
The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand


 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.1.1. Synthesis of Ligand
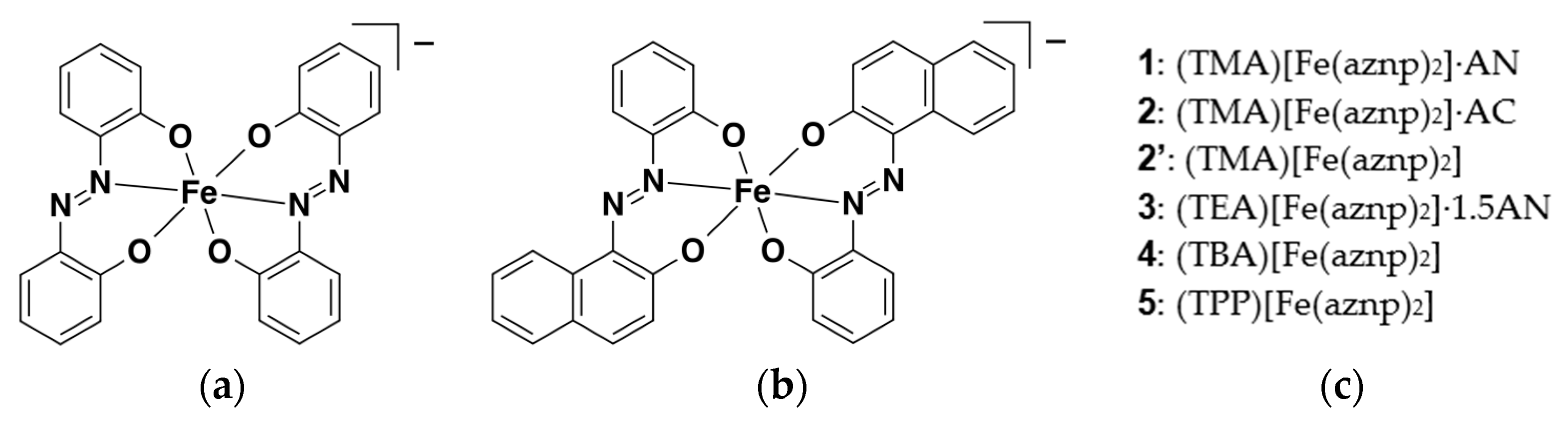
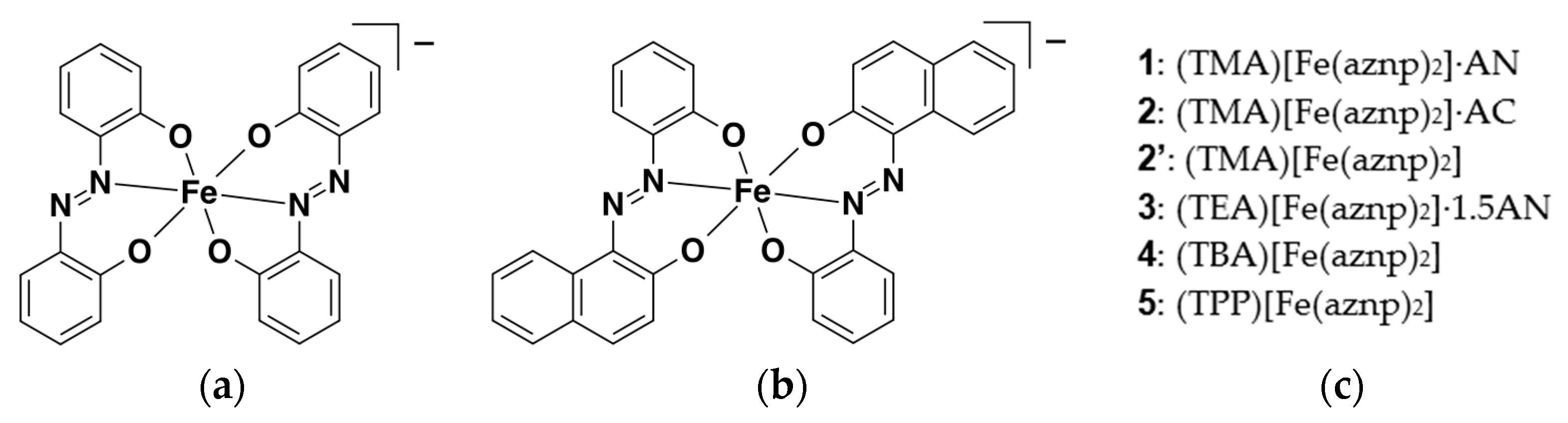
2.1.2. Synthesis of Complexes 1–5
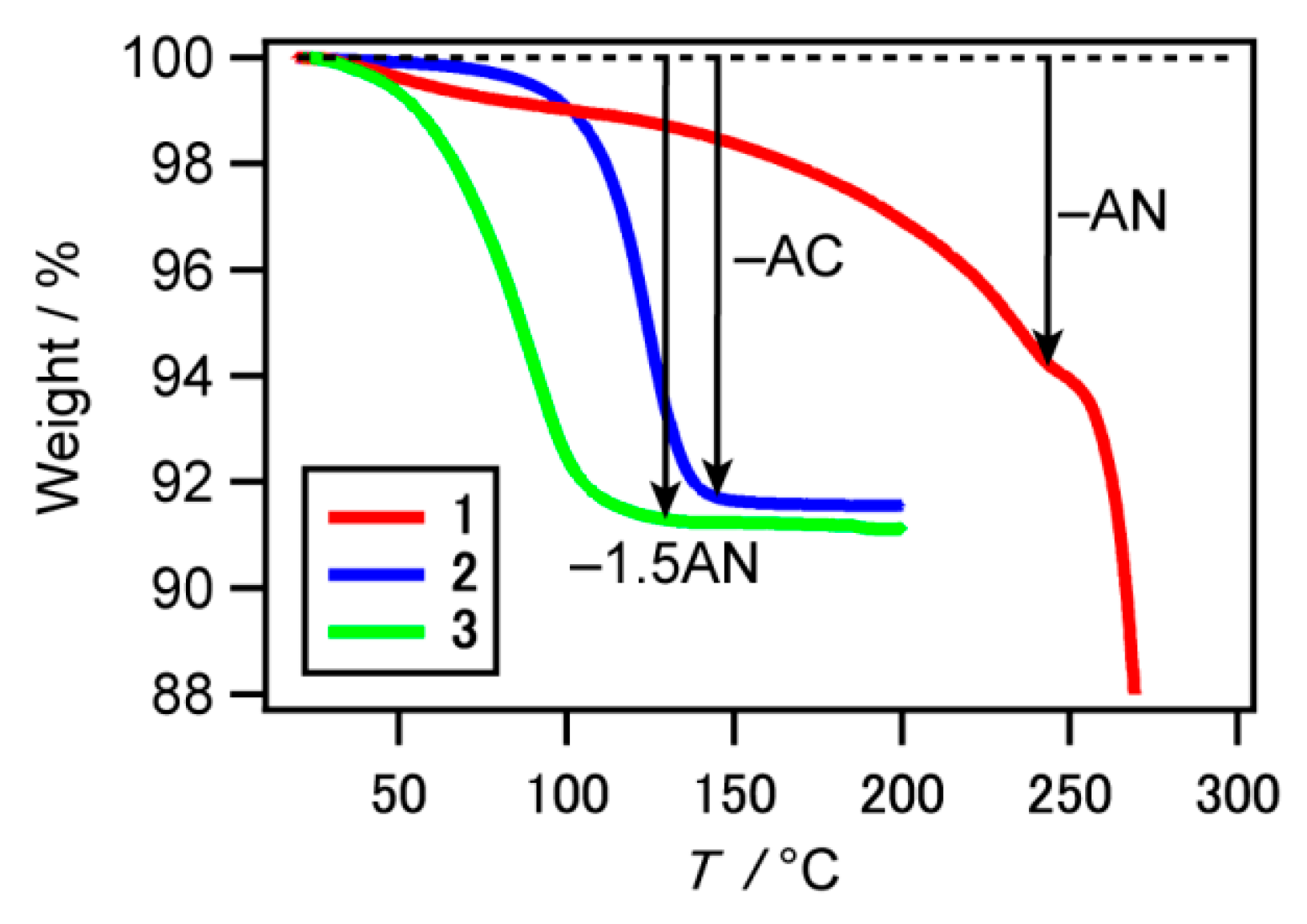
2.2. Thermogravimetry-Differential Thermal Analysis (TG-DTA) for 1–3
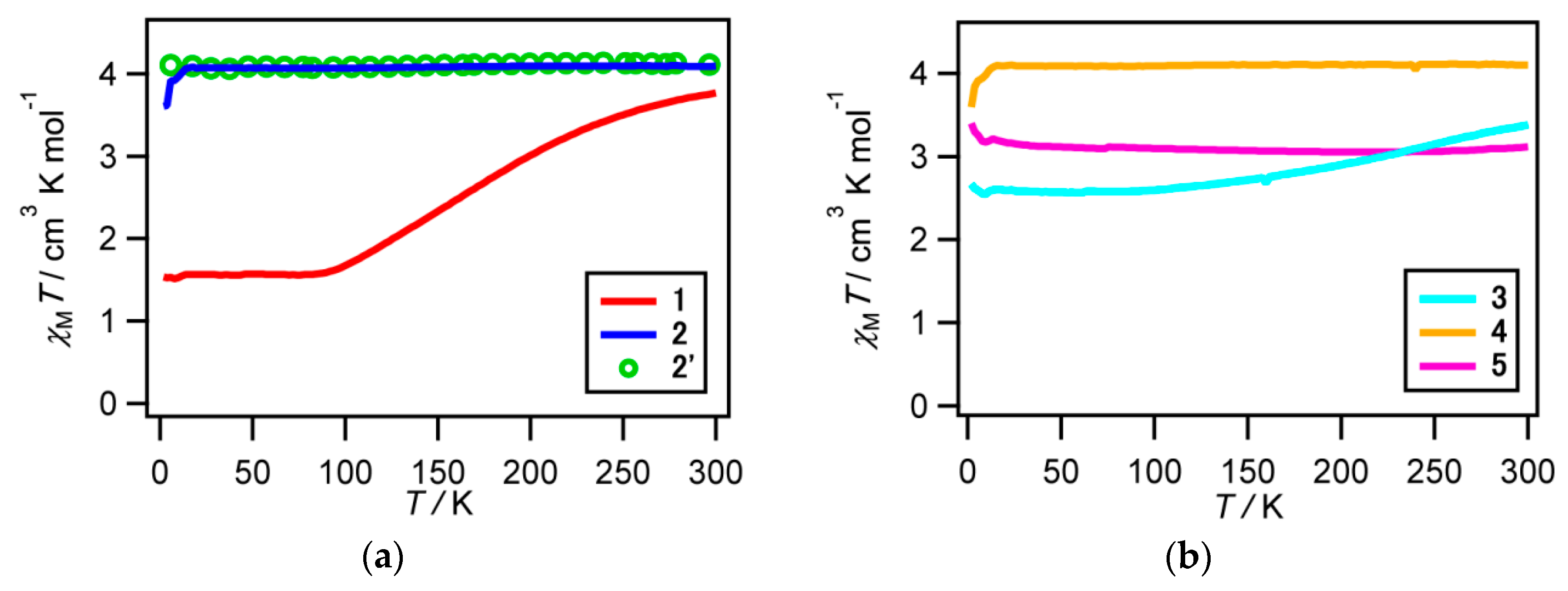
2.3. Magnetic Susceptibility for 1–5
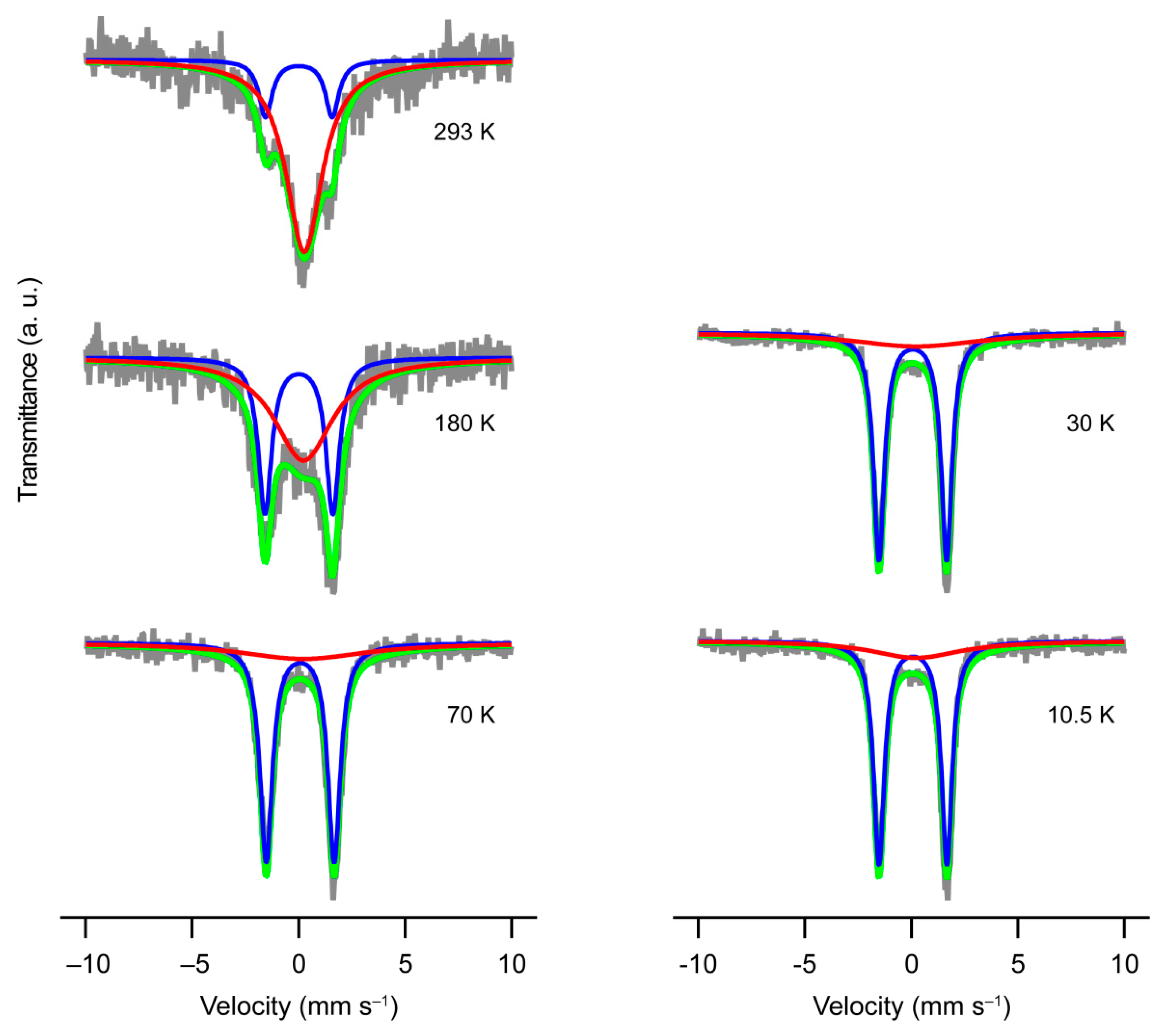
2.4. Mössbauer Spectroscopy for 1
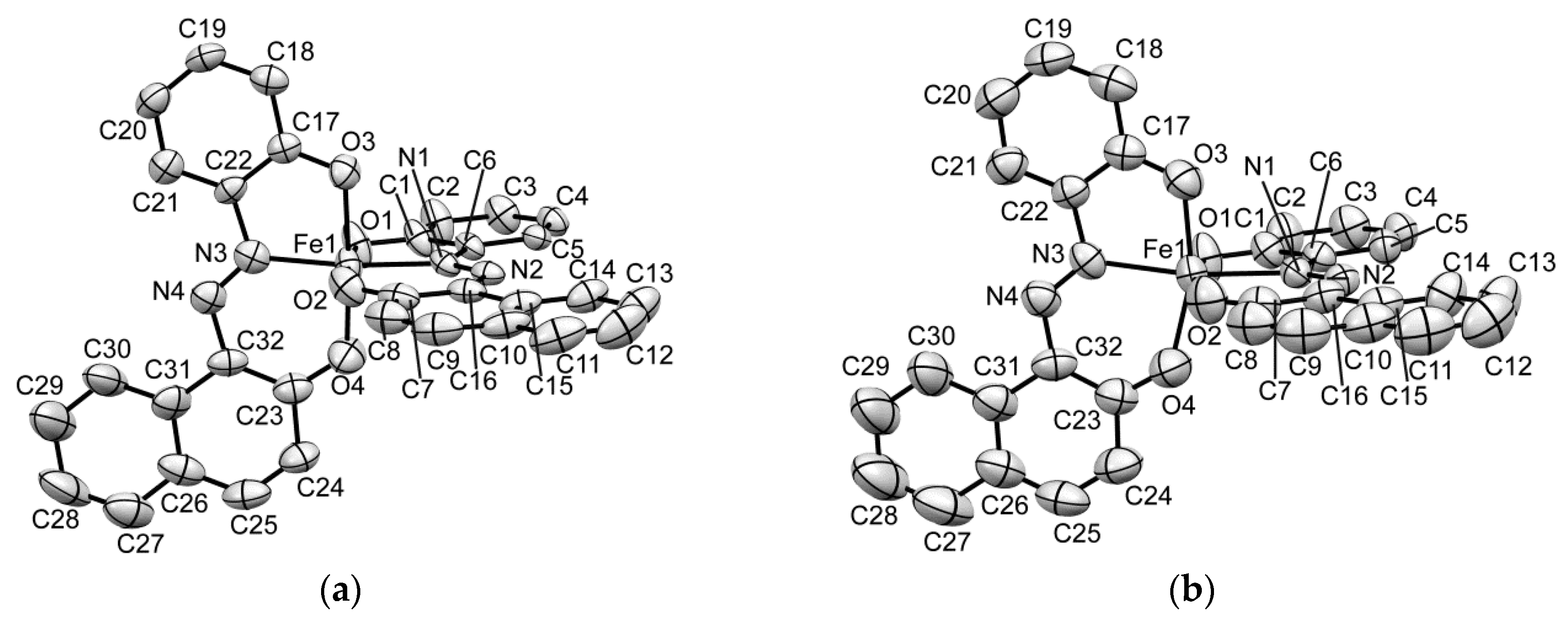
2.5. Crystal Structures of 1 and 2
2.5.1. Molecular Structure Description of the [Fe(aznp)2] Anion in 1, 2, and 2’
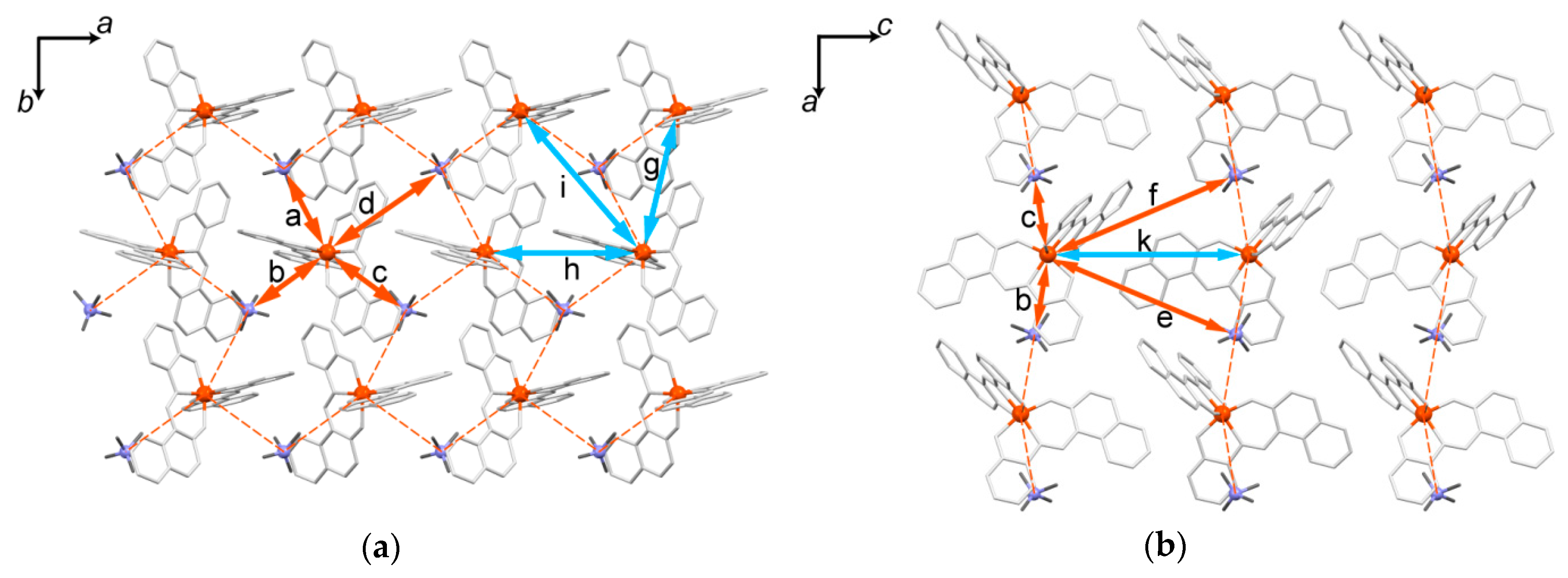
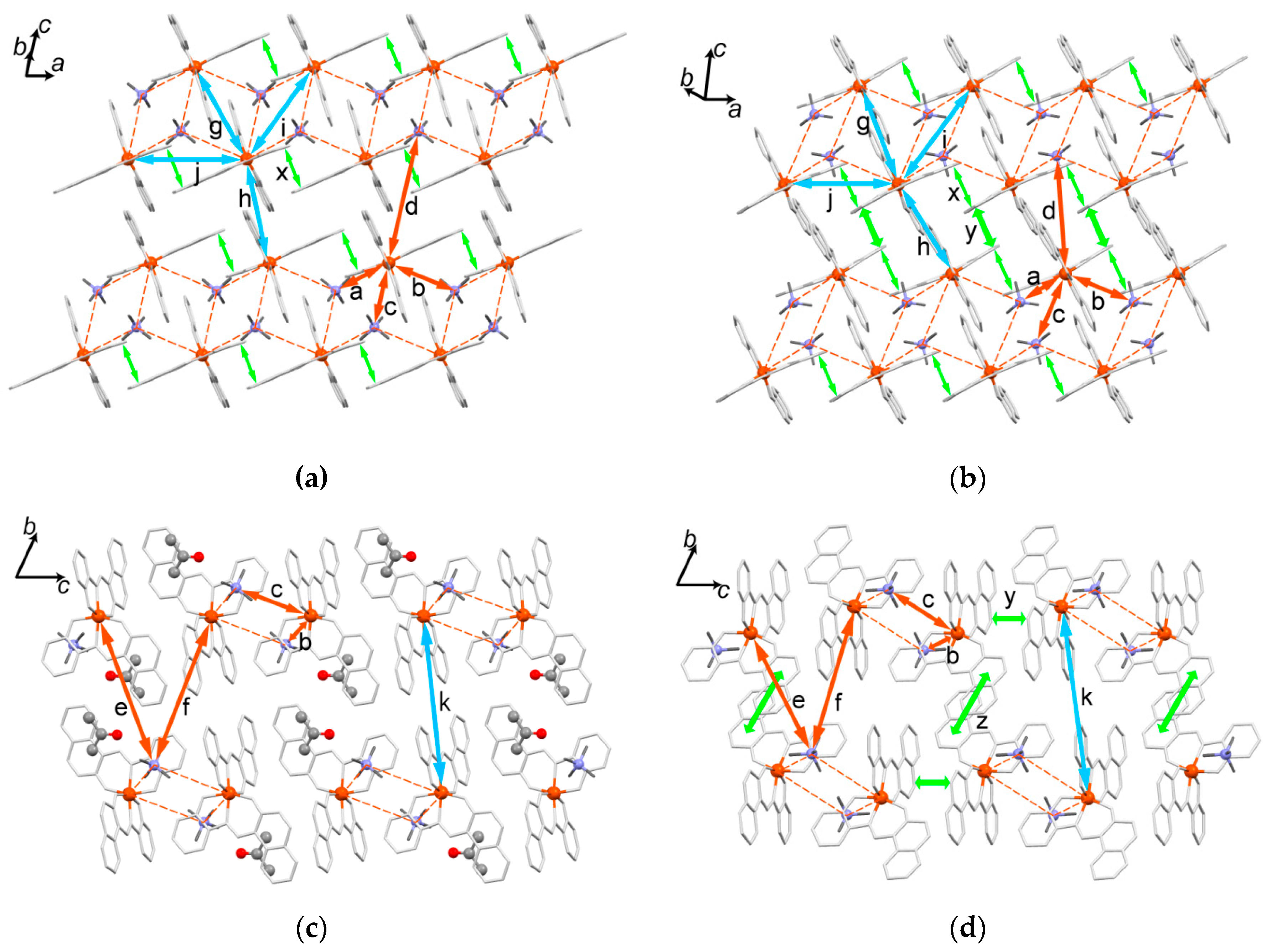
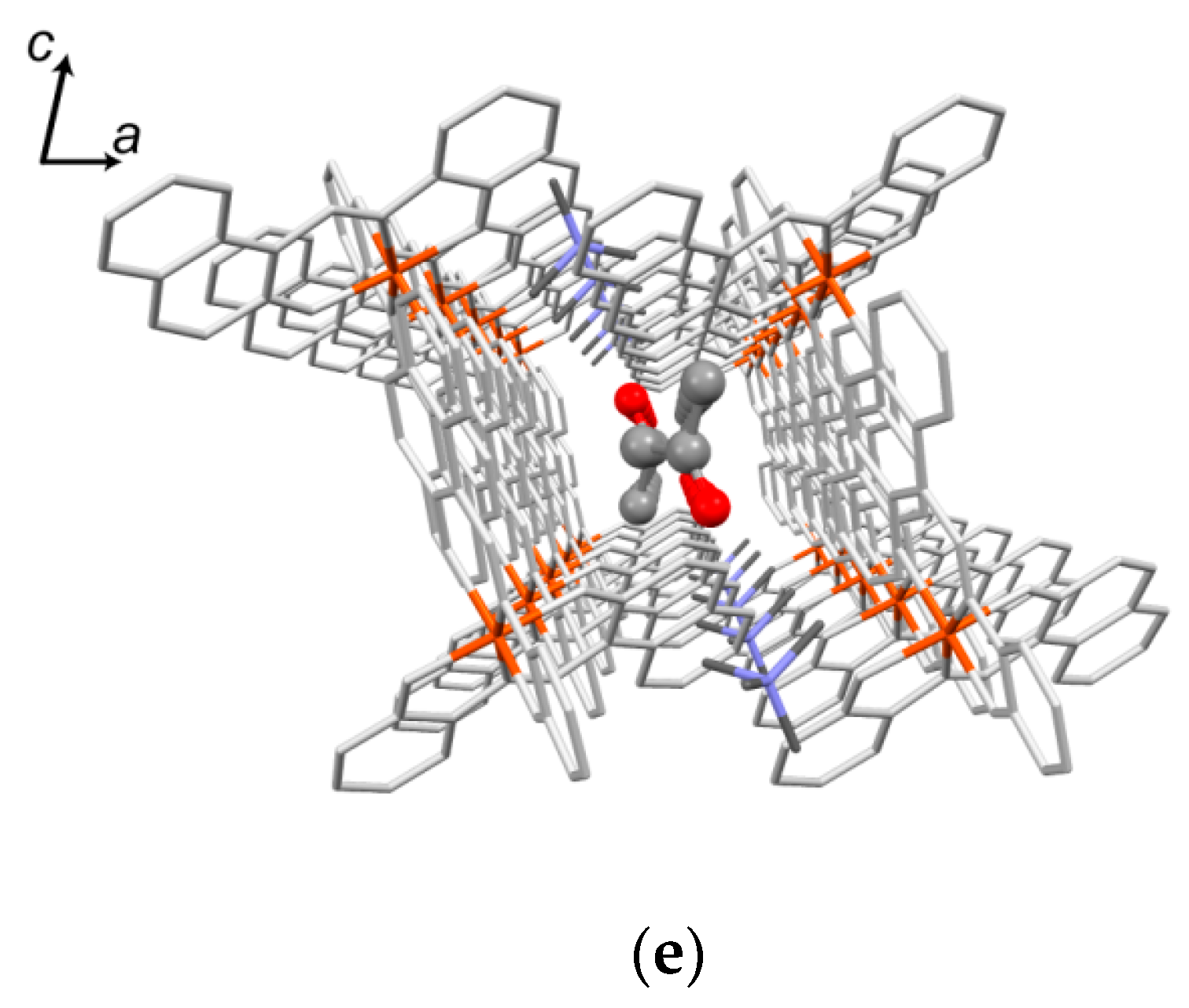
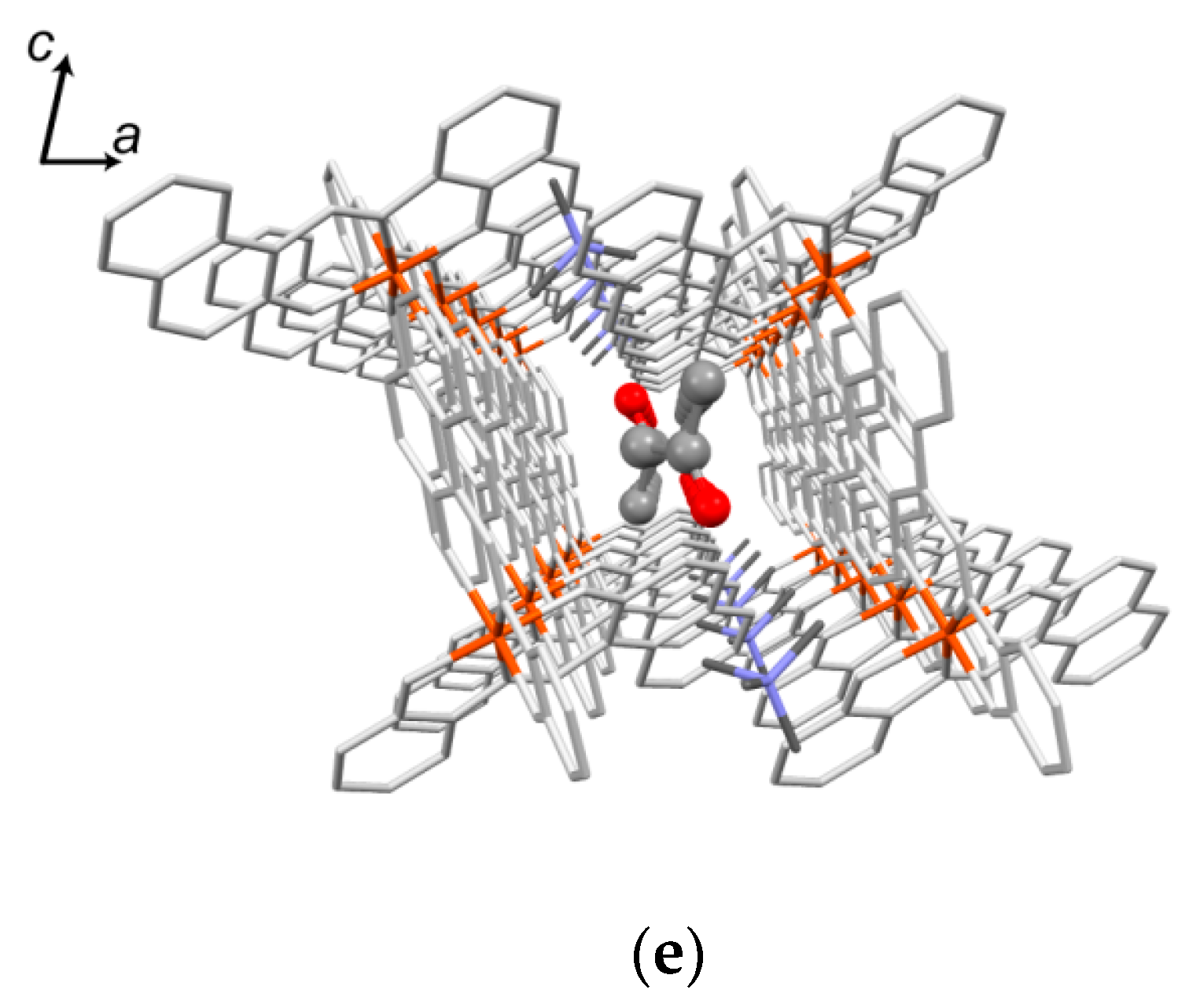
2.5.2. Crystal Description of 1
2.5.3. Crystal Description of 2
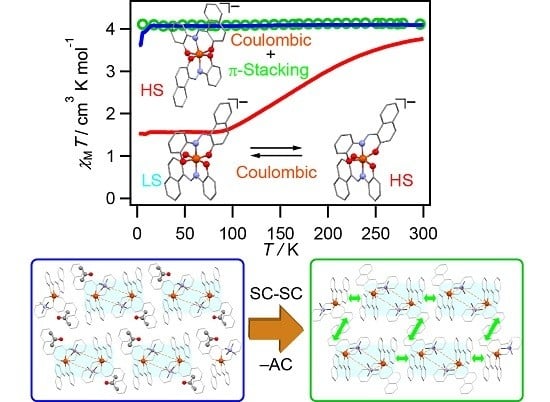
2.5.4. Desolvation-Induced Crystal Structure Transformation from 2 to 2’
2.5.5. Structural Comparison between 1, 2, and 2’
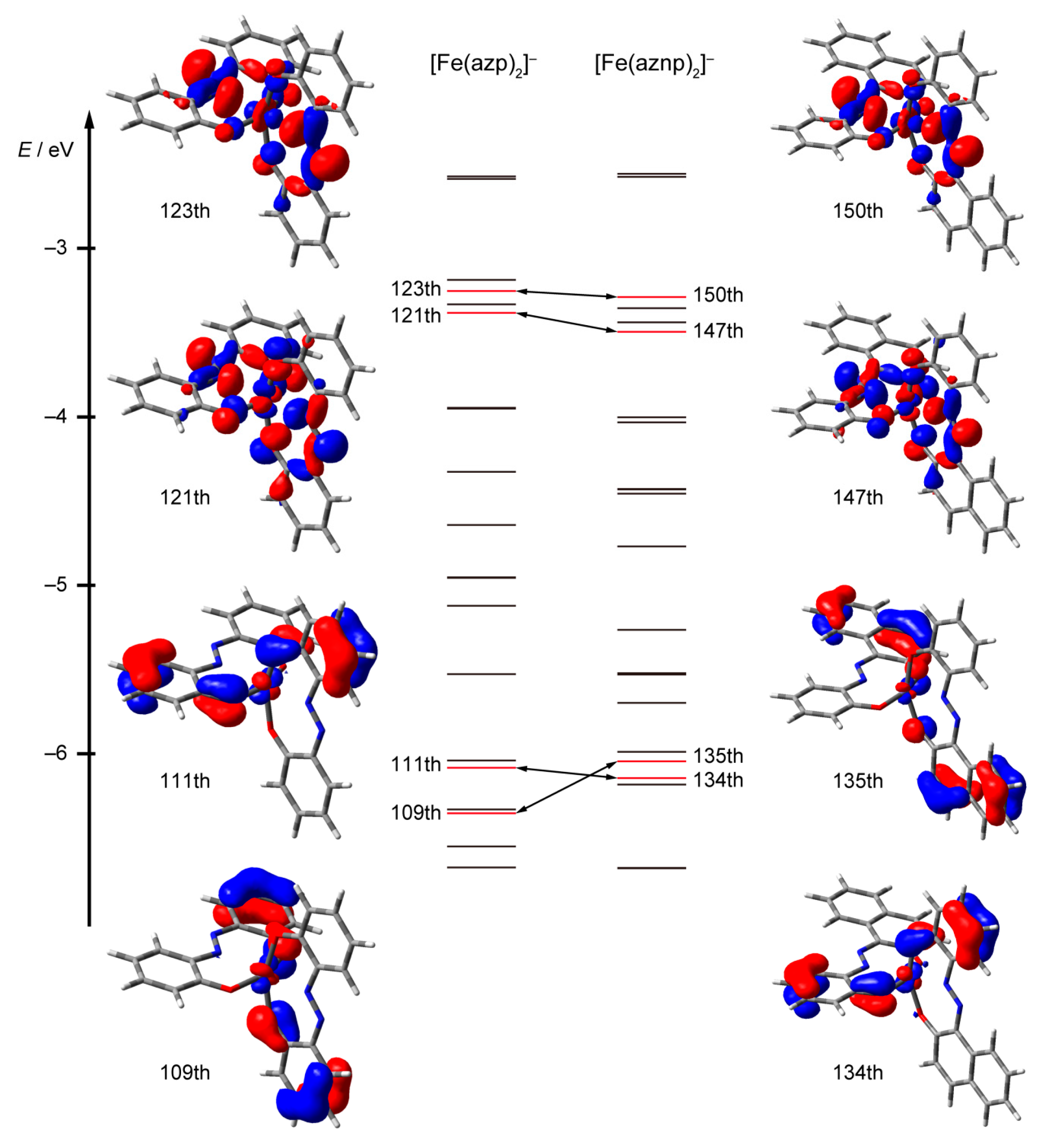
2.6. Density Functional Theory (DFT) Calculations
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of (2’-methoxyphenylazo)-2-hydroxynaphthalene (MeHaznp)
3.1.2. Synthesis of H2aznp
3.1.3. General Synthetic Procedure of the [Fe(aznp)2] Compounds (1–5)
3.2. Physical Measurements
3.3. Crystal Structure Determinations
3.4. DFT Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gütlich, P.; Goodwin, H.A. Spin crossover—An overall perspective. In Spin Crossover in Transition Metal Compounds; Gütlich, P., Goodwin, H.A., Eds.; Springer-Verlag: Berlin, Heidelberg, Germany, 2004; Volume 1, pp. 1–47. [Google Scholar]
- Halcrow, M.A. (Ed.) Spin-Crossover Materials; John Wiley & Sons, Ltd.: Oxford, UK, 2013.
- Takahashi, K.; Cui, H.-B.; Okano, Y.; Kobayashi, H.; Einaga, Y.; Sato, O. Electrical conductivity modulation coupled to a high-spin—Low-spin conversion in the molecular system [FeIII(qsal)2][Ni(dmit)2]3·CH3CN·H2O. Inorg. Chem. 2006, 45, 5739–5741. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Cui, H.-B.; Okano, Y.; Kobayashi, H.; Mori, H.; Tajima, H.; Einaga, Y.; Sato, O. Evidence of the chemical uniaxial strain effect on electrical conductivity in the spin-crossover conducting molecular system: [FeIII(qnal)2][Pd(dmit)2]5·acetone. J. Am. Chem. Soc. 2008, 130, 6688–6689. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Lemaire, M.T. Hybrid Spin-crossover conductor exhibiting unusual variable-temperature electrical conductivity. Inorg. Chem. 2009, 48, 10489–10491. [Google Scholar] [CrossRef] [PubMed]
- Nihei, M.; Takahashi, N.; Nishikawa, H.; Oshio, H. Spin-crossover behavior and electrical conduction property in iron(II) complexes with tetrathiafulvalene moieties. Dalton Trans. 2011, 40, 2154–2156. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.; Benjamin, S.M.; Steven, E.; Brooks, J.S.; Shatruk, M. Photomagnetic response in highly conductive iron(II) spin- crossover complexes with TCNQ radicals. Angew. Chem. Int. Ed. 2015, 54, 823–827. [Google Scholar]
- Nihei, M.; Tahira, H.; Takahashi, N.; Otake, Y.; Yamamura, Y.; Saito, K.; Oshio, H. Multiple bistability and tristability with dual spin-state conversions in [Fe(dpp)2][Ni(mnt)2]2·MeNO2. J. Am. Chem. Soc. 2010, 132, 3553–3560. [Google Scholar] [CrossRef] [PubMed]
- Ohkoshi, S.; Imoto, K.; Tsunobuchi, Y.; Takano, S.; Tokoro, H. Light-induced spin-crossover magnet. Nat. Chem. 2011, 3, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ababei, R.; Pichon, C.; Roubeau, O.; Li, Y.-G.; Bréfuel, N.; Buisson, L.; Guionneau, P.; Mathonière, C.; Clérac, R. Rational design of a photomagnetic chain: Bridging single-molecule magnets with a spin-crossover complex. J. Am. Chem. Soc. 2013, 135, 14840–14853. [Google Scholar] [CrossRef] [PubMed]
- Fukuroi, K.; Takahashi, K.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Mori, H. Synergistic spin transition between spin crossover and spin-Peierls-like singlet formation in the halogen-bonded molecular hybrid system: [Fe(Iqsal)2][Ni(dmit)2]·CH3CN·H2O. Angew. Chem. Int. Ed. 2014, 53, 1983–1986. [Google Scholar] [CrossRef] [PubMed]
- Okai, M.; Takahashi, K.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y. Novel Fe(II) spin crossover complexes involving a chalcogen-bond and π-stacking interactions with a paramagnetic and nonmagnetic M(dmit)2 anion (M = Ni, Au; dmit = 4,5-dithiolato-1,3-dithiole-2-thione). J. Mater. Chem. C 2015, 3, 7858–7864. [Google Scholar] [CrossRef]
- Ohkoshi, S.; Takano, S.; Imoto, K.; Yoshikiyo, M.; Namai, A.; Tokoro, H. 90-degree optical switching of output second-harmonic light in chiral photomagnet. Nat. Photonics 2014, 8, 65–71. [Google Scholar] [CrossRef]
- Wang, C.F.; Li, R.-F.; Chen, X.-Y.; Wei, R.-J.; Zheng, L.-S.; Tao, J. Synergetic spin crossover and fluorescence in one-dimensional hybrid complexes. Angew. Chem. Int. Ed. 2015, 54, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Zelentsov, V.V. Spin transitions in iron(III) complexes with thiosemicarbazones of O-hydroxyaldehydes. Sov. Sci. Rev. B Chem. 1987, 10, 485–512. [Google Scholar]
- Floquet, S.; Boillot, M.-L.; Rivière, E.; Varret, F.; Boukheddaden, K.; Morineau, D.; Négrier, P. Spin transition with a large thermal hysteresis near room temperature in a water solvate of an iron(III) thiosemicarbazone complex. New J. Chem. 2003, 27, 341–348. [Google Scholar] [CrossRef]
- Floquet, S.; Guillou, N.; Négrier, P.; Rivière, E.; Boillot, M.-L. The crystallographic phase transition for a ferric thiosemicarbazone spin crossover complex studied by X-ray powder diffraction. New J. Chem. 2006, 30, 1621–1627. [Google Scholar] [CrossRef]
- Cook, C.; Habib, F.; Aharen, T.; Clérac, R.; Hu, A.; Murugesu, M. High-temperature spin crossover behavior in a nitrogen-rich FeIII-based system. Inorg. Chem. 2013, 52, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Hirosawa, N.; Oso, Y.; Ishida, T. Spin crossover and light-induced excited spin-state trapping observed for an iron(II) complex chelated with tripodal tetrakis(2-pyridyl)methane. Chem. Lett. 2012, 41, 716–718. [Google Scholar] [CrossRef]
- Takahashi, K.; Kawamukai, K.; Okai, M.; Mochida, T.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K. A new family of anionic FeIII spin crossover complexes featuring a weak-field N2O4 coordination octahedron. Chem. Eur. J. 2016, 22, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Drew, H.D.K.; Landquist, J.K. Structure of the cooper lakes of azo-dyes. J. Chem. Soc. 1938, 292–304. [Google Scholar] [CrossRef]
- Mitra, S.; Biswas, H.; Bandyopadhyay, P. Synthesis, spectral properties and redox behaviour of cis-dioxo-molybdenum(IV) complexes with tridentate arylazo ligands. Polyhedron 1995, 14, 1581–1584. [Google Scholar] [CrossRef]
- Schweig, A.; Baumgartl, H.; Schulz, R. IR and UV matrix photochemistry and solvent effects: The isomerization of diazocyclohexadienones (ortho quinone diazides)—Detection of molecules with the 1,2,3-benzoxadiazole structure. A UV/Vis and IR absorption and UV photoelectron spectroscopic investigation. J. Mol. Struct. 1991, 247, 135–171. [Google Scholar]
- Schetty, G. Neuartige Isomeriefälle bei 1:2-CrIII- und CoIII-Komplexen von o,o’-Dihydroxyazoverbindungen: Pyramidal gebundener Stickstoff mit hoher Inversionsbarriere? Helv. Chim. Acta 1970, 53, 1437–1459. [Google Scholar] [CrossRef]
- Haghbeen, K.; Tan, E.W. Facile synthesis of catechol azo dyes. J. Org. Chem. 1998, 63, 4503–4505. [Google Scholar] [CrossRef]
- Wignall, J.W.G. Mössbauer line broadening in trivalent iron compounds. J. Chem. Phys. 1966, 44, 2462–2467. [Google Scholar] [CrossRef]
- Sano, H. On the spin relaxation of 57Fe(III) species produced through EC-decay in diamagnetic 57Co-labelled Co(III) compounds. J. Radioanal. Chem. 1977, 36, 105–111. [Google Scholar] [CrossRef]
- Hayami, S.; Gu, Z.; Shiro, M.; Einaga, Y.; Fujishima, A.; Sato, O. First observation of light-induced excited spin state trapping for an Iron(III) complex. J. Am. Chem. Soc. 2000, 122, 7126–7127. [Google Scholar] [CrossRef]
- Hayami, S.; Gu, Z.; Yoshiki, H.; Fujishima, A.; Sato, O. Iron(III) spin-crossover compounds with a wide apparent thermal hysteresis around room temperature. J. Am. Chem. Soc. 2001, 123, 11644–11650. [Google Scholar] [CrossRef] [PubMed]
- König, E. Landolt-Börnstein Neue Serie Gruppe II; Hellwege, K.-H., Hellwege, A.M., Eds.; Springer-Verlag: Berlin, Heidelberg, Germany; New York, NY, USA, 1966; Volume 2, pp. 1-16–1-18. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. A new mixing of hartree-fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian basis set for molecular wavefunctions containing third-row atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Hay, P.J. Gaussian basis sets for molecular calculations. The representation of 3D orbitals in transition-metal atoms. J. Chem. Phys. 1977, 66, 4377–4384. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T/K | Spin-State | Ratio | IS 1/mm·s−1 | QS 2/mm·s−1 | LW 3/mm·s−1 |
|---|---|---|---|---|---|
| 293 | HS 4 | 82.1% | 0.259(17) | − | 2.06(11) |
| LS 5 | 17.9% | 0.01(2) | 3.13(5) | 0.76(11) | |
| 180 | HS | 59.6% | 0.22(4) | − | 3.4(2) |
| LS | 40.4% | 0.007(9) | 3.180(18) | 0.76(4) | |
| 70 | HS | 27.8% | 0.21(18) | − | 7.3(11) |
| LS | 72.2% | 0.07(3) | 3.190(6) | 0.703(14) | |
| 30 | HS | 26.1% | 0.2(6) | − | 7.2(9) |
| LS | 73.9% | 0.0640(17) | 3.171(4) | 0.621(8) | |
| 10.5 | HS | 24.2% | 0.2(7) | − | 5.1(6) |
| LS | 75.8% | 0.070(2) | 3.183(4) | 0.609(8) |
| 1 | 2 | 2’ | ||
|---|---|---|---|---|
| Formula | C38H35FeN6O4 | C39H38FeN5O5 | C36H32FeN5O4 | |
| Formula Weight | 695.57 | 712.59 | 654.51 | |
| Color | black | black | black | |
| Dimension/mm | 0.40 × 0.20 × 0.05 | 0.12 × 0.07 × 0.01 | 0.10 × 0.10 × 0.02 | |
| T/K | 90 | 273 | 90 | 296 |
| Crystal system | orthorhombic | orthorhombic | triclinic | triclinic |
| Space Group | Pbca | Pbca | P-1 | P-1 |
| a/Å | 18.337(4) | 18.323(3) | 9.105(4) | 9.885(6) |
| b/Å | 16.237(3) | 16.463(2) | 14.122(6) | 12.558(7) |
| c/Å | 22.673(4) | 23.132(3) | 15.504(6) | 14.709(8) |
| α/° | 90 | 90 | 64.034(5) | 65.497(7) |
| β/° | 90 | 90 | 77.865(6) | 76.767(7) |
| γ/° | 90 | 90 | 81.570(6) | 88.750(8) |
| V/Å3 | 6751(2) | 6977.8(17) | 1748.9(13) | 1611.8(15) |
| Z | 8 | 8 | 2 | 2 |
| ρcalcd/gcm−3 | 1.369 | 1.324 | 1.353 | 1.349 |
| μ (Mo-Kα) | 0.497 | 0.481 | 0.483 | 0.515 |
| 2θmax/° | 54.96 | 54.96 | 52.74 | 38.06 |
| No. Reflections (Rint) | 37753 (0.0804) | 38952 (0.0702) | 9261 (0.0245) | 4190 (0.0502) |
| No. Observations (I > 2.00σ(I)) | 7705 (5600) | 7969 (5419) | 6964 (4929) | 2592 (1423) |
| No. Variables | 488 | 488 | 457 | 419 |
| R1 (I > 2.00σ(I)) | 0.0878 | 0.0679 | 0.0609 | 0.0821 |
| R (all data) | 0.1136 | 0.0960 | 0.0972 | 0.1483 |
| wR2 (all data) | 0.2246 | 0.1989 | 0.1579 | 0.2391 |
| Residual electron density/eÅ−3 | 0.776 −0.755 | 0.691 −0.453 | 0.768 −0.399 | 0.717 −0.284 |
| Goodness of fit | 1.062 | 1.039 | 1.029 | 1.051 |
| 1 | 1 | 2 | 2’ | |
|---|---|---|---|---|
| T/K | 90 | 273 | 90 | 296 |
| Fe1-O1/Å | 1.913(3) | 1.960(2) | 1.999(3) | 1.983(9) |
| Fe1-O2/Å | 1.878(3) | 1.965(3) | 1.979(3) | 1.959(10) |
| Fe1-N1/Å | 1.952(3) | 2.110(2) | 2.161(3) | 2.146(12) |
| Fe1-O3/Å | 1.936(3) | 1.975(3) | 1.994(2) | 2.009(7) |
| Fe1-O4/Å | 1.884(4) | 1.969(3) | 1.983(2) | 1.998(8) |
| Fe1-N3/Å | 1.967(4) | 2.136(3) | 2.156(3) | 2.130(10) |
| Σ 1/° | 27.63(15) | 80.24(11) | 99.67(10) | 91.4(4) |
| Θ 2/° | 46.72(13) | 151.24(9) | 185.12(9) | 172.3(3) |
| φ 3/° | 172.44(15) | 168.96(10) | 168.30(9) | 165.0(4) |
| θ4/° | 87.60 | 87.51 | 88.97 | 87.6 |
| 1 | 1 | 2 | 2’ | |
|---|---|---|---|---|
| T/K | 90 | 273 | 90 | 296 |
| Fe···N/Å | ||||
| a | 5.429(5) | 5.549(4) | 4.982(3) | 5.552(12) |
| b | 5.784(4) | 5.724(3) | 5.649(4) | 5.661(12) |
| c | 5.756(4) | 5.736(3) | 5.767(4) | 5.831(11) |
| d | 8.202(4) | 8.275(4) | 10.299(5) | 10.202(12) |
| e | 11.368(6) | 11.810(5) | 11.381(5) | 9.526(12) |
| f | 11.581(6) | 11.760(5) | 13.357(5) | 11.847(14) |
| Fe···Fe/Å | ||||
| g | 8.5622(15) | 8.6246(11) | 8.183(3) | 8.252(5) |
| h | 9.3266(17) | 9.2947(13) | 8.235(3) | 7.925(4) |
| i | 10.7535(16) | 10.8840(12) | 8.758(3) | 9.655(6) |
| j | – | – | 9.105(4) | 9.885(6) |
| k | 11.611(2) | 11.7504(16) | 14.060(5) | 13.936(7) |
| π-distances/Å | ||||
| x | – | – | 3.44 | 3.52 |
| y | – | – | – | 3.43 |
| z | – | – | – | 3.44 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murata, S.; Takahashi, K.; Sakurai, T.; Ohta, H.; Yamamoto, T.; Einaga, Y.; Shiota, Y.; Yoshizawa, K. The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand. Crystals 2016, 6, 49. https://doi.org/10.3390/cryst6050049
Murata S, Takahashi K, Sakurai T, Ohta H, Yamamoto T, Einaga Y, Shiota Y, Yoshizawa K. The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand. Crystals. 2016; 6(5):49. https://doi.org/10.3390/cryst6050049
Chicago/Turabian StyleMurata, Suguru, Kazuyuki Takahashi, Takahiro Sakurai, Hitoshi Ohta, Takashi Yamamoto, Yasuaki Einaga, Yoshihito Shiota, and Kazunari Yoshizawa. 2016. "The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand" Crystals 6, no. 5: 49. https://doi.org/10.3390/cryst6050049
APA StyleMurata, S., Takahashi, K., Sakurai, T., Ohta, H., Yamamoto, T., Einaga, Y., Shiota, Y., & Yoshizawa, K. (2016). The Role of Coulomb Interactions for Spin Crossover Behaviors and Crystal Structural Transformation in Novel Anionic Fe(III) Complexes from a π-Extended ONO Ligand. Crystals, 6(5), 49. https://doi.org/10.3390/cryst6050049