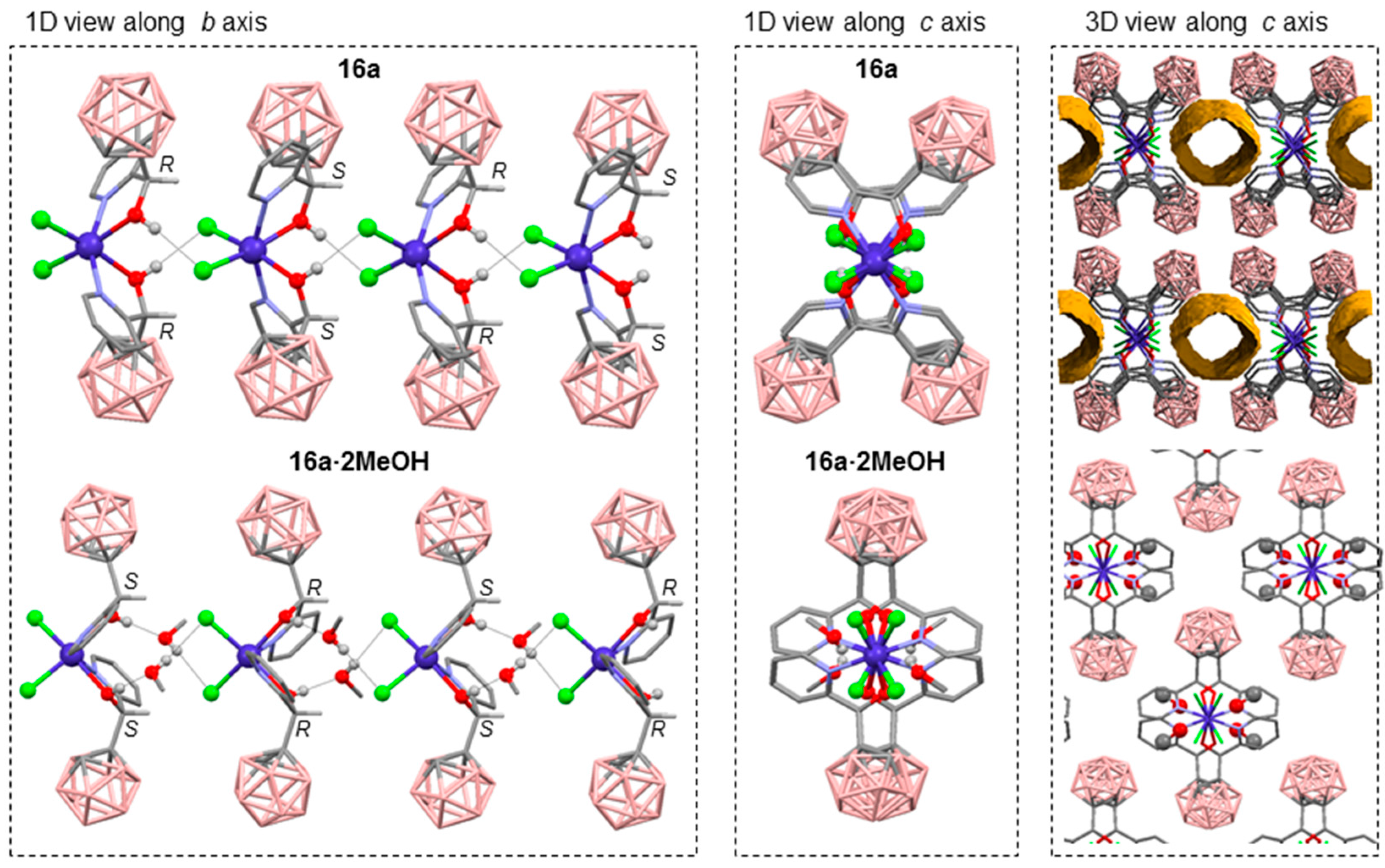
- Di Salvo, F.; Teixidor, F.; Viñas, C.; Giner Planas, J.; Light, M.E.; Hursthouse, M.B.; Aliaga-Alcalde, N. Metallosupramolecular Chemistry of Novel Chiral closo-o-Carboranylalcohol Pyridine and Quinoline Ligands: Syntheses, Characterization, and Properties of Cobalt Complexes. Cryst. Growth Des. 2012, 12, 5720–5736. [Google Scholar] [CrossRef]
- Ching, H.Y.V.; Clarke, R.J.; Rendina, L.M. Supramolecular β-Cyclodextrin Adducts of Boron-Rich DNA Metallointercalators Containing Dicarba-closo-dodecaborane(12). Inorg. Chem. 2013, 52, 10356–10367. [Google Scholar] [CrossRef] [PubMed]
- Teixidor, F.; Benakki, R.; Viñas, C.; Kivekäs, R.; Sillanpää, R. Partial Degradation of the New exo-Heterodisubstituted Carborane Derivatives with d10 Transition Metal Ions (Cu, Au). Inorg. Chem. 1999, 38, 5916–5919. [Google Scholar] [CrossRef]
- Viñas, C.; Mar Abad, M.; Teixidor, F.; Sillanpää, R.; Kivekäs, R. Reactions of Pd(II) with closo-1,2-dicarbadodecaborane-1,2-diphosphines. J. Organomet. Chem. 1998, 555, 17–23. [Google Scholar] [CrossRef]
- Teixidor, F.; Viñas, C.; Abad, M.M.; Kivekäs, R.; Sillanpää, R. The formation of nido [7,8-(PR2)2–7,8-C2B9H10]− from closo 1,2−(PR2)2−1,2−C2B10H10 (R = Ph, Et, iPr or OEt): A process enhanced by complexation. J. Organomet. Chem. 1996, 509, 139–150. [Google Scholar] [CrossRef]
- Teixidor, F.; Viñas, C.; Sillanpaa, R.; Kivekas, R.; Casabo, J. Nido-Carborane-Containing Compounds Resulting from the Reaction of closo-Carboranes with Transition Metal Complexes. Inorg. Chem. 1994, 33, 2645–2650. [Google Scholar] [CrossRef]
- Teixidor, F.; Viñas, C.; Mar Abad, M.; Lopez, M.; Casabo, J. Synthesis of [7,8-(PPh2)2–7,8-C2B9H10]-: A ligand analogous to 1,2-bis(diphenylphosphino)ethane with a “built-in” negative charge. Organometallics 1993, 12, 3766–3768. [Google Scholar] [CrossRef]
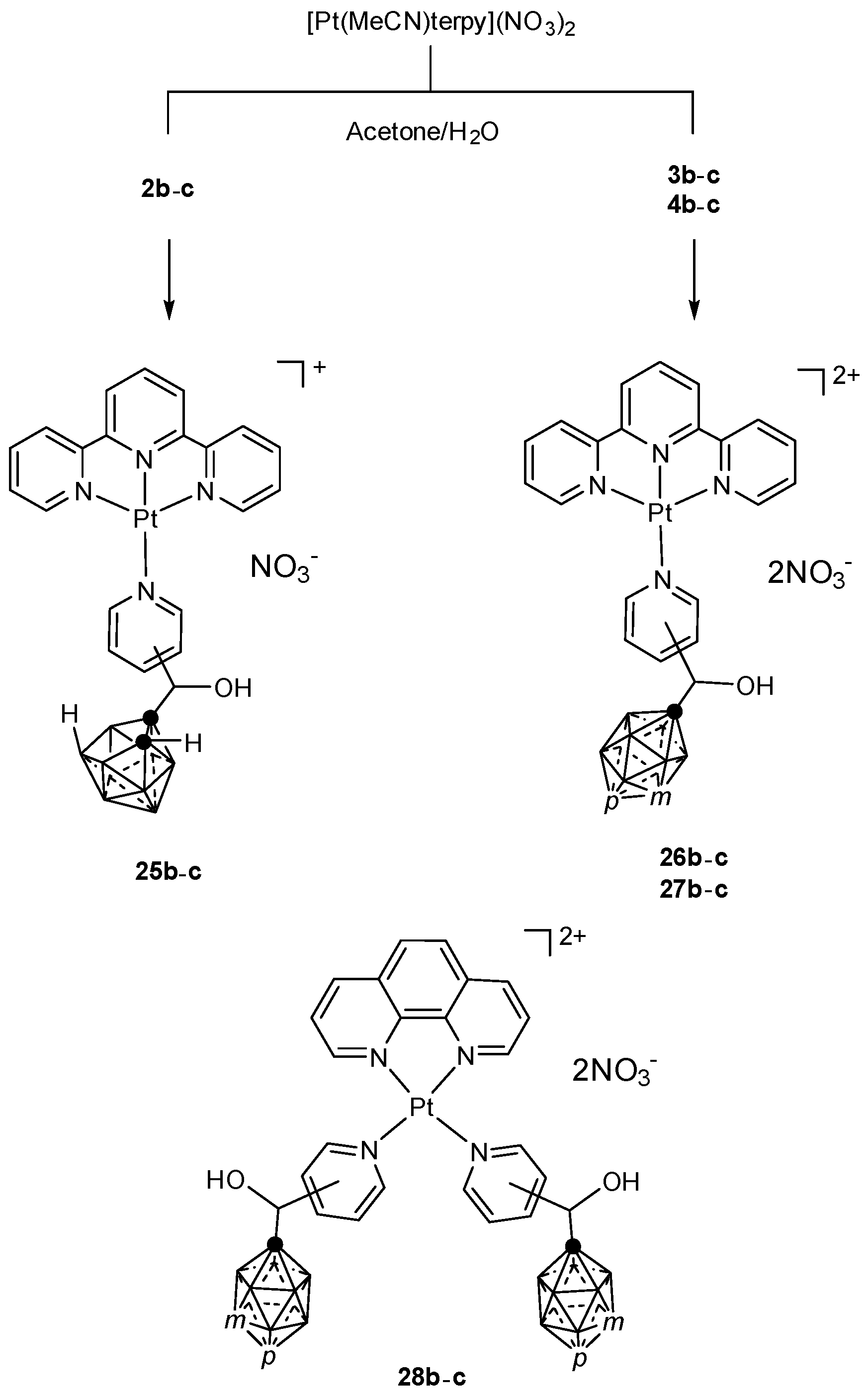
- Todd, J.A.; Rendina, L.M. The first examples of platinum(II)-amine complexes containing 1,2-dicarba-closo-dodecaborane(12). Inorg. Chem. Commun. 2004, 7, 289–291. [Google Scholar] [CrossRef]
- Yoo, J.; Do, Y. Synthesis of stable platinum complexes containing carborane in a carrier group for potential BNCT agents. Dalton Trans. 2009, 4978–4986. [Google Scholar] [CrossRef] [PubMed]
- Suttil, J.A.; McGuinness, D.S.; Gardiner, M.G.; Evans, S.J. Ethylene polymerisation and oligomerisation with arene-substituted phenoxy-imine complexes of titanium: Investigation of multi-mechanism catalytic behaviour. Dalton Trans. 2013, 42, 4185–4196. [Google Scholar] [CrossRef] [PubMed]
- Terao, H.; Ishii, S.; Mitani, M.; Tanaka, H.; Fujita, T. Ethylene/Polar Monomer Copolymerization Behavior of Bis(phenoxy–imine)Ti Complexes: Formation of Polar Monomer Copolymers. J. Am. Chem. Soc. 2008, 130, 17636–17637. [Google Scholar] [CrossRef] [PubMed]
- Di Salvo, F.; Teixidor, F.; Viñas, C.; Giner Planas, J. A Distinct Tetradentate N2O2-type Ligand: (o-Carboranyl)bis(2-hydroxymethyl)pyridine. Z. Anorg. Allg. Chem. 2013, 639, 1194–1198. [Google Scholar] [CrossRef]
- Knight, P.D.; Scott, P. Predetermination of chirality at octahedral centres with tetradentate ligands: Prospects for enantioselective catalysis. Coord. Chem. Rev. 2003, 242, 125–143. [Google Scholar] [CrossRef]
- Knof, U.; von Zelewsky, A. Predetermined Chirality at Metal Centers. Angew. Chem. Int. Ed. 1999, 38, 302–322. [Google Scholar] [CrossRef]
- Vigato, P.A.; Peruzzo, V.; Tamburini, S. Acyclic and cyclic compartmental ligands: Recent results and perspectives. Coord. Chem. Rev. 2012, 256, 953–1114. [Google Scholar] [CrossRef]
- Kanazawa, Y.; Tsuchiya, Y.; Kobayashi, K.; Shiomi, T.; Itoh, J.-I.; Kikuchi, M.; Yamamoto, Y.; Nishiyama, H. Asymmetric Conjugate Reduction of α,β-Unsaturated Ketones and Esters with Chiral Rhodium(2,6-bisoxazolinylphenyl) Catalysts. Chem. Eur. J. 2006, 12, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H. Synthesis and use of bisoxazolinyl-phenyl pincers. Chem. Soc. Rev. 2007, 36, 1133–1141. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Planas, J.G.; Teixidor, F.; Viñas, C. N,O-Type Carborane-Based Materials. Crystals 2016, 6, 50. https://doi.org/10.3390/cryst6050050
Planas JG, Teixidor F, Viñas C. N,O-Type Carborane-Based Materials. Crystals. 2016; 6(5):50. https://doi.org/10.3390/cryst6050050
Chicago/Turabian StylePlanas, José Giner, Francesc Teixidor, and Clara Viñas. 2016. "N,O-Type Carborane-Based Materials" Crystals 6, no. 5: 50. https://doi.org/10.3390/cryst6050050
APA StylePlanas, J. G., Teixidor, F., & Viñas, C. (2016). N,O-Type Carborane-Based Materials. Crystals, 6(5), 50. https://doi.org/10.3390/cryst6050050