Abstract
The enthalpies of formation of transition metals diborides in various structures have been obtained from density functional theory (DFT) calculations in order to determine the ground state at T = 0 K and p = 0. The evolution of the enthalpies of formation along the 3D, 4D, and 5D series has been correlated to the considered crystal structures. In the whole, the calculated values of the enthalpies of formation of the diborides in their ground state are in good agreement with the experimental ones when available. The calculated values of the lattice parameters at T = 0 K of the ground state agree well with the experimental values. The total and partial electronic densities of states have been computed. Special features of the transition metal electronic partial density of states have been evidenced and correlated to the local environment of the atoms.
1. Introduction
Transition metal borides are of great technological interest due to their physical and chemical properties such as high melting point, hardness, thermal conductivity, and chemical inertness. Diborides of transition metal of the beginning of the series possess the layered hexagonal C32 structure of AlB2-type (hP3). Diborides of elements of Mn and Fe columns crystallize in the hexagonal ReB2-type structure (hP6) or in the orthorhombic RuB2-type structure (oP6). Elements of Cr column such as Mo and W adopt more complicated structures: MoB2-type (hR6) and WB2-type (hP12). The mechanical properties of these diborides have been extensively studied experimentally or theoretically. A comprehensive review about the properties of borides obtained from experiment and theoretical calculations was given by Ivanovskii [1,2].
The aim of the present work is to obtain from density functional theory (DFT) calculations the enthalpies of formation of diborides of transition metals of the beginning of the 3D, 4D, and 5D series. The structures which have been considered are hP3 (AlB2-type), hR6 (or hP18) (CaSi2-type or MoB2-type), hP12 (WB2-type), hP6 (ReB2-type), oP6 (RuB2-type). Additionally, we have included in our study the following two structures: hR3 (CaSi2-type), and tI12 (ThSi2-type). This last structure has been included because first principles calculations have shown that this structure could be stable at high pressure in the cases of TiB2 [3] and MoB2 [4] compounds. The characteristics of the structures considered in the present work are given in Table 1 [5,6,7,8,9,10,11,12,13].

Table 1.
Structures of the diborides considered in the present work *.
| Compound | Pearson Symbol | Space Group | Lattice Parameters | Positions |
|---|---|---|---|---|
| TiB2 AlB2-type | hP3 | P6/mmm N°191 | Exp. [5] a = 3.032 Å c = 3.229 Å | B (2d) 1/3, 2/3, 1/2 Ti (1a) 0, 0, 0 |
| Exp. [6] a = 3.0316 Å c = 3.2301 Å | ||||
| Calc. * a = 3.0337 Å c = 3.2260 Å | ||||
| MoB2 Prot MoB2 or Prot CaSi2 6-lay | hP18 | R(-)3m N°166 | Exp. [7] a = 3.0119 Å c = 20.931 Å | B1 (6c) 0, 0, 0.33306 B2 (6c) 0, 0, 0.18155 Mo (6c) 0, 0, 0.0750 |
| hP18 | Exp. [8] a = 3.0136 Å c = 20.939 Å | B1 (6c) 0, 0, 0.33230 B2 (6c) 0, 0, 0.18184 Mo (6c) 0, 0, 0.07569 | ||
| hP18 | Calc. * a = 3.0201 Å c = 21.0353 Å | B1 (6c) 0, 0, 0.33214 B2 (6c) 0, 0, 0.18147 Mo (6c) 0, 0, 0.07599 | ||
| hR6 | R(-)3m N°166 | Calc. * a = 7.2266 Å α = 24.1286 Å | B1 (2c) 0.33219, 0.33219, 0.33219 B2 (2c) 0.18146, 0.18146, 0.18146 Mo (2c) 0.07595, 0.07595, 0.07595 | |
| Prot WB2 HT | hP12 | P63/mmc N° 194 | Exp. [7] [Frotscher] a = 2.9864 Å c = 13.896 Å | B (2b)0, 0, 1/4 B (2c) 1/3, 2/3, 1/4 B (4f) 1/3, 2/3, 0.0243 Mo (4f) 1/3, 2/3, 0.63759 |
| Exp. [9] a = 2.9872 Å c = 13.8823 Å | B (2b)0, 0, 1/4 B (2c) 1/3, 2/3, 1/4 B (4f) 1/3, 2/3, 0.0243 Mo (4f) 1/3, 2/3, 0.63759 | |||
| Calc. * a = 3.0196 Å c = 14.0937 Å | B (2b)0, 0, 1/4 B (2c) 1/3, 2/3, 1/4 B (4f) 1/3, 2/3, 0.02276 Mo (4f) 1/3, 2/3, 0.63481 | |||
| Prot ReB2 | hP6 | P63/mmc N° 194 | Exp. [10] a = 2.9005 Å c = 7.4772 Å | B (4f) 1/3, 2/3, 0.54783 Re (2c) 1/3, 2/3, 1/4 |
| Exp. [11] a = 2.8982 Å c = 7.4723 Å | B (4f) 1/3, 2/3, 0.547 Re (2c) 1/3, 2/3, 1/4 | |||
| Calc. * a = 2.9178 Å c = 7.5014 Å | B (4f) 1/3, 2/3, 0.5478 Re (2c) 1/3, 2/3, 1/4 | |||
| Prot RuB2 | oP6 | Pmmn N° 59 | Exp. [10] a = 2.8651 Å b = 4.6448 Å c = 4.0456 Å | B (4e) 1/4, 0.0544, 0.1385 Ru (2a) 1/4, 1/4, 0.6505 |
| Exp. [12] a = 2.8657 Å b = 4.6457 Å c = 4.0462 Å | B (4e) 1/4, 0.063, 0.138 Ru (2a) 1/4, 1/4, 0.649 | |||
| Calc. * a = 2.8804 Å b = 4.6637 Å c = 4.0582 Å | B (4e) 1/4, 0.0544, 0.1364 Ru (2a) 1/4, 1/4, 0.6518 | |||
| Prot ThSi2 | tI12 | I41/amd N° 141 | Exp. [13] a = 4.127 Å c = 14.194 Å | Si (8e) 0, 1/4, 0.2915 Th (4a) 0, 3/4, 1/8 |
| Prot CaSi2 3-lay | hR9 | R(-3)m N° 166 | Exp. [13] a = 3.8295 Å c = 15.904 Å | Si (6c) 0, 0, 0.19733 Ca (3a) 0, 0, 0 |
The calculations have been performed for elements of Ti to Fe columns of the periodic table. For the transition metals belonging to the Co and Ni columns, no TMB2 compound is stable. In the case of the Sc column, no LaB2 compound is stable. This can be verified in the publication of Van Der Geest and Kolmogorov [14]. Indeed, these authors have recently investigated a large number of binary boron alloys. The convex hull of enthalpies of formation at zero temperature and pressure has been proposed for a large number of binary boron-based systems.
Investigations of the evolution of the enthalpies of formation of 4D and 5D transition metal diborides have been published [15,16,17,18,19,20,21,22,23,24,25,26,27]. Some of these studies are restricted to the AlB2-type structure [15,16,17,18,19]. In others, the enthalpies of formation of the diborides in the hR6, hP12, hP6, and oP6 structures have been calculated either in 4D [20,21,22] or in the 5D series [23,24,25,26,27].
2. Computational Details
The DFT calculations were performed with the Vienna ab initio simulation package (VASP) [28,29], making use of the projector augmented waves (PAW) technique [30,31]. For the generalized gradient approximation (GGA) exchange correlation functional, we have used the Perdew-Burke-Ernzerhof parameterization (PBE) [32]. A plane-wave cutoff energy of 400 eV for each element has been taken. For the Brillouin-zone integration, the Methfessel-Paxton [33] technique with a modest smearing of the one-electron levels (0.2 eV) was used. The structures are fully relaxed. Care is taken that no additional symmetries are gained during the relaxation procedure. For all the hexagonal structures, a gamma centered k-point grid is used. For the other structures, a Monkhorst-Pack [34] grid is chosen. In each case, the number of k points has been determined such a way that the number of irreducible k points multiplied by the number of atoms in the cell is at least greater than 500. In this way, there is convergence of the values of the lattice parameters and of the total energy. All calculations are performed using the “accurate” setting within VASP to avoid wrap-around errors. With the chosen plane-wave cutoff and k-point sampling, the reported enthalpies of formation are estimated to be converged with a precision better than ±0.2 kJ/mol of atoms. Spin-polarized calculations have been performed in the cases of CrB2, MnB2, and FeB2 compounds.
The energy of formation per atom, , is obtained from the minimum total energy of the compound (expressed per atom) by subtracting the composition-weighted minimum total energies of pure TM element in its stable state at T=0K and α rhombohedral boron (hR 12):
At T = 0 K and p = 0 Pa, the formation enthalpy equals the calculated formation energy when the zero-vibration contribution which is much smaller is ignored.
We chose the α rhombohedral boron (hR 12) as reference state. The reference state usually taken in the databases [35] is β rhombohedral boron (hR 105 without defects). Two recent works [36,37] have shown that α rhombohedral boron is the most stable structure at T = 0K. This point has been discussed in a previous publication [38].
3. Results
The enthalpies of formation of the diborides in the various structures we have considered in the present work are displayed in Figure S1 and Table S1 of the supplementary data. The values reported for CrB2, MnB2, and FeB2 compounds correspond to spin-polarized calculations: anti-ferromagnetic states in the case of CrB2 and MnB2, and ferromagnetic state in the case of FeB2. In the case of MnB2 where the magnetic effects are important the values of the enthalpies of formation of the various structures in the non-magnetic and anti-ferro magnetic state are presented in the supplementary data. The first observation is that three sets of values are clearly defined. The first set corresponding to the C32 and ThSi2-type structures, the second set to the hP12 (WB2-type) and hR6 (CaSi2-type) structures, and the third set to hP6 (ReB2-type), oP6 (RuB2-type), and hR3 (CaSi2-type) structures. In each set, the values of the enthalpies of formation are very close. The enthalpies of formation of the diborides in the hP3 and tI12 structures present a regular increase with the atomic number of the transition metal. The enthalpies of formation of the hP12 and hR6 structures present an increase with the atomic number in the 3D series, a decrease from Zr to Nb or from Hf to Ta and then a regular increase with the atomic number. In the hP6, oP6, and hR3 structures, one observes a minimum of the enthalpy of formation for the transition metals of the Mn column. Inspection of the values of the enthalpies of formation allows to obtain the ground state for each diboride at T = 0K and under zero pressure: C32 for Ti, V, Zr, Nb, Hf, hP12 (WB2-type) for Ta and Cr, hR6 (CaSi2-type) for Mo and W (in these two cases the enthalpy of formation of the hP12 structure is less stable than the hR6 one of less 1 kJ), hP6 (ReB2-type) for Mn, Tc, and Re, oP6 (RuB2-type) for Ru and Os diborides. A comparison with the experimental information may be found in Table 2. In the case of the FeB2 compound we reported in Table 2 the ground state found by Kolmogorov et al. [39] and confirmed later by Li et al. [40]. Indeed, Kolmogorov et al. [39] found that in the case of FeB2 an orthorhombic structure, oP12, is more stable than the other structures. The results presented in Table 2 are in agreement with the results published by Van der Geest and Kolmogorov [14]. The discrepancies observed with the experimental investigations have already been discussed by these authors. In the case of MnB2 compound, all the GGA calculations show that the hP6 structure is the most stable one. Recently, Fan et al [41] have shown that hP3-MnB2 is stable and hP6-MnB2 is not stable at high temperature and pressure. Therefore, in the experimental conditions, only hP3-MnB2 can be synthesized. More recently, Gou et al. [42] performed calculations using the GGA+U functional and have shown that hP3-MnB2 is more stable than hP6-MnB2. As conclusion, further investigations are needed in order to clarify the problem.
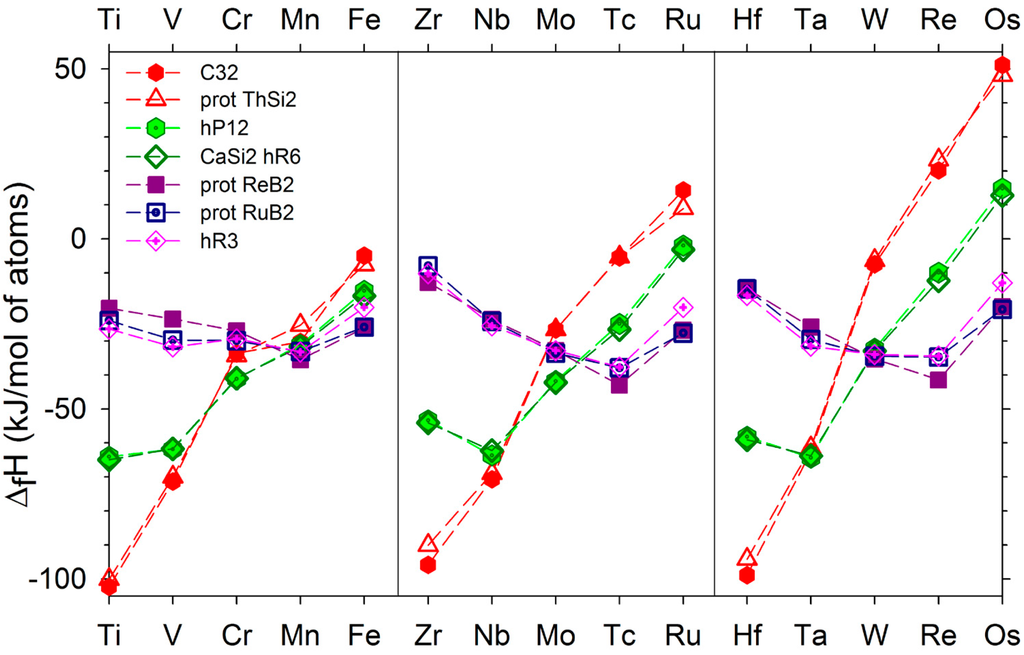
Figure 1.
Enthalpies of formation of the TMB2 compounds in the hP3 (AlB2-type), tI12 (α-ThSi2-type), hP12 (WB2-type), hR6 (MoB2-type), hP6 (ReB2-type), oP6 (RuB2-type), and hR3 (CaSi2-type) structures.
Figure 1.
Enthalpies of formation of the TMB2 compounds in the hP3 (AlB2-type), tI12 (α-ThSi2-type), hP12 (WB2-type), hR6 (MoB2-type), hP6 (ReB2-type), oP6 (RuB2-type), and hR3 (CaSi2-type) structures.
Table 2.
Structure of the early transition metal diborides observed experimentally [13] and calculated in the present work as being the ground state or only slightly metastable at zero temperature and pressure.
| Compound | TiB2 | VB2 | CrB2 | MnB2 | FeB2 |
|---|---|---|---|---|---|
| Experimental | hP3 | hP3 | hP3 | hP3 | hP3 |
| Ab-initio | hP3 | hP3 | hP12 | hP6 | oP12 |
| Compound | ZrB2 | NbB2 | MoB2 | TcB2 | RuB2 |
| Experimental | hP3 | hP3 | hR6, hP3(HT) | hP6 | oP6 |
| Ab-initio | hP3 | hP3 | hR6, hP12 | hP6 | oP6 |
| Compound | HfB2 | TaB2 | WB2 | ReB2 | OsB2 |
| Experimental | hP3 | hP3 | hP3, hP12 | hP6 | oP6 |
| Ab-initio | hP3 | hP12 | hP6 | hP6 | oP6, hP6 |
The available experimental values of the enthalpies of formation of the transition metal diborides are given in Table 3 [43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63].We have included in this table the experimental and calculated values of the formation enthalpies of C32-ScB2 and C32-YB2. The experimental data are scarce and the more often old. They present a large scatter when several determinations have been performed in a system. The calculated values are given for the ground state structure and also for the nearest metastable structure. In the whole, there is an agreement between the experimental and the calculated values and we can emphasize the nice agreement with the calorimetric values of the Kleppa’s group [45,48,49,57,58,59,60,61,64]. This agreement is very good for diborides of transition metals of Ti and V columns.
The lattice parameters corresponding to the prototypes are indicated in Table 1. The agreement between experimental and calculated values is good. One may notice that as usual the GGA values of the lattice parameters are slightly higher than the experimental ones. Additionally, those corresponding to the ground state and near stable structures are reported in Table S2 of the Supplementary data. The values of the volume per atom of each compound in each structure are displayed in Figure S2 and in Table S1 of the Supplementary data. The atomic volumes of the compounds present the same evolution with the atomic number as those of the pure transition metals (see Figure S1 of supplementary data). Inspection of Figure 2 gives the possibility of predicting high pressure transitions. Indeed a pressure increase favors the structure possessing the smallest atomic volume. In the case of TiB2, one observes that the atomic volume of the tI12 structure is smaller than the one of hP3 structure. Therefore a transition hP3-TiB2 to tI12-TiB2 could be possible at high pressure. This prediction is in agreement with the theoretical work of Zhang et al. [3]. The same observation can be made in the case of MoB2 compounds [4]. In the case of OsB2 compound, the atomic volume of the hP6 structure is smaller than the one of oP6 structure. Therefore a transition oP6-OsB2 to hP6-OsB2 could be expected at high pressure. This was theoretically predicted by Ren et al. [65], and recently confirmed experimentally [66]. Finally one observes that the molar volume of the hP3 structure is always smaller than the one of the hR6, hP12 structures. This could explain why the hP3 structure is often observed experimentally while the calculations show that this structure is not the ground state.
Table 3.
Experimental and calculated values of the formation enthalpies of TMB2 compounds.
| Compound | Experimental T = 298 K∆fH (kJ/mol of Atoms) | Method | Calculated T = 0 K Present Work (kJ/mol of Atoms) | |
|---|---|---|---|---|
| ScB2 | −102.3 | SSD [43] | hP3: −80.99 | |
| TiB2 | −69.7 | Calo. [44] | hP3: −102.30 | |
| −107.9 | OBC [45] | |||
| −109.5 | Calo. [46] | |||
| −98 | Calo. [47] | |||
| −105 | Equil. [48] | |||
| −109.5 | SSD [43] | |||
| −107.3, −107.8 | EMF [49] | |||
| VB2 | −67.9 | Equil. [50] | hP3: −71.33 | |
| −70.7 | SSD [43] | |||
| CrB2 | −39.8 | SSD [51] | hP3: −31.90 | |
| hP12: −41.13 | ||||
| MnB2 | −21.1 | SC [52] | hP3: −14.06 | |
| hP6: −35.52 | ||||
| YB2 | −35.7 | DSC [53] | hP3: −54.59 | |
| ZrB2 | −97.6 | Vap. Press. [54] | hP3: −95.87 | |
| −107.7 | OBC [55] | |||
| −103.3 | Vap. Press. [56] | |||
| −108.9 | FBC [57] | |||
| −93 | Calo. [47] | |||
| NbB2 | −82.4 | OBC [58] | hP3: −70.71 | |
| −65.9 | Calo. [59] | |||
| −85.3 | FBC [60] | |||
| −73 | Calo. [47] | |||
| −60.3 | DSC [61] | |||
| HfB2 | −106.55 | Vap. Press. [62] | hP3: −98.90 | |
| −109.5 | FBC [57] | |||
| −85 | Calo. [47] | |||
| TaB2 | −62.9 | FBC [60] | hP3: −62.58, | |
| −64.9 | Calo. [47] | hP12: −64.32 | ||
| −53.3 | DSC [53] | |||
| ReB2 | −21.5 | DSC [61] | hP6: −41.50 | |
| OsB2.5 | −11.4 | DSC [63] | oP6: −20.77 | |
SC: Solution calorimetry, DSC: Direct synthesis calorimetry, SSD: solute+solvent drop calorimetry, vap.press: vapour pressure, Calo: calorimetry, FBC: fluorine bomb calorimetry, OBC: oxygene bomb calorimetry.
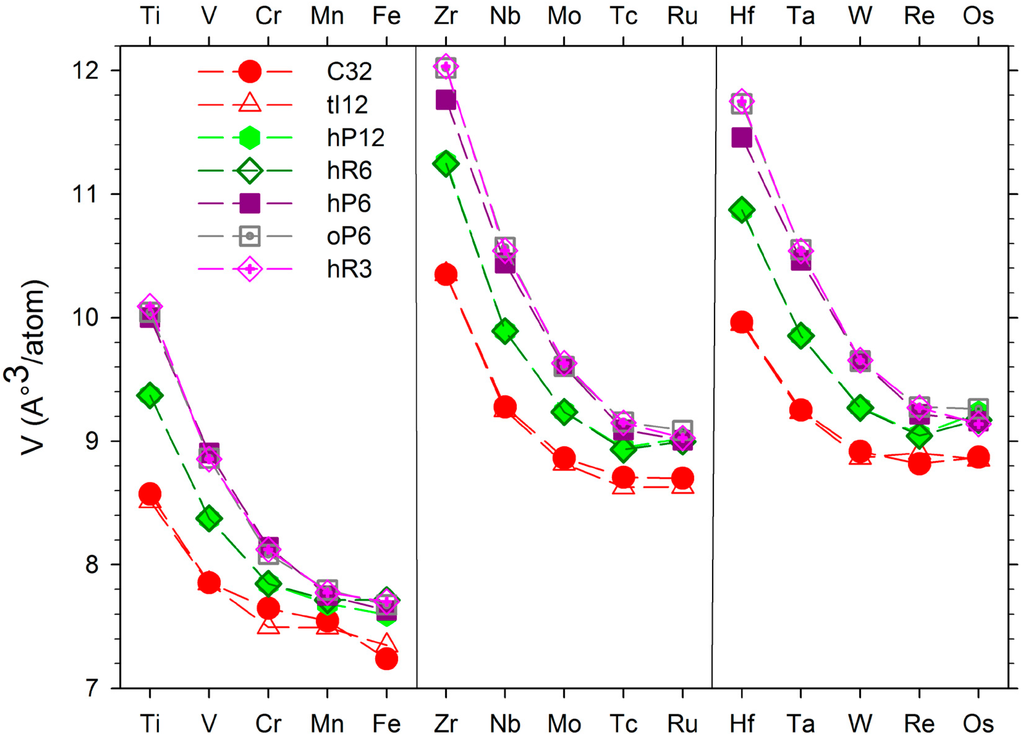
Figure 2.
Atomic volumes of the TMB2 compounds in the hP3 (AlB2-type), tI12 (α-ThSi2-type), hP12 (WB2-type), hR6 (MoB2-type), hP6 (ReB2-type), oP6 (RuB2-type), and hR3 (CaSi2-type) structures.
Figure 2.
Atomic volumes of the TMB2 compounds in the hP3 (AlB2-type), tI12 (α-ThSi2-type), hP12 (WB2-type), hR6 (MoB2-type), hP6 (ReB2-type), oP6 (RuB2-type), and hR3 (CaSi2-type) structures.
4. Discussion
The hP3 (C32) structure is a hexagonal layered structure in which the transition metal atoms are in the plan z = 0 and the B atoms form hexagons in the plan z = 1/2. Figure 3 presents the environment of the transition metal and the B atoms. The transition metal atoms are surrounded by twelve B atoms at a distance, where a and c are the hP3 lattice parameters. The B atoms are surrounded by three B atoms at a distance very short distance (typically 1.7 Å) and by six transition metal atoms. The bond lengths reported in Table 4 are those calculated for hP3-TiB2 compound.
Table 4.
Environment of the TM and B atoms in the hP3 structure.
| Atom | Env. | Nb. | Distance (Å) |
|---|---|---|---|
| B | B | 3 | 1.751 |
| Ti | 6 | 2.381 | |
| Ti | B | 12 | 2.381 |
In the ThSi2-type structure (Figure 4), the environments of the transition metal and B atoms is identical than the one in the hP3 structure. More the bond-lengths are very near the ones in the hP3 structure.
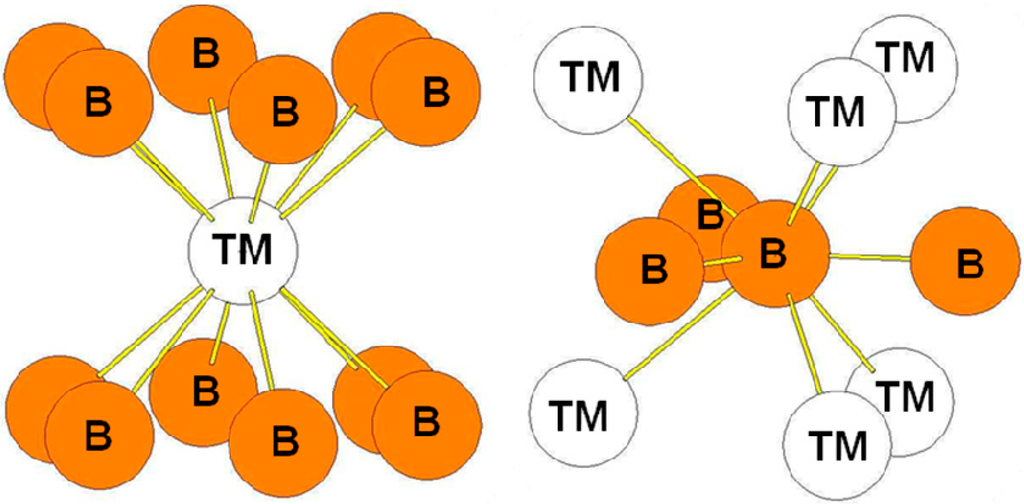
Figure 3.
Environment of TM and B atoms in the hP3 (AlB2-type) structure.
Figure 3.
Environment of TM and B atoms in the hP3 (AlB2-type) structure.
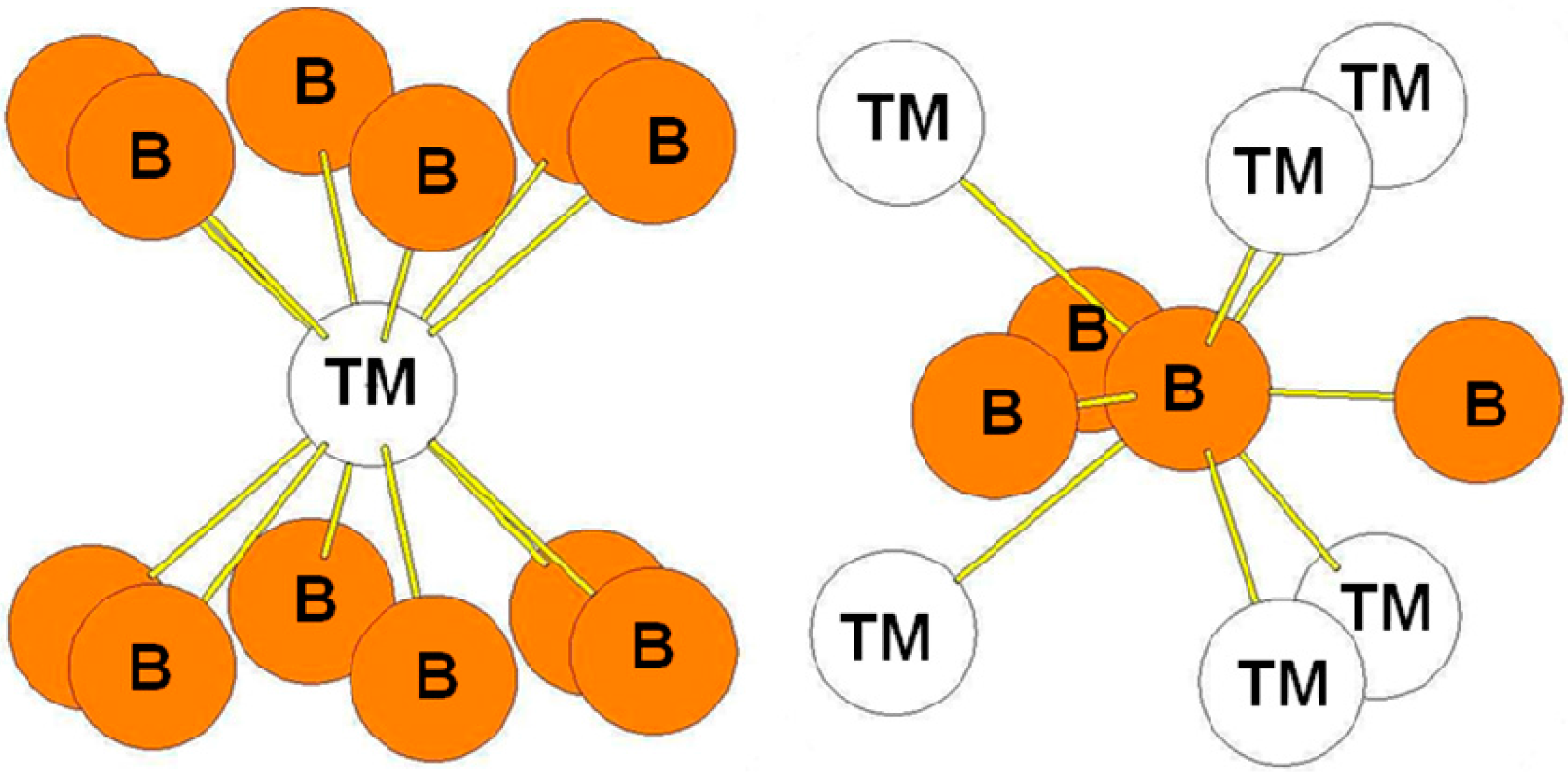
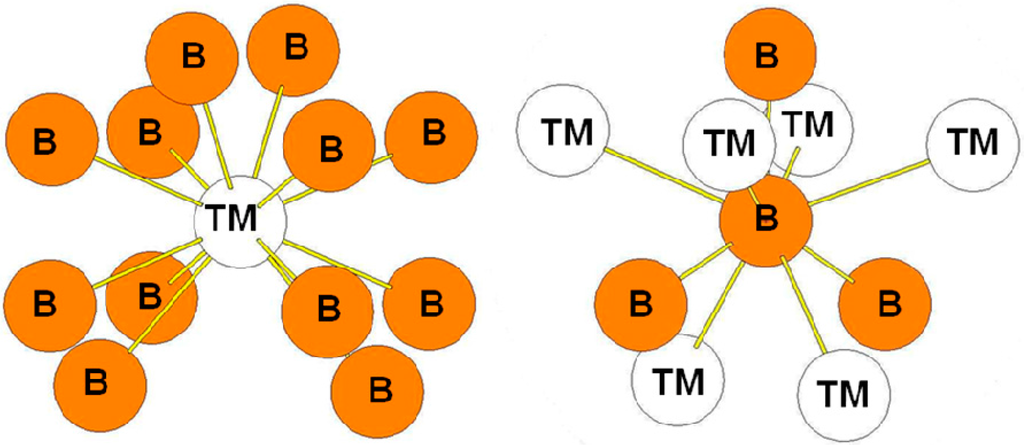
Figure 4.
The environment of TM and B atoms in the tI12 (α-ThSi2-type) structure.
Figure 4.
The environment of TM and B atoms in the tI12 (α-ThSi2-type) structure.
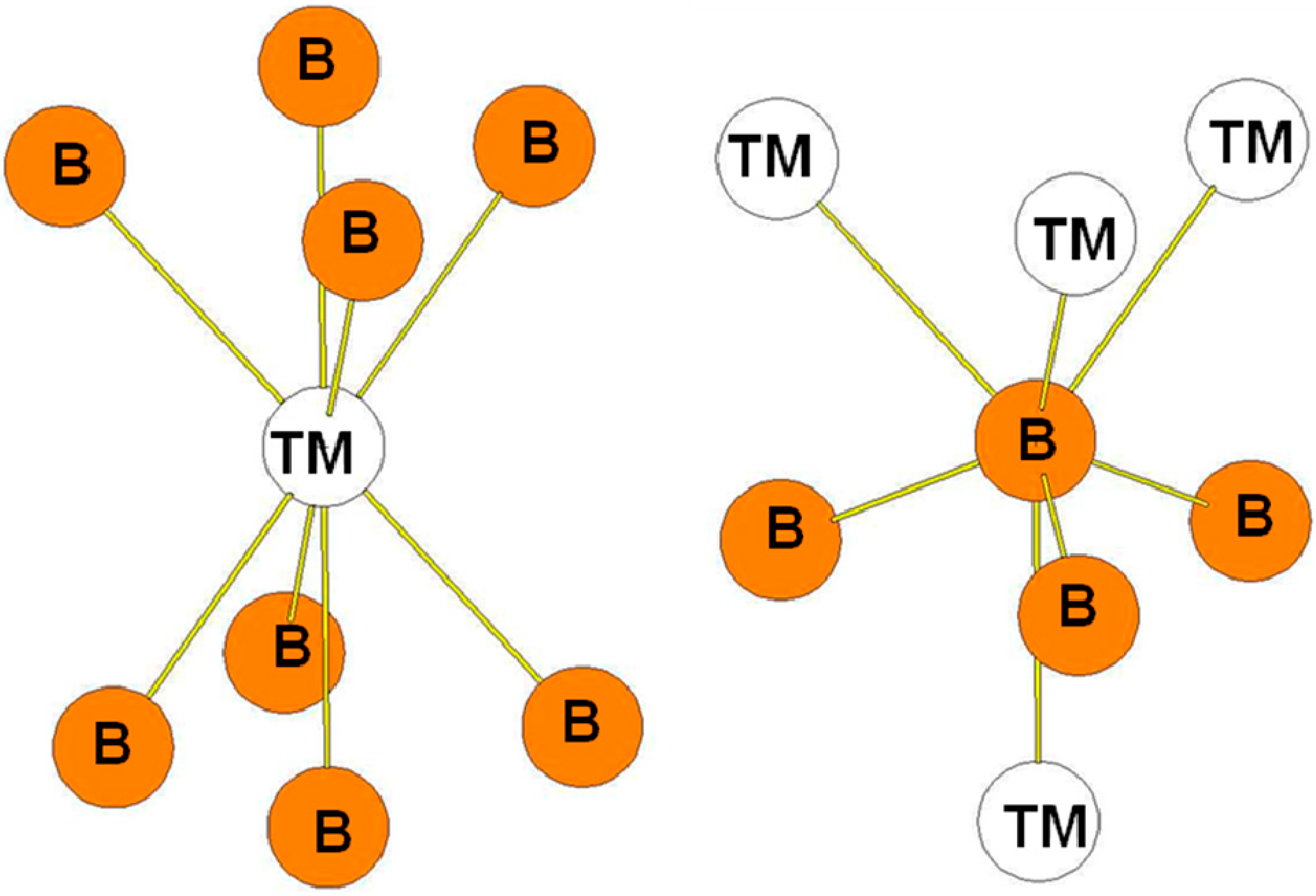
In the hR6 (hP18) structure (MoB2-type), the B atoms occupy two 2c Wyckoff positions (B1: 0.3322, 0.3322, 0.3322 and B2: 0.1815, 0.1815, 0.1815) while the transition metal atoms occupy other 2c positions (TM: 0.076, 0.076, 0.076) (see Figure 5a). The transition metal atoms are surrounded by six B1 atoms and four B2 atoms (see Figure 6). The B1 atoms in 2c positions with x = 0.3322 are surrounded by six transition metals atoms and three B1 atoms at a short distance. The B2 atoms in the 2c positions with x = 0.1815 are surrounded by four TM atoms and by three B2 atoms also at a short distance. Therefore the two different B atoms have different coordination numbers (CN = 9 for B1 atoms and CN = 7 for B2 atoms). The environment of the B1 atoms is the same as the one of the B atoms in the hP3 structure. The bond lengths reported in Table 5 are those calculated for hR6-MoB2 compound.
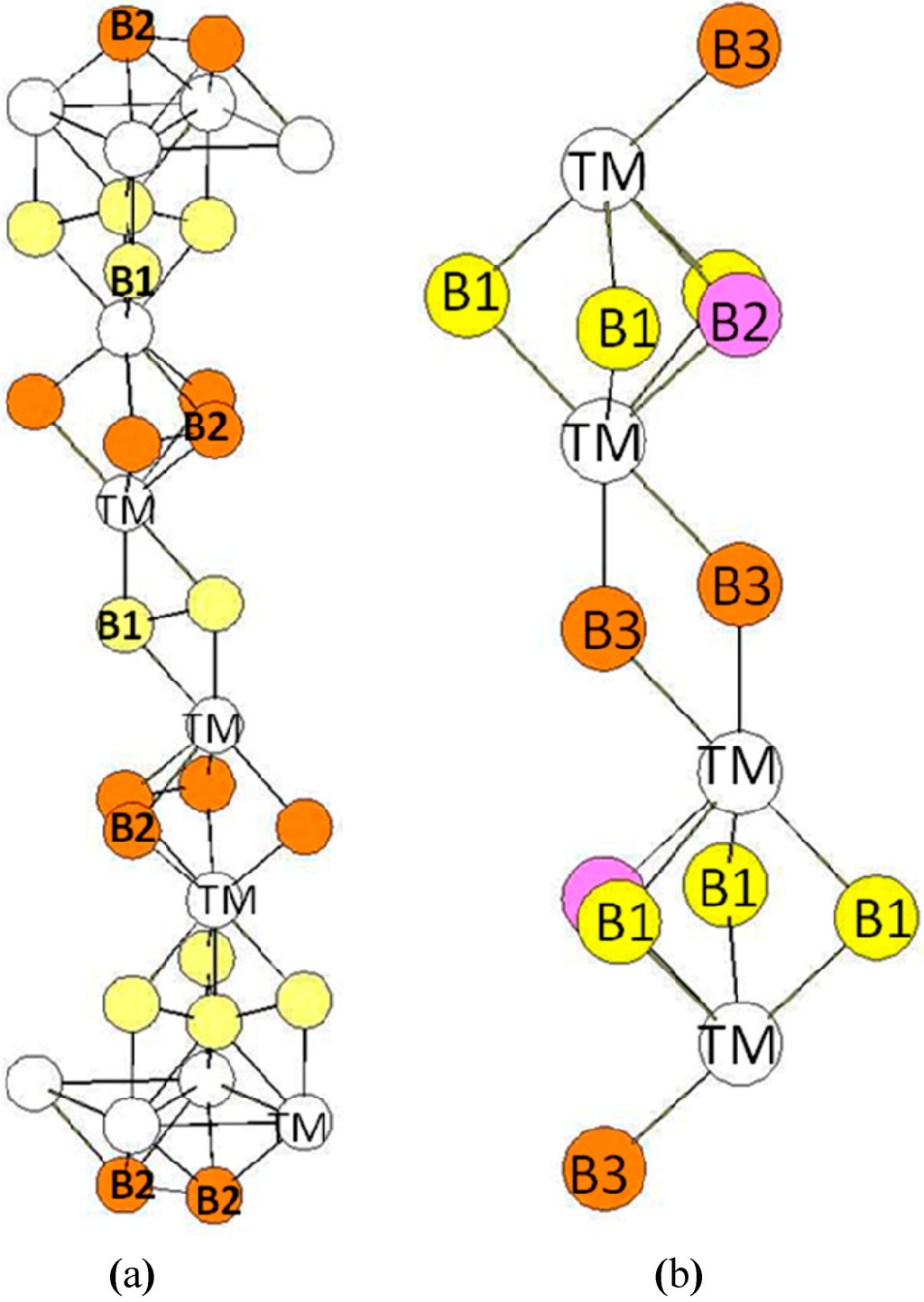
Figure 5.
(a) hR6 (MoB2-type) structure. The Wyckoff positions of B1 and B2 atoms in the R(-)3m group space are 2c (x = y = z = 0.33219) and 2c (x = y = z = 0.18146); (b) hP12 (WB2-type) structure. The Wyckoff positions of B1, B2, and B3 atoms in the P63/mmc group space are 2b, 2c, and 4f (see Table 1).
Figure 5.
(a) hR6 (MoB2-type) structure. The Wyckoff positions of B1 and B2 atoms in the R(-)3m group space are 2c (x = y = z = 0.33219) and 2c (x = y = z = 0.18146); (b) hP12 (WB2-type) structure. The Wyckoff positions of B1, B2, and B3 atoms in the P63/mmc group space are 2b, 2c, and 4f (see Table 1).
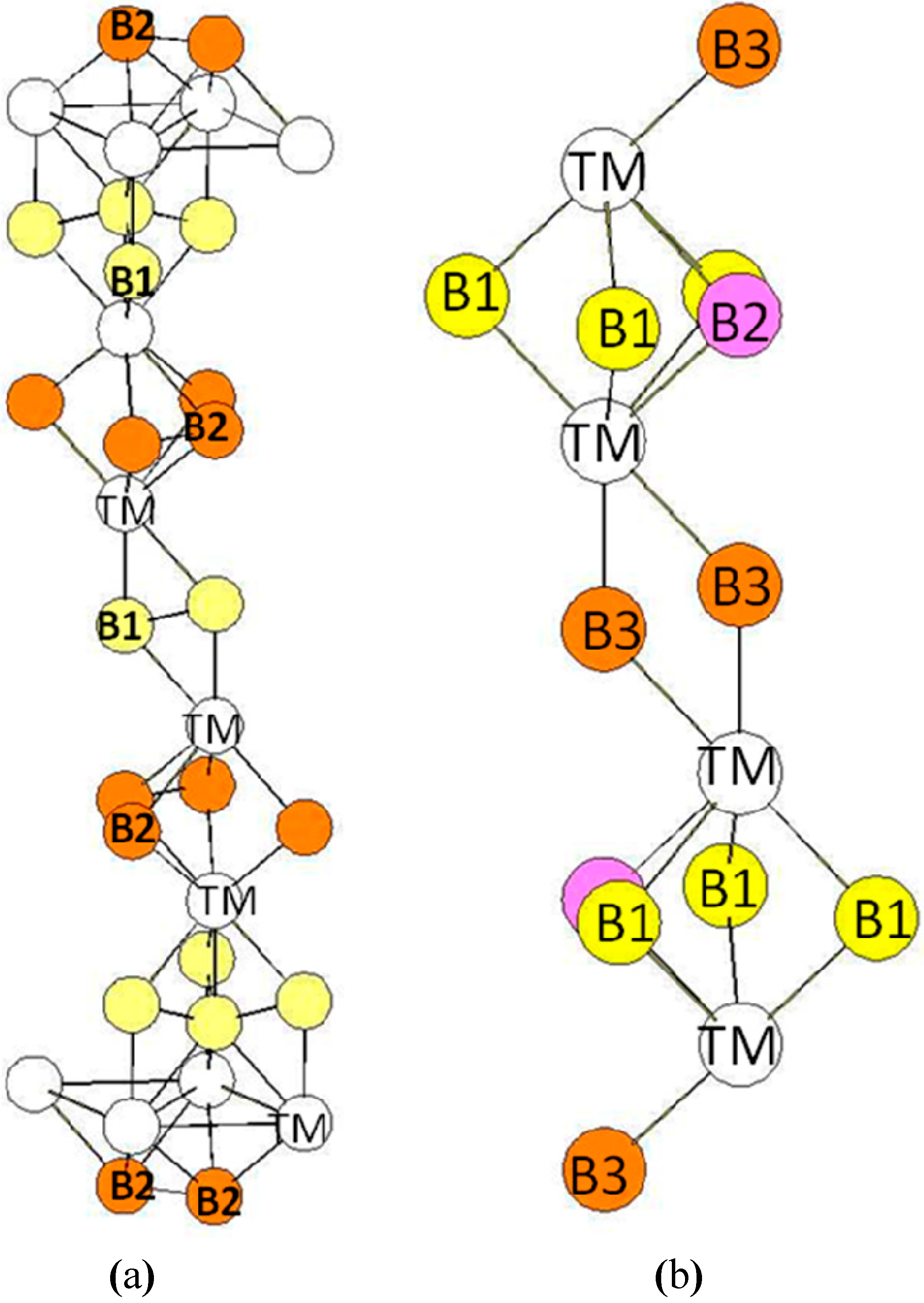
Table 5.
Bond lengths in the hR6-MoB2 compound. The Wyckoff positions of the B1 and B2 atoms are indicated in Table 1.
| Atom | Env. | Number | Distance(Å) |
|---|---|---|---|
| B1 | B1 | 3 | 1.745 |
| Mo | 3 | 2.382 | |
| Mo | 3 | 2.349 | |
| B2 | B2 | 3 | 1.852 |
| Mo | 1 | 2.22 | |
| Mo | 3 | 2.365 | |
| Mo | B1 | 3 | 2.382 |
| B1 | 3 | 2.349 | |
| B2 | 1 | 2.22 | |
| B2 | 3 | 2.365 | |
| Mo | 6 | 3.021 |
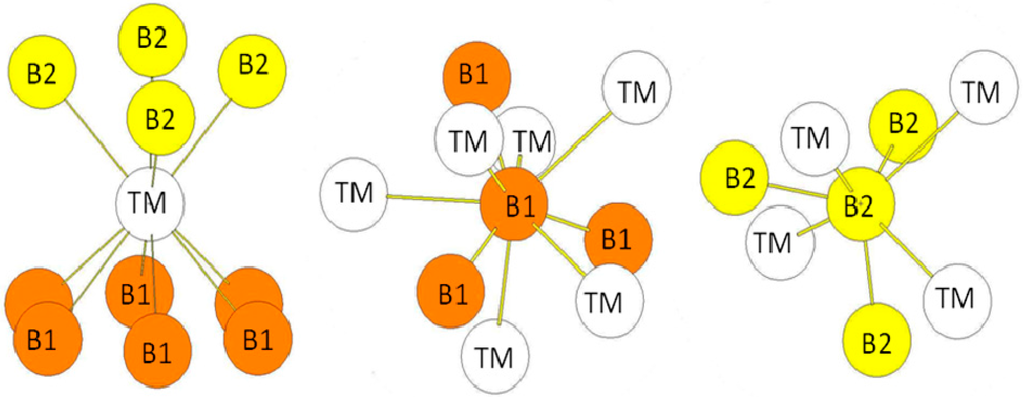
Figure 6.
Environment of TM and B atoms in the hR6 (MoB2-type) structure. The Wyckoff positions of B1 and B2 atoms in the R(-)3m group space are 2c (x = y = z = 0.33219) and 2c (x = y = z = 0.18146) (see Table 1).
Figure 6.
Environment of TM and B atoms in the hR6 (MoB2-type) structure. The Wyckoff positions of B1 and B2 atoms in the R(-)3m group space are 2c (x = y = z = 0.33219) and 2c (x = y = z = 0.18146) (see Table 1).
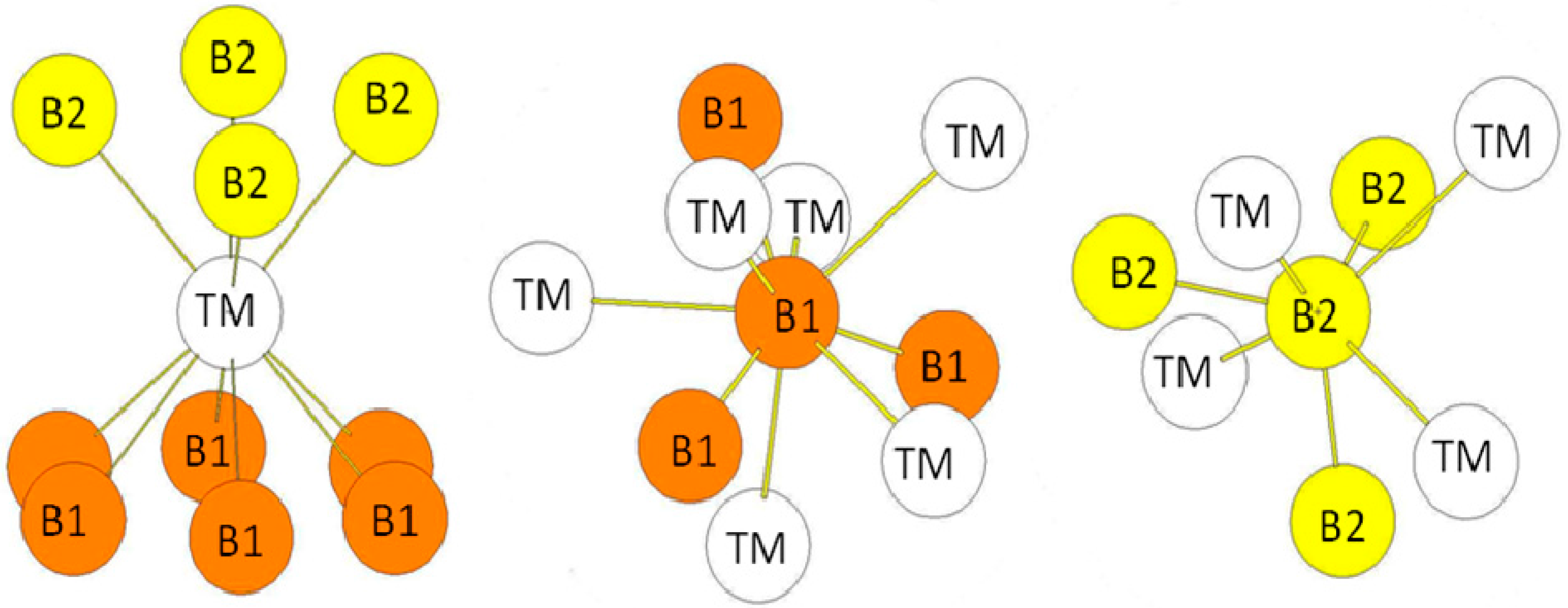
In the hP12 structure (WB2-type), the B atoms occupy the 2b, 2c, and 4f (z = 0.022762) Wyckoff positions while the transition metal atoms occupy other 4f Wyckoff positions with z = 0.634809 (see Figure 5b). The environment of the atoms is presented in Figure 7. The transition metal atoms are surrounded by ten B atoms divided in three B atoms (B1 in the 2b Wyckoff positions), three B atoms (B2 in the 2c Wyckoff positions), and four B atoms (B3 in the 4f Wyckoff positions). The B1 atoms are surrounded by three B2 atoms and six TM atoms. Similarly, the B2 atoms are surrounded by three B1 atoms and six TM atoms. The B3 atoms (4f positions) are surrounded by four TM atoms and three B3 atoms. The B-B bond-lengths are very short as in the hR6 structure. The environment of the B3 atoms is clearly different from the ones of B1 and B2 atoms. One may also quote that the environment of the B1 and B2 atoms is the same as the one of the B atoms in the hP3 structure. The bond lengths reported in Table 6 are those calculated in the case of the hP12-WB2 compound.
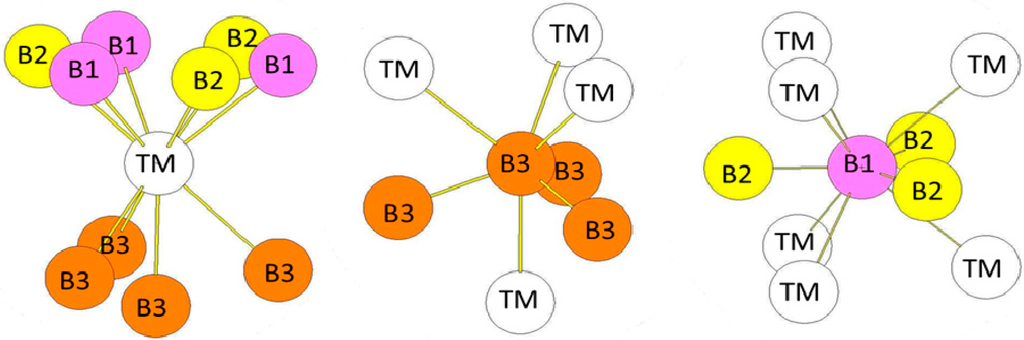
Figure 7.
The environment of TM and B atoms in the hP12 (WB2-type) structure. The Wyckoff positions of B1, B2, and B3 atoms in the P63/mmc group space are 2b, 2c, and 4f respectively (see Table 1).
Figure 7.
The environment of TM and B atoms in the hP12 (WB2-type) structure. The Wyckoff positions of B1, B2, and B3 atoms in the P63/mmc group space are 2b, 2c, and 4f respectively (see Table 1).
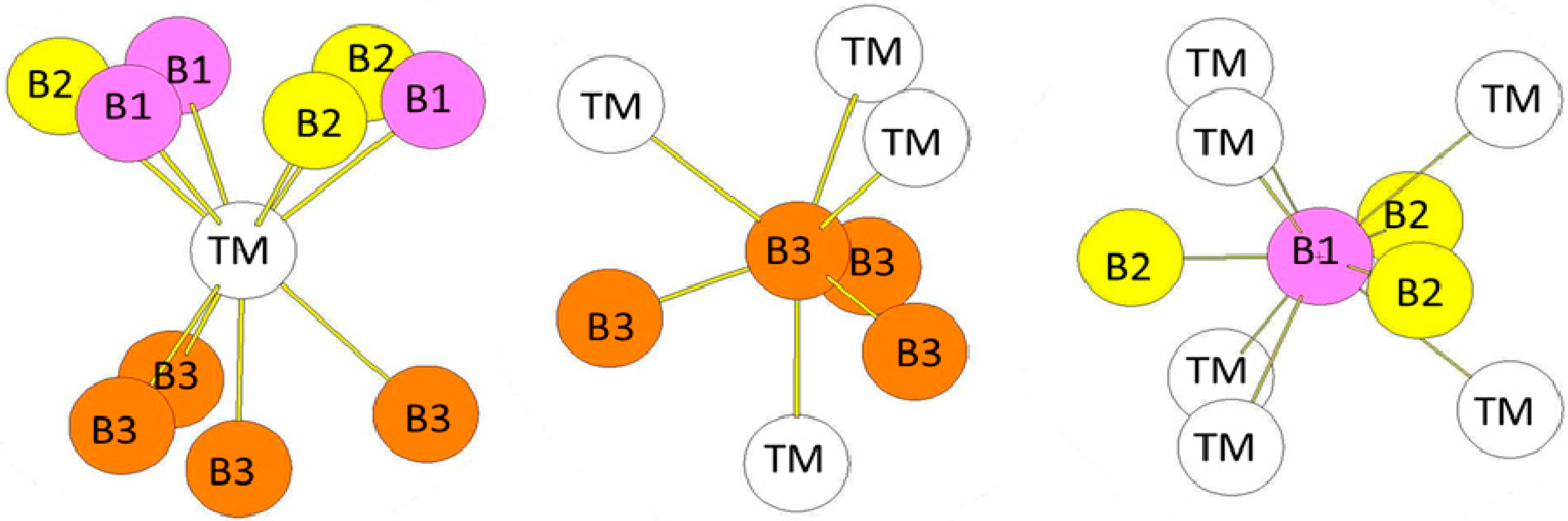
Table 6.
Bond lengths in the hP12 WB2 compound.
| Atom | Env. | Nb. | Distance(Å) |
|---|---|---|---|
| B (2b) | B (2c) | 3 | 1.743 |
| W (4f) | 6 | 2.382 | |
| B (2c) | B (2b) | 3 | 1.743 |
| W (4f) | 6 | 2.382 | |
| B (4f) | B (4f) | 2 | 1.858 |
| W (4f) | 1 | 2.221 | |
| W (4f) | 3 | 2.352 | |
| W (4f) | B (4f) | 1 | 2.221 |
| B (4f) | 3 | 2.352 | |
| B (2b) | 3 | 2.382 | |
| B (2c) | 3 | 2.382 |
In the ReB2-type structure, the B atoms occupy the 4f Wyckoff positions while the transition atoms occupy the 2c ones. The surroundings of the B and Re atoms in the structure are presented in Figure 8. The transition metal atoms are surrounded by eight B atoms while the B atoms are surrounded by three B atoms at a short distance and by four transition metal atoms. The bond lengths reported in Table 7 are those calculated for the hP6-ReB2 compound. The environment of the B atoms is the same as the one of the B2 atoms in the case of the hR6 structure and as the one of B3 atoms in the case of the hP12 structure.
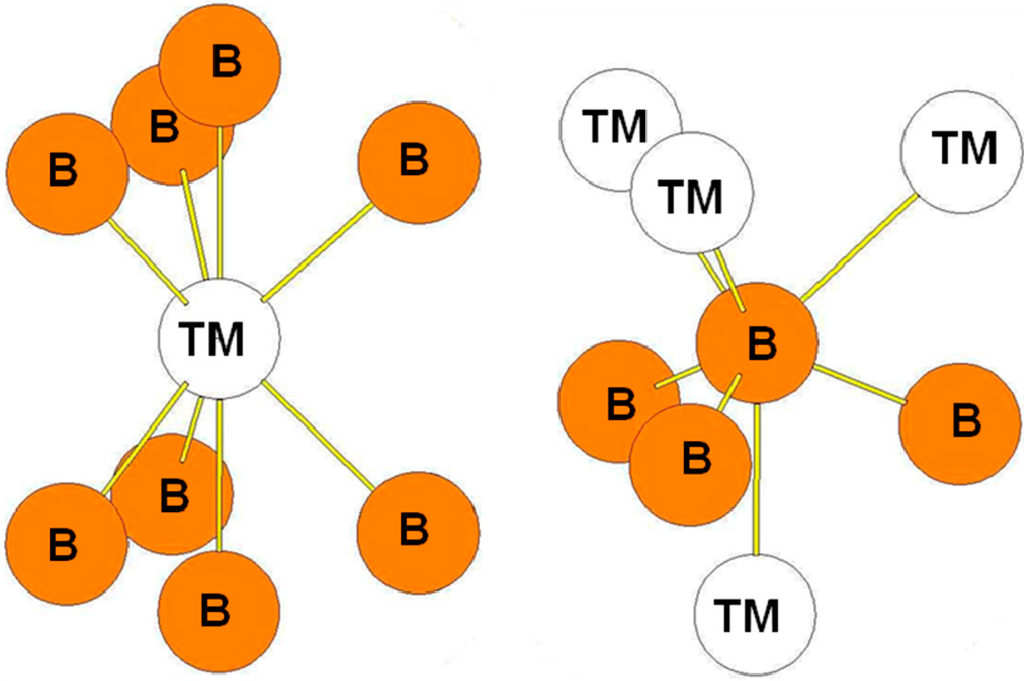
Figure 8.
Environment of TM and B atoms in the hP6 (ReB2-type) structure.
Figure 8.
Environment of TM and B atoms in the hP6 (ReB2-type) structure.
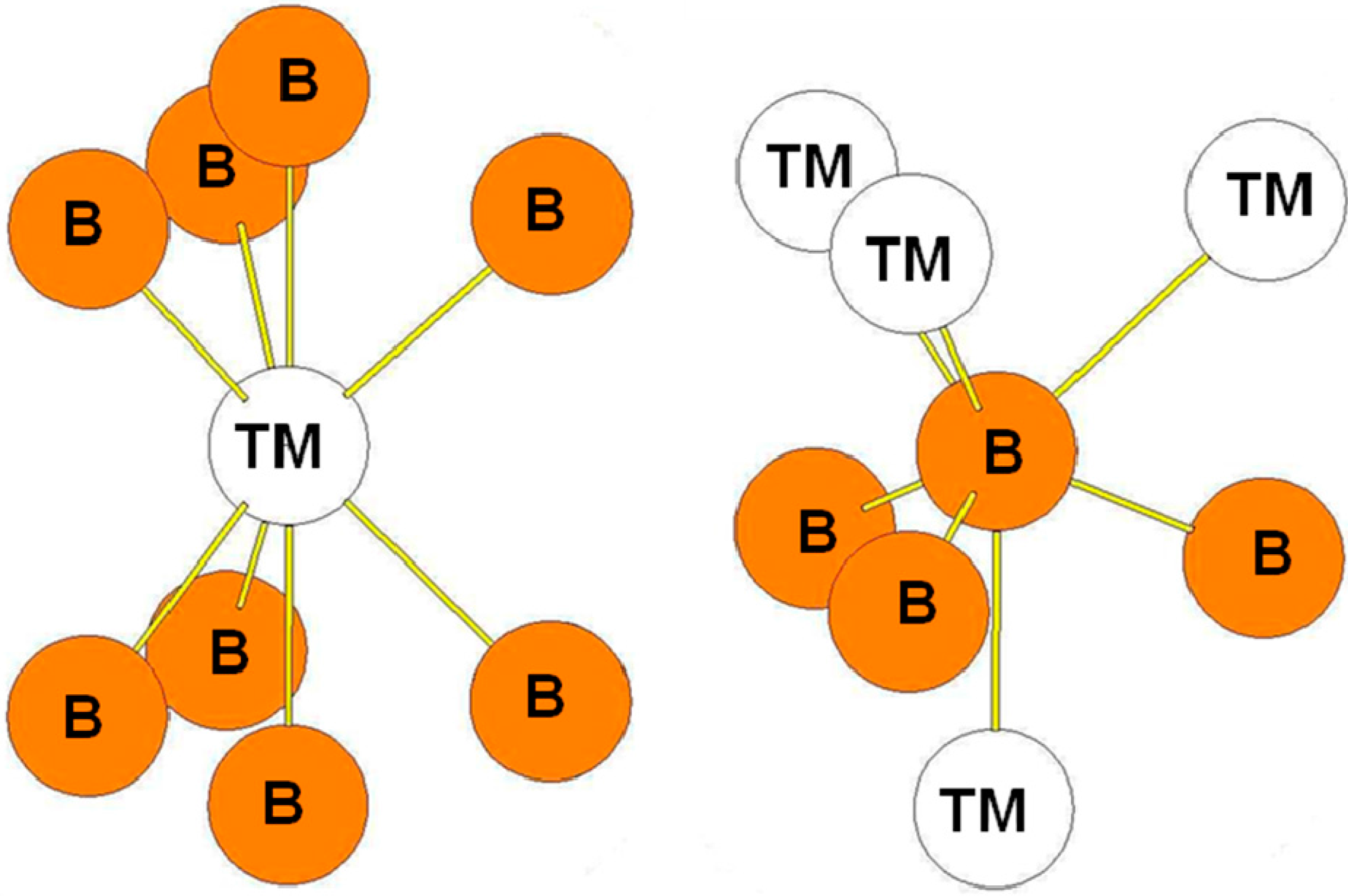
Table 7.
Bond lengths in the hP6-ReB2 compound.
| Atom | Env. | Nb. | Distance(Å) |
|---|---|---|---|
| B | B | 3 | 1.831 |
| Re | 1 | 2.234 | |
| Re | 3 | 2.267 | |
| Re | B | 2 | 2.234 |
| B | 6 | 2.267 |
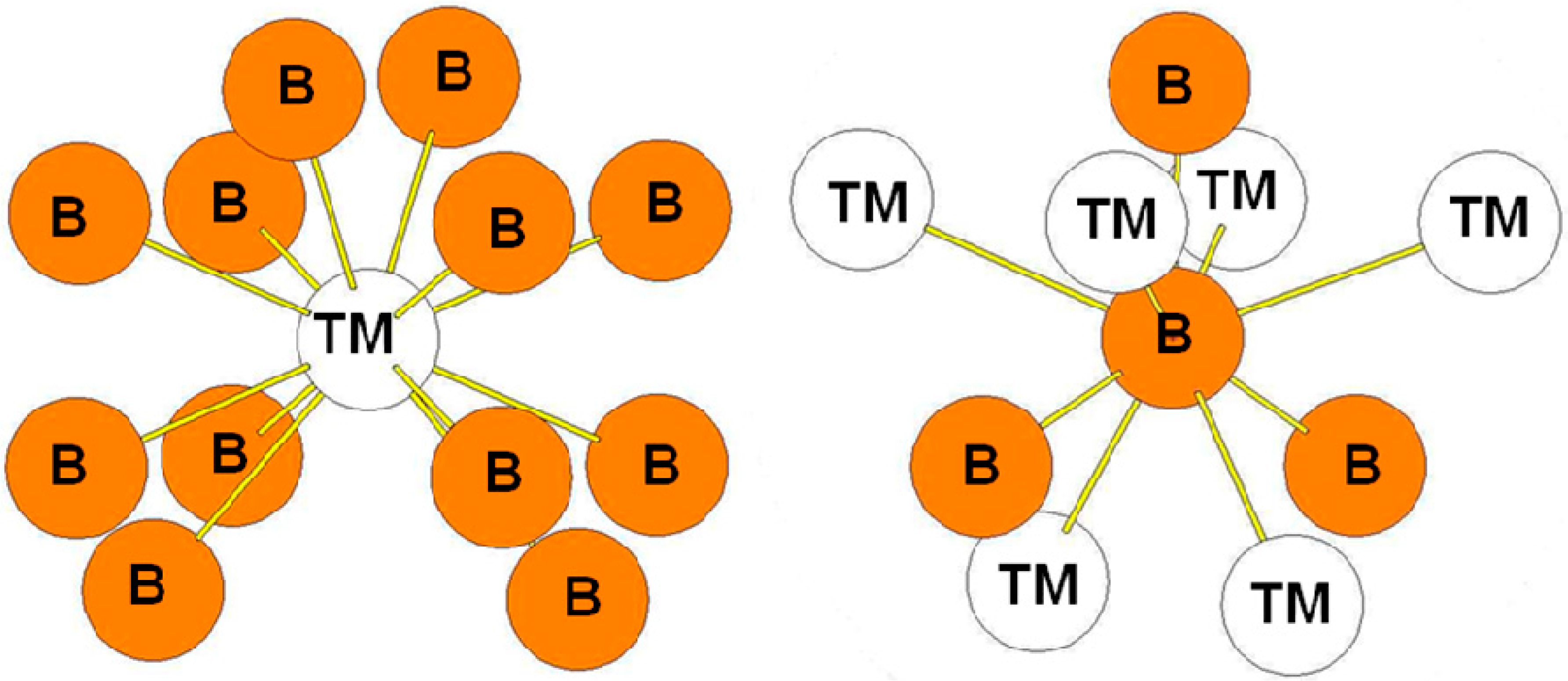
In the RuB2-type structure, the B atoms occupy the 4e positions while the Ru atoms occupy the 2a positions. The surroundings of the B and Ru atoms in the structure are presented in Figure 9. As in the case of ReB2-type structure, the transition metal atoms are surrounded by eight B atoms while the B atoms are surrounded by three B atoms at a short distance and by four transition metal atoms. The bond lengths reported in Table 8 are those calculated for the oP6-RuB2 compound. The environment of the B atoms in the oP6 structure is the same as the one of the B2 atoms in the hR6 structure and as the one of B3 atoms in the hP12 structure.
We also present in Figure 10 the environment of the atoms in the hR3 (hP9) CaSi2-type structure in which the B atoms occupy the 2c positions while the transition metal occupies the 1a positions (in the hR3 description). We can observe that the surrounding of the atoms is very similar to the one in the ReB2 and RuB2 type structures.
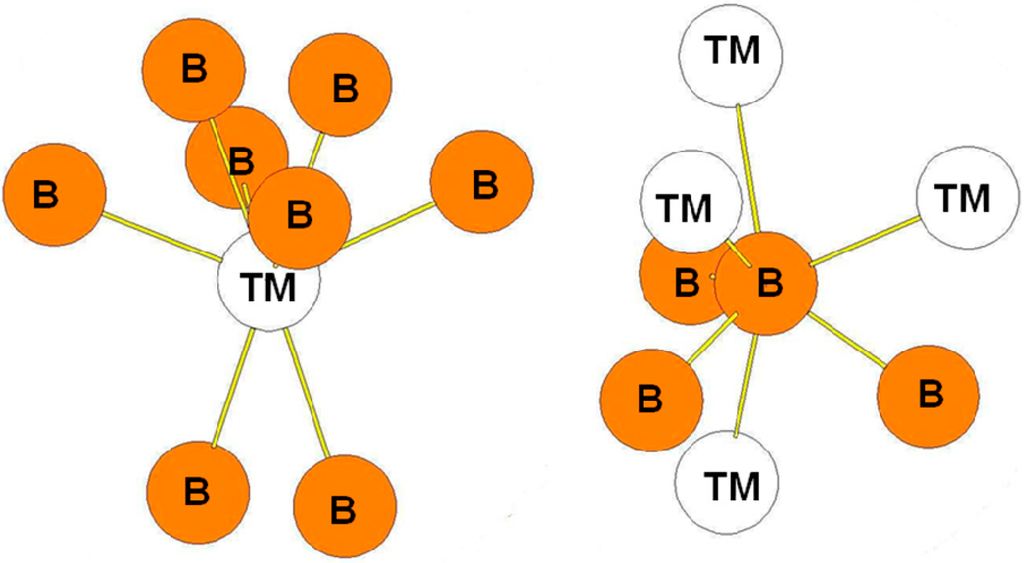
Figure 9.
Environment of TM and B atoms in the oP6 (RuB2-type) structure.
Figure 9.
Environment of TM and B atoms in the oP6 (RuB2-type) structure.
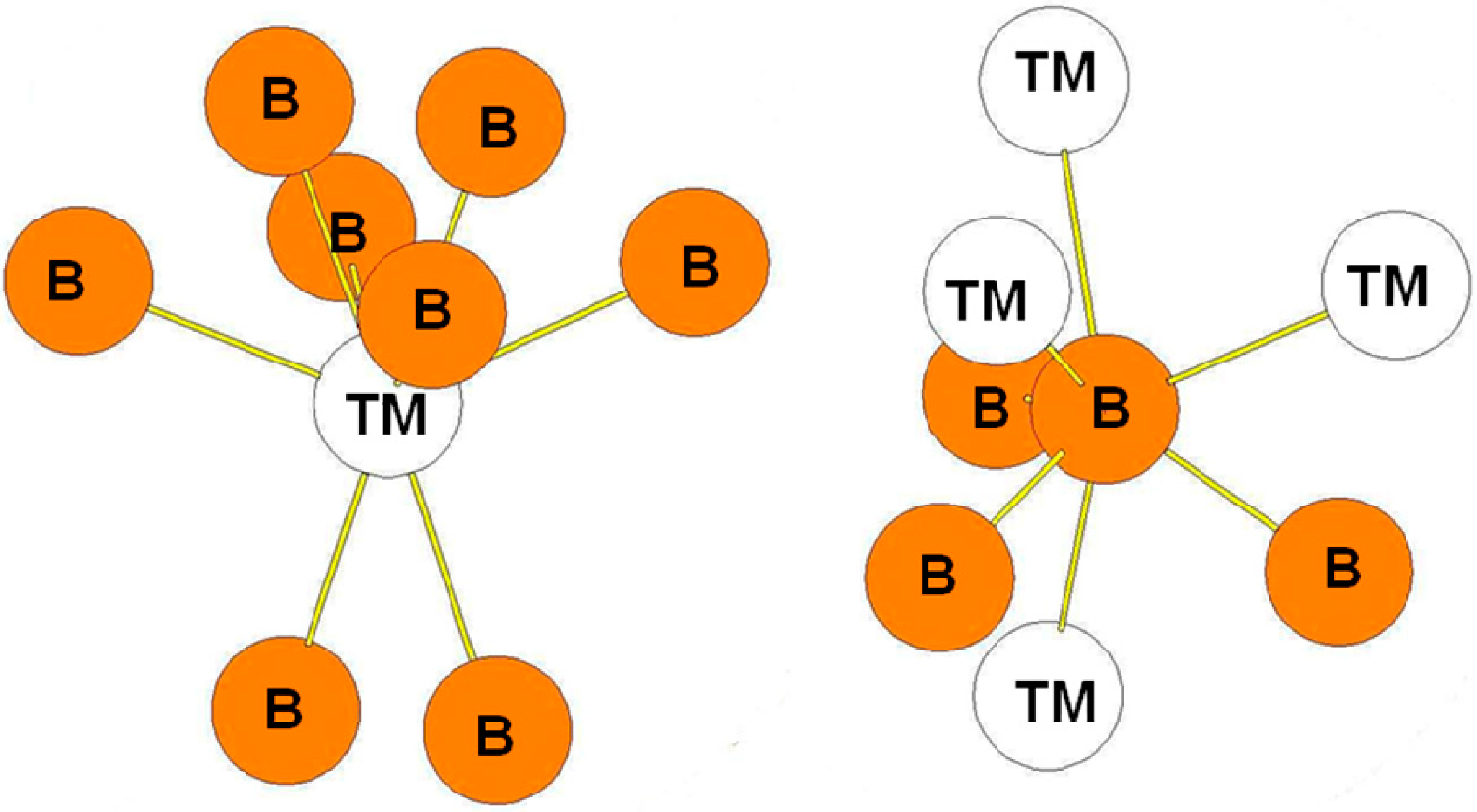
Table 8.
Bond lengths in the oP6-RuB2 compound.
| Atom | Env. | Nb. | Distance (Å) |
|---|---|---|---|
| B | B | 1 | 1.824 |
| B | 2 | 1.886 | |
| Ru | 1 | 2.168 | |
| Ru | 1 | 2.282 | |
| Ru | 4 | 2.197 | |
| Ru | B | 2 | 2.282 |
| B | 2 | 2.168 | |
| B | 4 | 2.197 |
Table 9 summarizes the environment of the transition and boron atoms in the considered structures. There is a reduction of the coordination numbers of both the transition metal and of the B atoms from the hP3 to the hP6 or oP6 structures. The hR6 and hP12 structures are intermediate between them. This probably explains why the formation enthalpies obtained for these two structures are intermediate except in the cases where they correspond to the ground state (Cr column). All the structures considered in the present work are characterized by short B-B bonds. The number of these short bonds remains identical for all the considered structures in the present work. In the last column of Table 9, the environment of the transition and boron atoms in the oP12 structure is indicated. One may remark that in the case of oP12-FeB2, the TM atoms are surrounded by four B atoms two at a very short distance (1.737, 1.870 Å) and two at a less short distance (2.159 Å).
Table 9.
Coordination numbers of the TM and B atoms in the hP3, tI12, hR6, hP12, hP6, oP6, hR3, and oP12 structures.
| Structure | hP3, tI12 | hR6, hP12 | hP6, oP6, hR3 | oP12 |
|---|---|---|---|---|
| CN (TM) | 12 B | 10 B | 8 B | 10 B |
| CN (B) | 6 TM, 3 B | 6 TM, 3B | 4 TM, 3B | 5 TM, 4 B |
| CN(B) | -- | 4 TM, 3B | -- | -- |
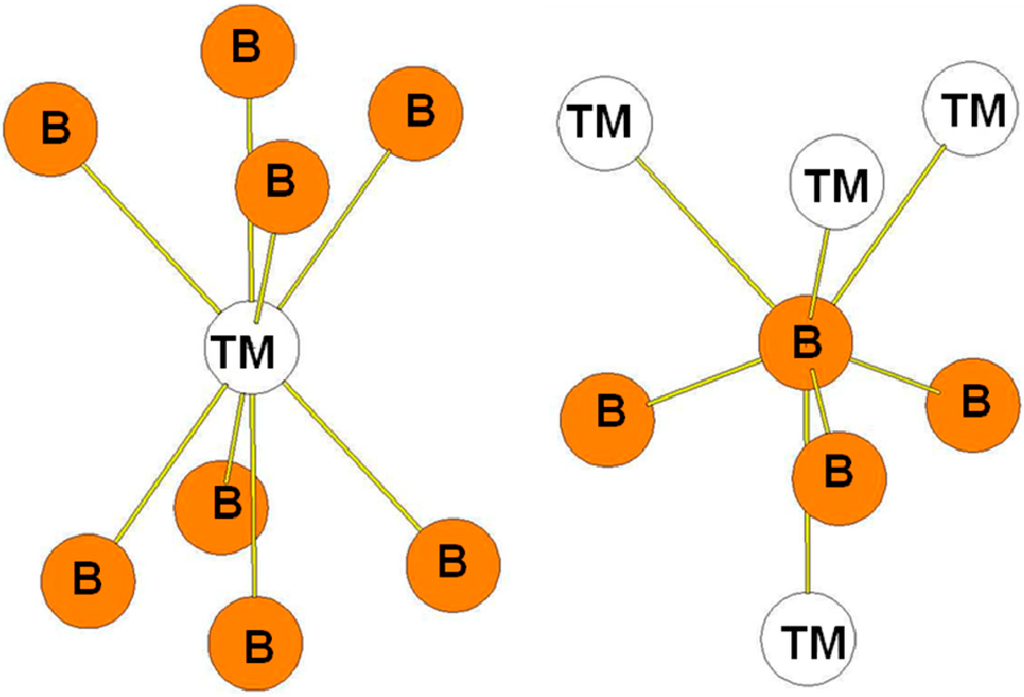
Figure 10.
Environment of TM and B atoms in the hR3 (CaSi2-type) structure.
Figure 10.
Environment of TM and B atoms in the hR3 (CaSi2-type) structure.
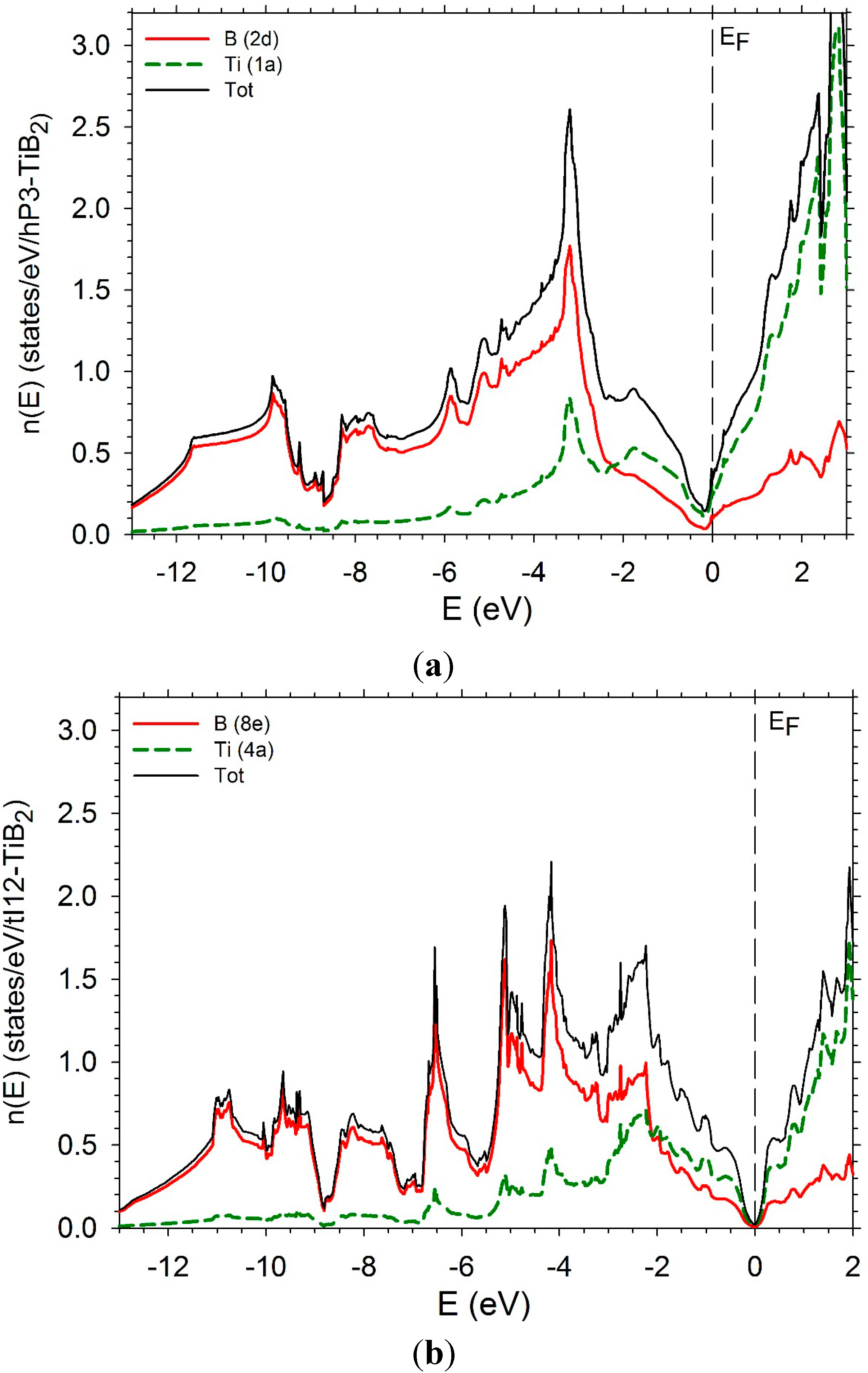
In order to complete this study, we have computed the electronic densities of states (DOS) as well the partial densities of states of the transition metal and boron of several diborides. The DOS of hP3-TiB2 is displayed in Figure 11a. The Fermi level is in the bottom of the pseudo-gap which is very deep. The DOS of tI12-TiB2 (Figure 11 b) also presents a pseudo gap with the Fermi level in the bottom with a number of states near zero. Some differences appear below the Fermi level. The PDOS of B and Ti present a peak around −3 eV due to a strong hybridization between the B p states and the Ti d states. The peak is less high and larger in the case of the tI12 structure. An increase of the number of d electrons leads to a shift of the Fermi-level on the right of the bottom of the pseudo-gap. The DOS of hP3-VB2 may be found in reference [37].
The DOS of hR6-MoB2 and hP12-WB2 are presented in Figure 11b,c. They are very similar with a large pseudo-gap and the Fermi-level in the middle of the pseudo-gap. They are relatively similar. In the case of the hR6 structure, the B1 and B2 PDOS are very different. The PDOS of the B1 atoms present more structure than the one of the B2 atoms which is more flat. At the Fermi level the Mo d states dominate. In the hP12 structure, the PDOS of the B(2b) and B(2c) can be perfectly superimposed. They have a flat shape. The PDOS of B (4f) atoms is different with a sharp peak at between −4 and −3 eV. At the same energy, the PDOS of W presents also a sharp peak. These peaks correspond to the hybridization between B p states of B (4f) and d states of W. At the Fermi level and above, the d states of W dominate.
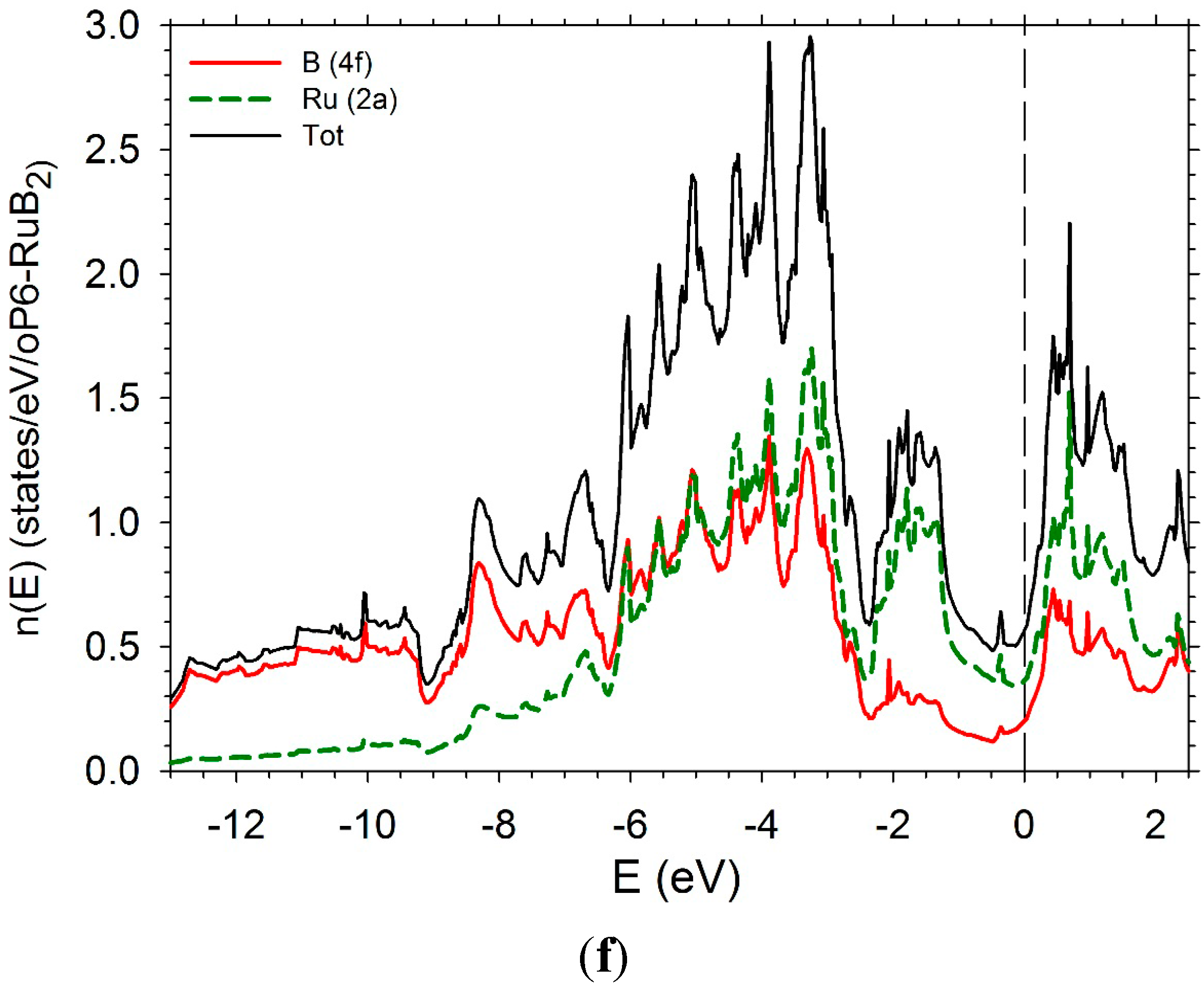
The DOS of hP6-ReB2 and oP6-RuB2 (Figure 11e,f) present a large pseudo-gap. In the case of hP6-ReB2, the Fermi-level is in the middle of the pseudo-gap while it is on the right in the case of oP6-RuB2 due to a greater number of d electrons.
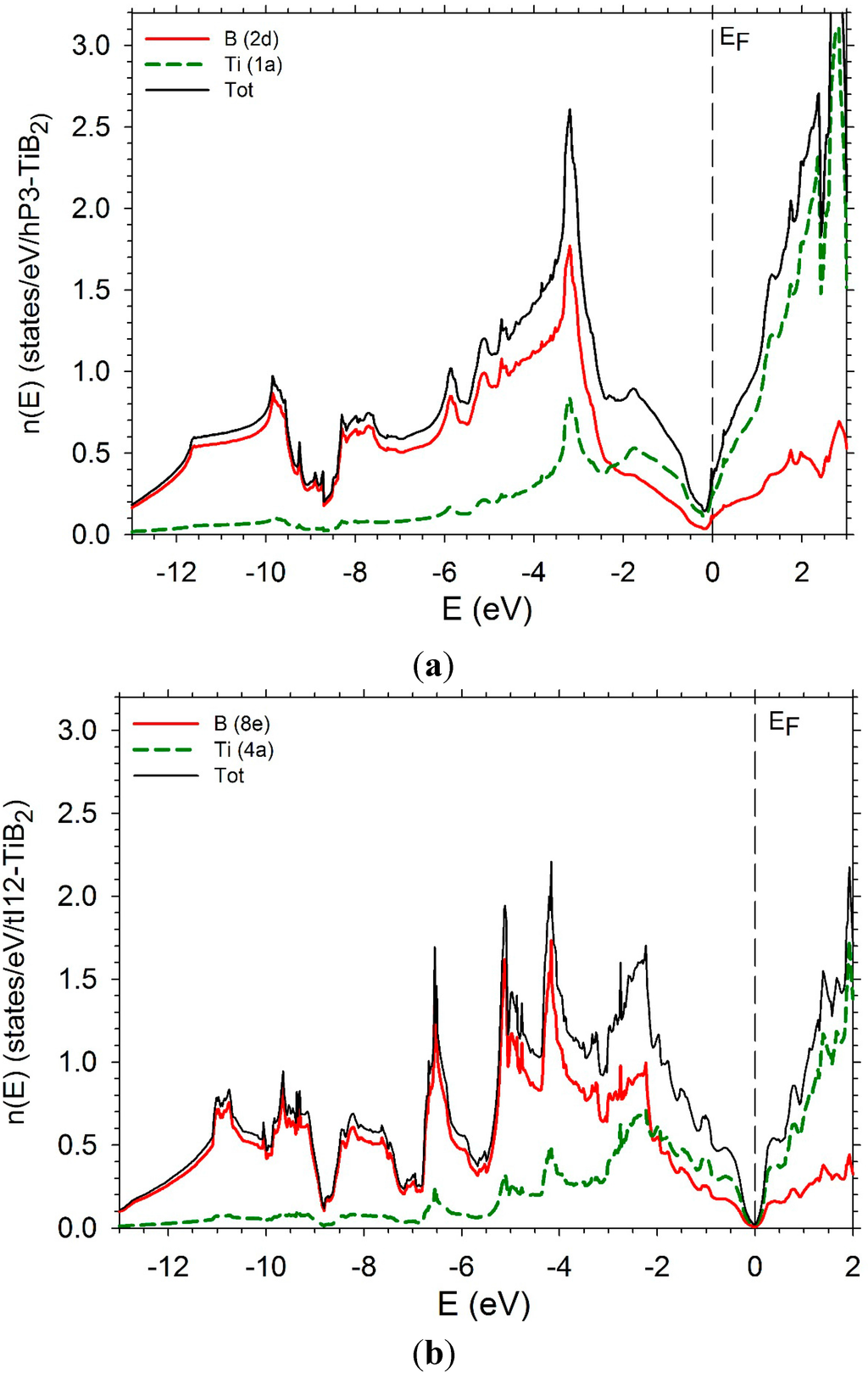
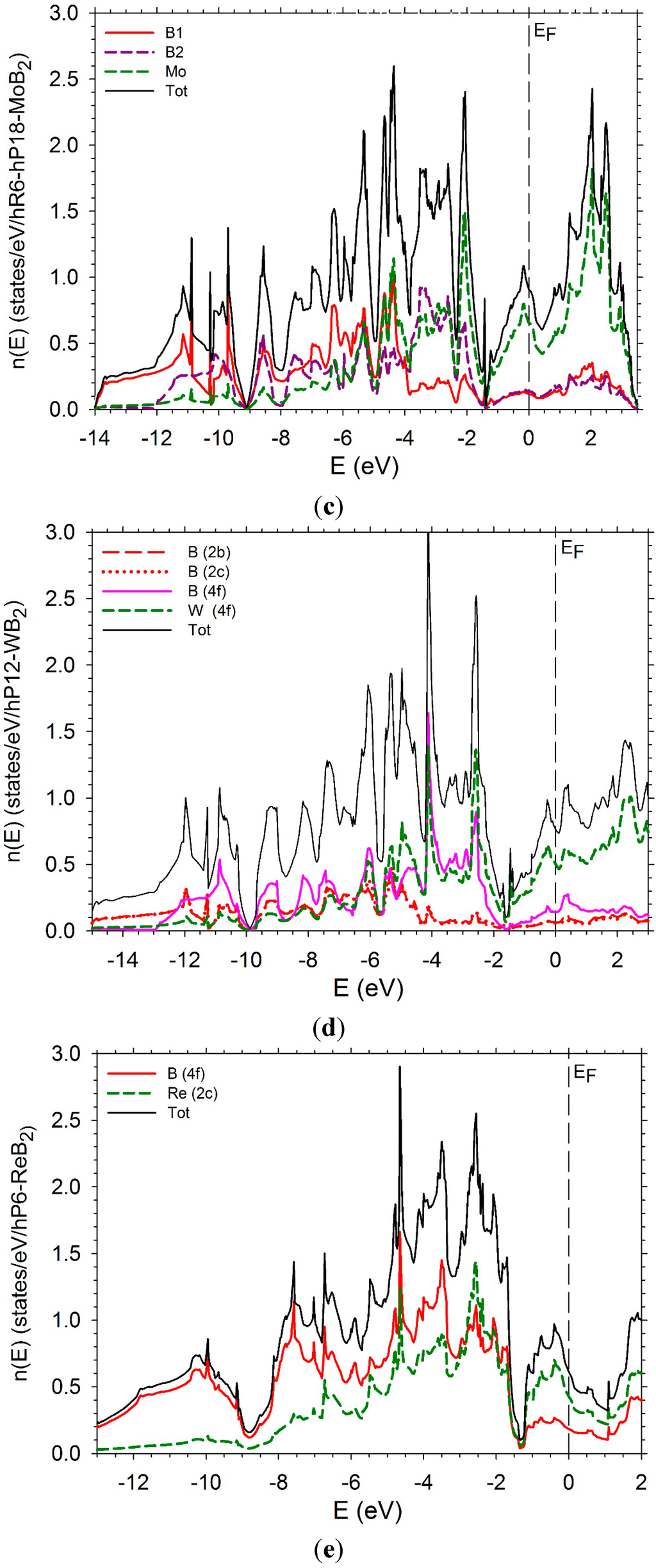
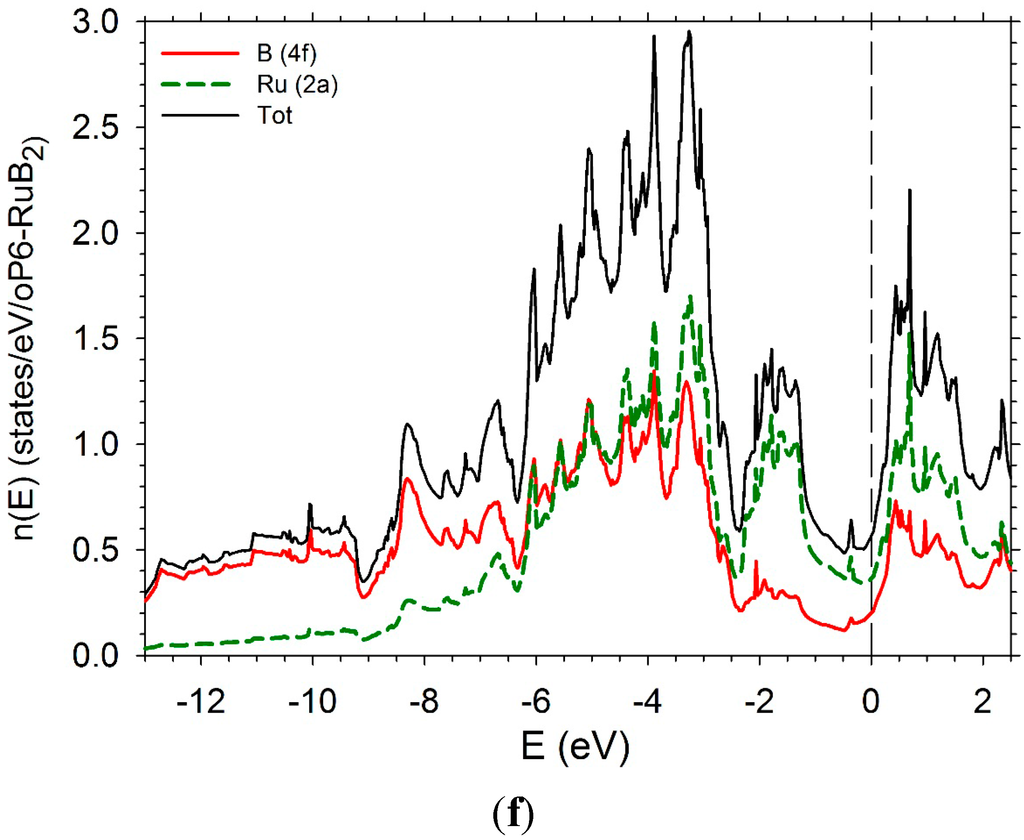
Figure 11.
Electronic densities of states. (a) hP3-TiB2; (b) tI12-TiB2; (c) hR6-MoB2; (d) hP12-WB2; (e) hP6-ReB2; (f) oP6-RuB2.
Figure 11.
Electronic densities of states. (a) hP3-TiB2; (b) tI12-TiB2; (c) hR6-MoB2; (d) hP12-WB2; (e) hP6-ReB2; (f) oP6-RuB2.
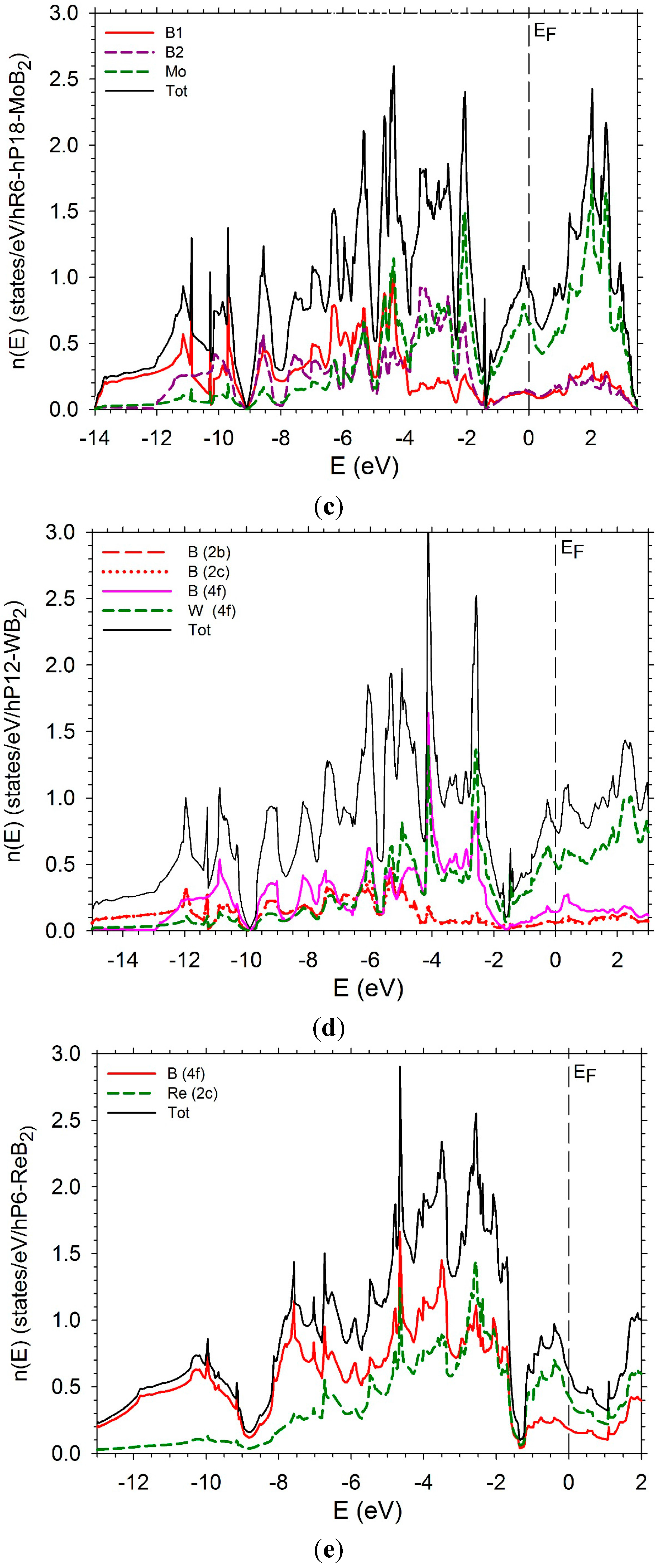
5. Conclusions
The enthalpies of formation of TMB2 compounds where TM is a transition element of the beginning of the series 3D, 4D, and 5D have been calculated in the structures hP3 (AlB2-type), hR6 (or hP18) (CaSi2-type), hP12 (WB2-type), hP6 (ReB2-type), oP6 (RuB2-type). Three sets of structures can be distinguished. In the first one, hP3 and tI12, the enthalpies of formation are very similar, the hP3 structure being slightly more stable than the tI12 one at least at ordinary pressure. In these two structures, the TM atoms are surrounded by 12 B atoms and the B atoms are surrounded by 3B atoms and 6 TM atoms. The electronic densities of states of the two structures present the same shape with a deep pseudogap. The Fermi level is in the bottom of the pseudo gap in the cases of Ti, Zr, and Hf. With an increase of the number of d electrons, the Fermi level is shifted on the right of the pseudogap. In the second set of structures, hP6, oP6, and hR3, the TM atoms are surrounded by 8 B atoms while the B atoms are surrounded by 3 B atoms and 4 TM atoms. The third set of structures, hR6 and hP12, represent an intermediate case: the TM atoms are surrounded by 10 B atoms. Again the B atoms are surrounded by 3 B atoms. But half of the B atoms are surrounded by 4 TM atoms as in the hP6, oP6, and hR3, while the other half of the B atoms are surrounded by 6 TM atoms as in the hP3 and tI12 structures. Let us recall that all the considered structures are characterized by short B-B bonds. The order of magnitude of the bond length is 1.7 to 1.8 Å.
The formation enthalpies of the hP3 and tI12 structures display a drastic increase in the 3D, or 4D, or 5D series while those of hP6, oP6, or hR3 are near constant with the atomic number in the transition series. The behavior of the enthalpies of formation of the compounds in the hR6 and hP12 series is in between these two sets. The crossing between the three sets of formation enthalpies occurs around Mn in the 3D series, Mo in the 4D series, and between Ta and W in the 5D series.
Supplementary Files
Supplementary File 1Author Contributions
Catherine Colinet conceived the project and all authors contributed in the manuscript preparation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ivanovskii, A.L. Mechanical and electronic properties of diborides of transition 3D-5D metals from first principles: Toward search of novel ultra-incompressible and superhard materials. Progr. Mater. Sci. 2012, 57, 184–228. [Google Scholar] [CrossRef]
- Ivanovskii, A.L. Hardness of hexagonal AlB2-like diborides of s, p and d metals from semi-empirical estimations. Int. J. Refract. Metals Hard Mater. 2013, 36, 179–182. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, H.; Wang, H.; Zhang, X.; Litaka, T.; Ma, Y. First-Principles prediction on the high-pressure structures of transition metal diborides (TMB2, TM = Sc, Ti, Y, Zr). Inorg. Chem. 2010, 49, 6859–6864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yan, H.; Wei, Q.; Wang, H. Pressure-induced phase transition and mechanical properties of molybdenum diboride: First principles calculations. J. Appl. Phys. 2012, 112, 013522. [Google Scholar] [CrossRef]
- Waskowska, A.; Lyaschenko, A.; Gerward, L.; Olsen, J.S.; Babu, K.R.; Vaitheeswaran, G.; Kanchana, V.; Svane, A.; Filipov, V.B.; Levchenko, G. Thermoelastic properties of ScB2, TiB2, YB4 and HoB4: Experimental and theoretical studies. Acta Mater. 2011, 59, 4886–4894. [Google Scholar] [CrossRef]
- Aviles, M.A.; Cordoba, J.M.; Sayagués, M.J.; Gotor, F.J. Mechanochemical synthesis of Ti1-xZrxB2 and Ti1-xHfxB2 solid solutions. Ceram. Int. 2011, 37, 1895–1904. [Google Scholar] [CrossRef]
- Frotscher, M.; Klein, W.; Bauer, J.; Fang, C.-M.; Halet, J.-F.; Senyshyn, A.; Baehtz, C.; Albert, B. M2B5 or M2B4 ? A reinvestigation of the Mo/B and W/B system. Z. Anorg. Allg. Chem. 2007, 633, 2626–2630. [Google Scholar] [CrossRef]
- Kodess, B.N.; Butman, L.A.; Sambueva, S.R. Refinement of Mo2B5 structural type.
- Leithe Jasper, A.; Klesnar, H.P.; Rogl, P. Reinvestigation of isothermal sections in M(M = Mo, W)-Fe-B ternary systems at 1323 K. Nippon Kinzoku Gakkaishi 2000, 64, 154–162. [Google Scholar]
- Frotscher, M.; Hölzel, M.; Albert, B. Crystal structures of the metal diborides ReB2, RuB2, and OsB2. Z. Anorg. Allg. Chem. 2010, 636, 1783–1786. [Google Scholar] [CrossRef]
- Zogal, O.J.; Fojud, Z.; Herzig, P.; Pietraszko, A.; Lyashchenko, A.B.; Jurga, S.; Paderno, V.N. Crystal structure, electric field gradient, and electronic charge densities in ReB2: A single crystal X-ray, 11B nuclear magnetic resonance, and first-principles study. J. Appl. Phys. 2009, 106, 033514. [Google Scholar] [CrossRef]
- Singh, Y.; Niazi, A.; Vannette, M.D.; Prozorov, R.; Johnston, D.C. Superconducting and normal-state properties of the layered boride OsB2. Phys. Rev. B. 2007, 76, 1–15. [Google Scholar] [CrossRef]
- Villars, P.; Cenzual, K. Pearson’s Crystal Data—Crystal Structure Database for Inorganic Compounds, Release 2014/15; ASM International: Materials Park, OH, USA, 2014. [Google Scholar]
- Van Der Geest, A.G.; Kolmogorov, A.N. Stability of 41 metal-boron systems at 0 GPa and 30 GPa from first principles. Calphad 2014, 46, 184–204. [Google Scholar] [CrossRef]
- Vajeeston, P.; Ravindran, P.; Ravi, C.; Asokamani, R. Electronic structure, bonding, and ground-state properties of AlB2-type transition-metal diborides. Phys. Rev. B 2001, 63, 045115. [Google Scholar] [CrossRef]
- Oguchi, T. Cohesion in AlB2-type diborides: A first-principles study. J. Phys. Soc. Jpn. 2002, 71, 1495–1500. [Google Scholar] [CrossRef]
- Xu, X.; Fu, K.; Yu, M.; Lu, Z.; Zhang, X.; Liu, G.; Tang, C. The thermodynamic, electronic and elastic properties of the early-transition-metal diborides with AlB2-type structure: A density functional theory study. J. Alloys Compd. 2014, 607, 198–206. [Google Scholar] [CrossRef]
- Zhou, Y.; Xiang, H.; Feng, Z.; Li, Z. General Trends in Electronic Structure, Stability, Chemical Bonding and Mechanical Properties of Ultrahigh Temperature Ceramics TMB2 (TM = transition metal). J. Mater. Sci. Technol. 2015, 31, 285–294. [Google Scholar] [CrossRef]
- Liang, Y.; Zhong, Z.; Zhang, W. A thermodynamic criterion for designing superhard transition-metal borides with ultimate boron content. Comput. Mater. Sci. 2013, 68, 222–228. [Google Scholar] [CrossRef]
- Pallas, A.; Larsson, K. Structure determination of the 4D metal diborides: A quantum mechanical study. J. Phys. Chem. B 2006, 110, 5367–5371. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jiang, J.Z. Elastic and electronic properties of low compressible 4D transition metal diborides: First principles calculations. Solid State Commun. 2010, 150, 2093–2096. [Google Scholar] [CrossRef]
- Ying, C.; Zhao, E.; Lin, L.; Hou, Q. Structural determination and physical properties of 4D transitional metal diborides by first-principles calculations. Modern Phys. Lett. B 2014, 28, 1450213. [Google Scholar] [CrossRef]
- Hao, X.; Wu, Z.; Xu, Y.; Zhou, D.; Liu, X.; Meng, J. Trends in elasticity and electronic structure of 5D transition metal diborides: First-principles calculations. J. Phys. Condens. Matter 2007, 19, 196212. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.-J. Mechanical and electronic properties of 5D transition metal diborides MB2 (M = Re, W, Os, Ru). J. Appl. Phys. 2009, 105, 083539. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Fu, C.L.; Krcmar, M.; Painter, G.S. Electronic and structural origin of ultraincompressibility of 5D transition-Metal diborides MB2 (M = W, Re, Os). Phys. Rev. Lett. 2008, 100, 196403. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-K.; Zhao, E.-J.; Wu, Z.-J. First-principles calculations of structural thermodynamic and mechanical properties of 5D transitional metal diborides. Wuli Xuebao/Acta Phys. Sinica 2013, 62, 046201. [Google Scholar]
- Zhong, M.-M.; Kuang, X.-Y.; Wang, Z.-H.; Shao, P.; Ding, L.-P.; Huang, X.-F. Phase stability, physical properties, and hardness of transition-metal diborides MB2 (M = Tc, W, Re, and Os): First-principles investigations. J. Phys. Chem. C 2013, 117, 10643–10652. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals an semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1998, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, S.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Methfessel, M.; Paxton, A.T. High precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone Integrations. Phys. Rev. B 1976, 135, 5188–5192. [Google Scholar] [CrossRef]
- Dinsdale, A.T. SGTE Data for Pure Elements. Calphad 1991, 15, 317–425. [Google Scholar] [CrossRef]
- Shang, S.; Wang, Y.; Arroyave, R.; Liu, Z.-K. PhaseStability in α- and β-Rhombohedral Boron. Phys. Rev. B 2007, 75, 092101. [Google Scholar] [CrossRef]
- Van Setten, M.J.; Uijttewaal, M.A.; de Wijs, G.A.; de Groot, R.A. Thermodynamic Stability of Boron. The Role of Defects and Zero Point Motion. J. Am. Chem. Soc. 2007, 129, 2458–2465. [Google Scholar] [CrossRef] [PubMed]
- Colinet, C.; Tedenac, J.-C. Enthalpies of formation and electronic densities of states of vanadium borides. J. Phase Equilib. Diffus. 2014, 35, 396–405. [Google Scholar] [CrossRef]
- Kolmogorov, A.N.; Shah, S.; Margine, E.R.; Bialon, A.F.; Hammerschmidt, T.; Drautz, R. New superconducting and semiconducting Fe-B compounds predicted with an Ab initio evolutionary search. Phys. Rev. Lett. 2010, 105, 217003. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-H.; Wang, W.-L.; Hu, L.; Wei, B.-B. First-principle calculations of structural, elastic, and thermodynamic properties of Fe-B compounds. Intermetallics 2014, 46, 211–221. [Google Scholar] [CrossRef]
- Fan, J.; Bao, K.; Jin, X.; Meng, X.; Duan, D.; Liu, B.; Cui, T. How to get superhard MnB2: A first-principles study. J. Mater. Chem. 2012, 22, 17630–17635. [Google Scholar] [CrossRef]
- Gou, H.; Steinle-Neumann, G.; Bykova, E.; Nakajima, Y.; Miyajima, N.; Li, Y.; Ovsyannikov, S.V.; Dubrovinsky, L.S.; Dubrovinskaia, N. Stability of MnB2 with AlB2-type structure revealed by first-principles calculations and experiments. Appl. Phys. Lett. 2013, 102, 061906. [Google Scholar] [CrossRef]
- Topor, L.; Kleppa, O.J. Enthalpies of Formation of First-Row Transition-Metal Diborides by a New Calorimetric Method. J. Chem. Thermodyn. 1985, 17, 1003–1016. [Google Scholar] [CrossRef]
- Lowell, C.E.; Williams, W.S. High-temperature calorimeter for the determination of heats of formation of refractory compounds. Rev. Sci. Instrum. 1961, 32, 1120–1123. [Google Scholar] [CrossRef]
- Huber, E.J., Jr. The Heat of Formation of Titanium Diboride. J. Chem. Eng. Data 1966, 11, 430–431. [Google Scholar] [CrossRef]
- Akhachinskij, V.V.; Chirin, N.A. Proceedings of Symposium on Thermodynamics of Nuclear Materials; Volume 2. International Atomic Energy Agency (IAEA): Vienna, Austria, 1975; pp. 467–472.
- Kirpichev, E.P.; Rubtsov, Yu.I.; Sorokina, T.V.; Prokudina, V.K. Standard enthalpy of formation of transition metal borides. Russian J. Phys. Chem. 1979, 53, 1128–1130. [Google Scholar]
- Yurick, T.J.; Spear, K.E. Thermodynamics of TiB2 from Ti–B–N studies. In Thermodynamics of Nuclear Materials; International Atomic Energy Agency (IAEA): Vienna, Australia, 1980; pp. 73–79. [Google Scholar]
- Jain, A.; Pankajavallia, R.; Anthonysamy, S.; Ananthasivana, K.; Babua, R.; Ganesana, V.; Gupta, G.S. Determination of the thermodynamic stability of TiB2. J. Alloys Compd. 2010, 491, 747–752. [Google Scholar] [CrossRef]
- Spear, K.E.; Schäfer, H.; Gilles, P.W. Thermodynamics of Vanadium Borides. In High Temperature Technology; Butterworths: London, UK, 1969; pp. 201–212. [Google Scholar]
- Topor, L.; Kleppa, O.J. Molar enthalpy of formation of CrB2 by high-temperature calorimetry. J. Chem. Thermodyn. 1985, 17, 109–116. [Google Scholar] [CrossRef]
- Kleppa, O.J.; Sato, S. New applications of high-temperature solution calorimetry III. Enthalpies of formation of Mn2B, MnB, and MnB2. J. Chem. Thermodyn. 1982, 14, 133–143. [Google Scholar] [CrossRef]
- Meschel, S.V.; Kleppa, O.J. Standard enthalpies of formation of some refractory borides of 4D and 5D elements from high-temperature direct synthesis calorimetry. J. Chem. Phys. 1993, 90, 349–354. [Google Scholar]
- Leitnaker, J.M.; Bowman, M.G.; Gilles, P.W. High-Temperature Evaporation and Thermodynamic Properties of Zirconium Diboride. J. Chem. Phys. 1962, 36, 350–358. [Google Scholar] [CrossRef]
- Huber, E.J., Jr.; Head, E.L.; Holley, C.E., Jr. The Heats of Formation of Zirconium Diboride and Dioxide. J. Phys. Chem. 1964, 68, 3040–3042. [Google Scholar] [CrossRef]
- Trulson, O.C.; Goldstein, H.W. Mass Spectrometric Study of Zirconium Diboride. J. Phys. Chem. 1965, 69, 2531–2536. [Google Scholar] [CrossRef]
- Johnson, G.K.; Greenberg, E.; Margrave, J.L.; Hubbard, W.N. Fluorine bomb calorimetry. Enthalpies of formation of the diborides of ziconium and hafnium. J. Chem. Eng. Data 1967, 12, 137–141. [Google Scholar] [CrossRef]
- Samsonov, G.V. Heats of formation of borides of some transition metals. Zhur. Fiz. Khim. 1956, 30, 2057–2060. [Google Scholar]
- Reznitskii, L.A. Determination of the heat of formation of the NbB1.82 from the NbB2 phase. Russian J. Phys. Chem. 1967, 41, 612–614. [Google Scholar]
- Johnson, G.K.; Greenberg, E.; Margrave, J.L.; Hubbard, W.N. Fluorine bomb calorimetry. Enthalpies of formation of the diborides of niobium and tantalum. J. Chem. Eng. Data 1967, 12, 597–600. [Google Scholar] [CrossRef]
- Meschel, S.V.; Kleppa, O.J. Standard enthalpies of formation of NbB2, MoB, and ReB2 by high-temperature direct synthesis calorimetry. Met. Trans. A 1993, 24, 947–950. [Google Scholar] [CrossRef]
- McClaire, L.A. Thermodynamic and Kinetic Studies for a Refractory Materials Program; Technical Documentary Report No. ASD-TDR-62–204, Part III; Wright Patterson Air Force Base: Dayton, OH, USA, 1964. [Google Scholar]
- Meschel, S.V.; Kleppa, O.J. Enthalpies of formation of refractory borides of 5D elements by high temperature direct synthesis calorimetry I. IrB1.35 and OsB2.5. J. Alloys Compd. 1991, 177, 159–166. [Google Scholar] [CrossRef]
- Meschel, S.V.; Kleppa, O.J. Thermochemistry of alloys of transition metals and lanthanide metals with some IIIB and IVB elements in the periodic table. J. Alloys Compd. 2001, 321, 183–200. [Google Scholar] [CrossRef]
- Ren, F.; Wang, Y.; Lo, V.C. Pressure induced structural phase transition of OsB2: First-principles calculations. J. Solid State Chem. 2010, 183, 915–919. [Google Scholar] [CrossRef]
- Xie, Z.; Graule, M.; Orlovskaya, N.; Payzant, E.A.; Cullen, D.A.; Blair, R.G. Novel high pressure hexagonal OsB2 by mechanochemistry. J. Solid State Chem. 2014, 215, 16–21. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).