Abstract
Three cyano-1-(phenylsulfonyl)indole derivatives, 3-cyano-1-(phenylsulfonyl)indole, (I), 2-cyano-1-(phenylsulfonyl)indole, (II), and 2,3-dicyano-1-(phenylsulfonyl)indole, (III), and a key synthetic precursor 1-(phenylsulfonyl)-1-(1,1-dimethylethyl)indole-3-carboxamide, (IV), have been synthesized and their structures determined by single crystal X-ray crystallography. (I), C15H10N2O2S, is orthorhombic with space group P 212121 and cell constants: a = 4.9459(3) Å, b = 10.5401(7) Å, c = 25.0813(14) Å, V = 1307.50(14) Å3 and Z = 4. (II), C15H10N2O2S, is monoclinic with space group C 2/c and cell constants: a = 18.062(2) Å, b = 11.293(2) Å, c = 15.922(3) Å, α = 90°, β = 124.49(2)°, γ = 90°, V = 2676.7 Å3 and Z = 8. (III), C16H9N3O2S, is triclinic with space group P-1 and cell constants: a = 8.1986(8) Å, b = 9.6381(11) Å, c = 9.8113(5) Å, α = 95.053(6)°, β = 101.441(6)°, γ = 108.071(9)°, V = 713.02(11) Å3 and Z = 2. (IV), C19H20N2O3S, is orthorhombic with space group P ccn and cell constants: a = 13.7605(8) Å, b = 27.3177(14) Å, c = 9.7584(6) Å, α = 90°, β = 90°, γ =90°, V = 3668.2(4) Å3 and Z = 8. All four compounds have the same indole nitrogen phenylsulfonyl substituent and (I), (II), and (III) are nitrile derivatives. (IV) is a tert-butylamide. In the crystals, the dihedral angle between the mean planes of the indole and phenylsulfonyl groups are 85.4(2)° (I), 87.2(7)° (II), 75.1(7)° (III), and 88.6(2)° (IV), respectively. Additionally, DFT geometry-optimized molecular orbital calculations were performed and frontier molecular orbitals of each compound are displayed. Correlation between the calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals to the electronic excitation transitions from the absorption spectra of each compound has been proposed.
1. Introduction
In connection with our interest in developing novel indole chemistry [1], and in view of the enormous recent interest in the synthesis and biological activity of 2- and 3-cyanoindoles [2,3,4,5], we have synthesized three cyano-1-(phenylsulfonyl)indoles (I–III) and the synthetic precursor (IV) (Figure 1) and characterized them with NMR, single-crystal X-ray diffraction, and DFT molecular orbital calculations. These three compounds and the heteroaryl and aryl nitriles are key precursors of aldehydes, amines, amidines, tetrazoles, amides, and other carbonyl compounds [6,7] and are often employed in the synthesis of pharmaceuticals, dyes, agrochemicals, and natural products [8,9]. We report here the synthesis, crystal structures, and theoretical calculations for three cyano indole compounds and a precursor, namely, 3-cyano-1-(phenylsulfonyl)indole (I), 2-cyano-1-(phenylsulfonyl)indole (II), 2,3-dicyano-1-(phenylsulfonyl)indole (III), and 1-(phenylsulfonyl)-1-(1,1-dimethylethyl)indole-3-carboxamide (IV).
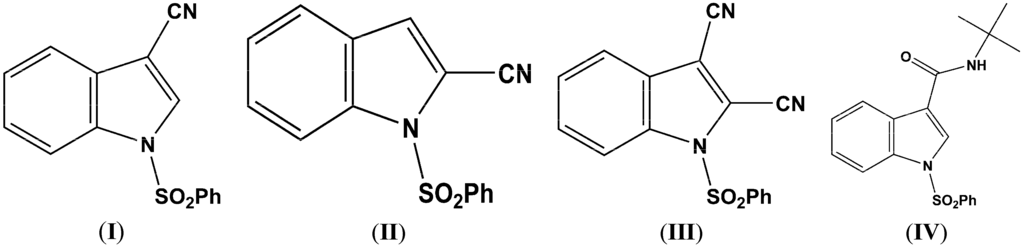
Figure 1.
The Molecular structures of C15H10N2O2S (I), C15H10N2O2S (II), C16H9N3O2S (III), and C19H20N2O3S (IV).
2. Results and Discussion
2.1. Structural Study of (I), (II), (III), and (IV)
The sulfonyl group in (I), (II), (III), and (IV) (Figure 2, Figure 3, Figure 4 and Figure 5) adopts the usual nitrogen-sulfonyl geometry seen in other 1-(phenylsulfonyl)indoles in which the nitrogen lone pair eclipses the two sulfur-oxygen bonds [10,11,12].
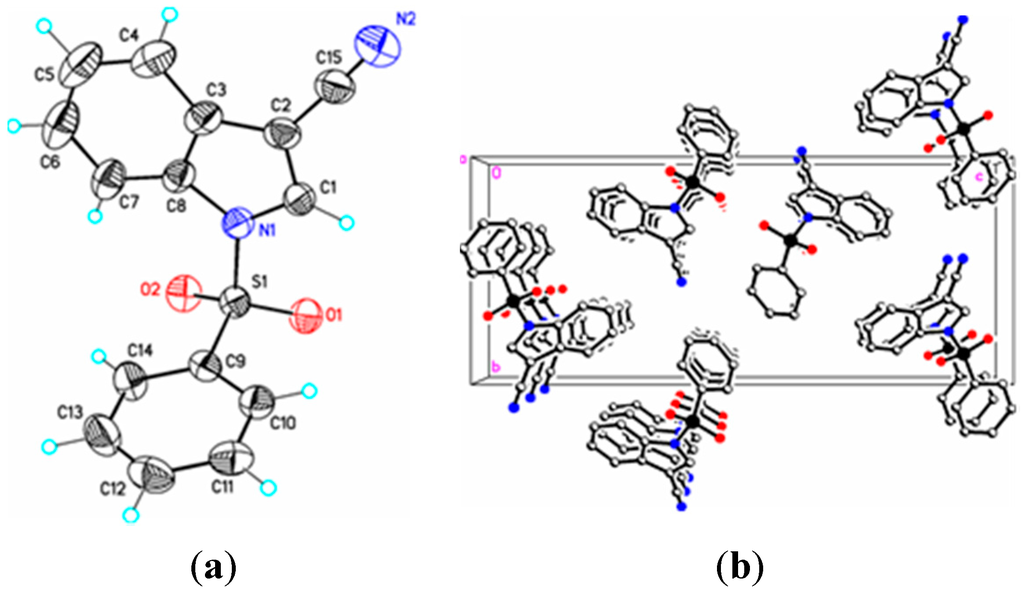
Figure 2.
(a) ORTEP drawing of (I) showing the atom numbering scheme and 50% probability displacement ellipsoids of non-H atoms; (b) The molecular packing for (I) viewed along the b axis. Hydrogen atoms not involved in hydrogen bonding have been removed for clarity.
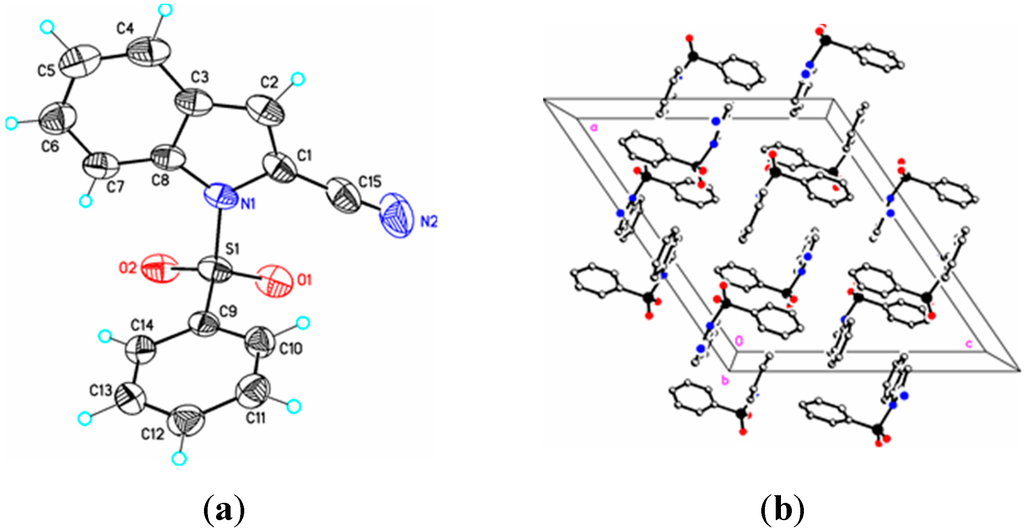
Figure 3.
(a) ORTEP drawing of (II) showing the atom numbering scheme and 50% probability displacement ellipsoids of non-H atoms; (b) The molecular packing for (II) viewed along the b axis. Hydrogen atoms not involved in hydrogen bonding have been removed for clarity.
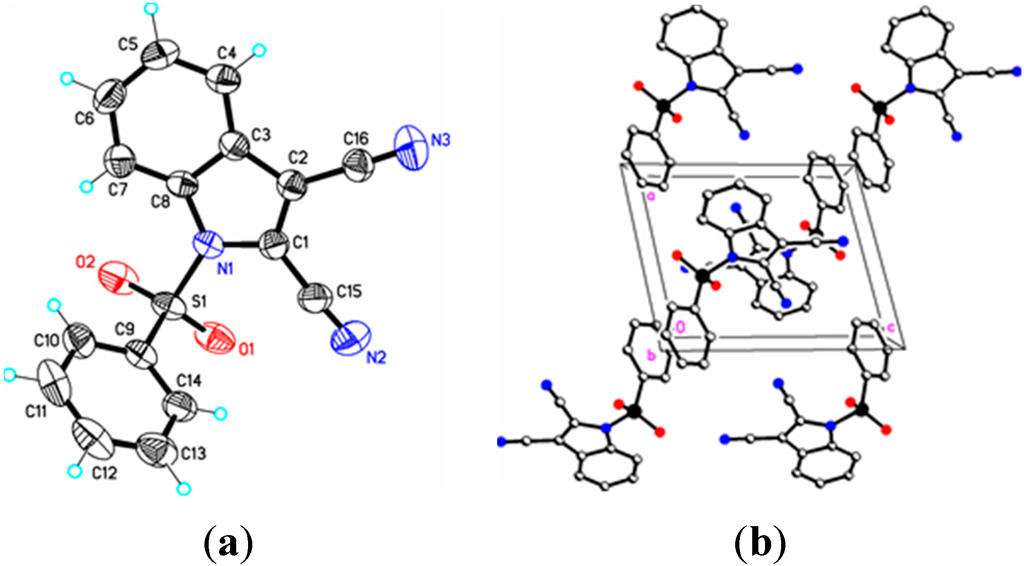
Figure 4.
(a) ORTEP drawing of (III) showing the atom numbering scheme and 50% probability displacement ellipsoids of non-H atoms; (b) The molecular packing for (III) viewed along the b axis. Hydrogen atoms not involved in hydrogen bonding have been removed for clarity.
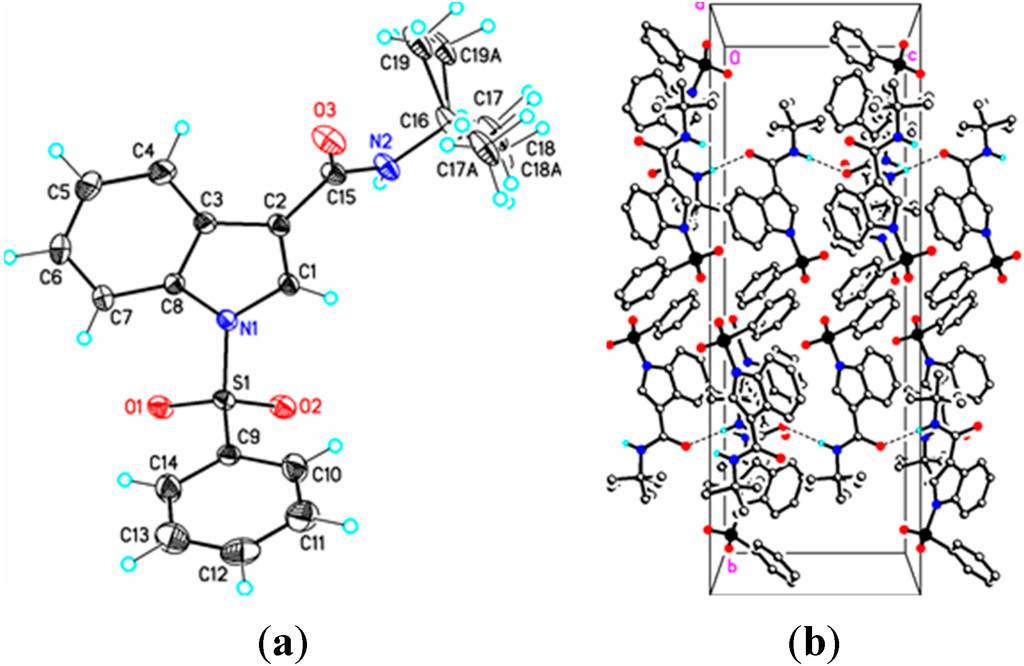
Figure 5.
(a) ORTEP drawing of (IV) showing the atom numbering scheme and 50% probability displacement ellipsoids of non-H atoms; (b) The molecular packing for (IV) viewed along the a axis. In (IVa) the tertiary butyl group is disordered over two sites in an occupancy ratio 0.544(10):0.456(10). Dashed lines in (IVb) indicate N2-H2N…O3 hydrogen bonding interactions. Hydrogen atoms not involved in hydrogen bonding have been removed for clarity.
Parameters are likewise in agreement with those described earlier for 1-(phenylsulfonyl)indoles. For example, the indole double bond length C1–C2 in (I) (1.355(3) Å), (II) (1.337(4) Å), (III) (1.361(5) Å), and (IV) (1.449(3) Å) are reasonably similar to that in 1-(phenylsulfonyl)indole (1.336(3) Å) [12], but indicative of some influence by the C–3 cyano substituent in (I) and (III) (Table 1). For these four compounds the sum of the angles around the indole nitrogen reveals the expected nearly ideal sp2-hybridization: (I), 358.8°; (II), 357.4°; (III), 359.9°; and (IV), 357.5°. The indole rings are essentially planar in the four compounds and the dihedral angles between the mean planes of the indole and phenylsulfonyl rings are 85.4(2)° (I), 87.2(7)° (II), 75.1(7)° (III), and 88.6(2)° (IV), respectively. For comparison, this angle is 94.0(2)° in 1-(phenylsulfonyl)indole [12].

Table 1.
Selected crystal and DFT bond lengths (Å), bond angles (°), and torsion angles (°) for (I) C15H10N2O2S, (II) C15H10N2O2S, (III) C16H9N3O2S and (IV) C19H20N2O3S.
| Atoms | Distance, Å | DFT, Å | Atoms | Distance, Å | DFT, Å | |
| (I) C15H10N2O2S | ||||||
| N1–S1 | 1.674(2) | *1.731 | N1–C1 | 1.374(3) | *1.386 | |
| N1–C8 | 1.409(3) | *1.410 | N2–C15 | 1.131(4) | *1.165 | |
| C15–C2 | 1.424(2) | *1.419 | C1–C2 | 1.355(3) | *1.372 | |
| S1–O1 | 1.4254(16) | *1.458 | S1–O2 | 1.4170(18) | *1.458 | |
| (II) C15H10N2O2S | ||||||
| S1–O2 | 1.4190(19) | *1.458 | S1–O1 | 1.4200(19) | *1.455 | |
| S1–N1 | 1.662(2) | *1.740 | S1–C9 | 1.751(2) | *1.789 | |
| N1–C8 | 1.407(3) | *1.405 | N1–C1 | 1.412(3) | *1.414 | |
| C1–C2 | 1.337(4) | *1.371 | C1–C15 | 1.427(3) | *1.418 | |
| C2–C3 | 1.414(4) | *1.428 | N2–C15 | 1.150(4) | *1.164 | |
| (III) C16H9N3O2S | ||||||
| S1–O2 | 1.414(3) | *1.457 | S1–O1 | 1.413(3) | *1.454 | |
| S1–N1 | 1.714(3) | *1.759 | S1–C9 | 1.754(3) | *1.786 | |
| N1–C1 | 1.388(4) | *1.400 | N1–C8 | 1.406(4) | *1.405 | |
| N2–C15 | 1.132(5) | *1.164 | N3–C16 | 1.135(5) | *1.163 | |
| C2–C16 | 1.436(5) | *1.419 | C1–C15 | 1.428(5) | *1.417 | |
| C1–C2 | 1.361(5) | *1.385 | C2–C3 | 1.429(5) | *1.437 | |
| (IV) C19H20N2O3S | ||||||
| S1–N1 | 1.6688(15) | *1.721 | S1–C9 | 1.7551(19) | *1.792 | |
| O3–C15 | 1.235(2) | *1.232 | N1–C1 | 1.397(2) | *1.396 | |
| N1–C8 | 1.413(2) | *1.410 | N2–C15 | 1.337(2) | *1.370 | |
| N2–C16 | 1.476(2) | *1.483 | C1–C2 | 1.349(3) | *1.367 | |
| C2–C3 | 1.449(3) | *1.450 | C2–C15 | 1.483(2) | *1.487 | |
| Atoms | Angles, ° | DFT, ° | Atoms | Angles, ° | DFT, ° | |
| (I) C15H10N2O2S | ||||||
| O2–S1–O1 | 121.64(10) | *123.10 | O2–S1–N1 | 105.80(10) | *105.81 | |
| O1–S1–N1 | 104.30(10) | *104.35 | O1–S1–C9 | 109.32(10) | *108.82 | |
| O2–S1–C9 | 109.63(10) | *108.98 | N1–S1–C9 | 104.66(10) | *104.05 | |
| C8–N1–S1 | 126.90(11) | *127.41 | C2–C1–N1 | 109.08(19) | *109.29 | |
| C1–N1–C8 | 108.98(19) | *109.19 | C1–N1–S1 | 122.94(15) | *122.12 | |
| (II) C15H10N2O2S | ||||||
| O2–S2–O1 | 121.13(12) | *122.53 | O2–S2–N1 | 106.31(11) | *105.05 | |
| O1–S1–N1 | 105.36(11) | *105.40 | O2–S1–C9 | 109.10(11) | *109.06 | |
| O1–S1–C9 | 108.75(11) | *109.12 | N1–S1–C9 | 104.99(10) | *104.02 | |
| C14–C9–S1 | 119.67(18) | *118.96 | C8–N1–S1 | 125.13(16) | *126.99 | |
| C1–N1–S1 | 125.38(19) | *124.26 | C2–C1–N1 | 109.2(3) | *109.28 | |
| C7–C8–N1 | 130.8(2) | *131.22 | C2–C1–C15 | 126.8(3) | *126.15 | |
| N2–C15–C1 | 174.5(3) | *175.57 | C4–C3–C2 | 133.7(2) | *132.52 | |
| N1–C1–C15 | 126.8(3) | *124.56 | C1–C2–C3 | 109.0(2) | *107.89 | |
| (III) C16H9N3O2S | ||||||
| O2–S–O1 | 122.22(18) | *123.96 | O2–S1–N1 | 105.18(15) | *126.71 | |
| O2–S1–N1 | 104.36(15) | *104.55 | O2–S1–C9 | 109.80(17) | *109.46 | |
| O1–S1–C9 | 109.43(17) | *109.55 | N1–S1–C9 | 104.13(14) | *103.49 | |
| N1–C1–C15 | 124.7(3) | *124.63 | C8–N1–C1 | 108.7(3) | *108.56 | |
| C3–C2–C16 | 126.01(3) | *126.15 | C4–C3–C2 | 132.6(3) | *132.20 | |
| C2–C1–N1 | 109.4(3) | *109.02 | C1–C2–C16 | 126.0(3) | *126.33 | |
| C8–N1–S1 | 126.3(2) | *127.08 | C2–C1–C15 | 125.9(3) | *126.33 | |
| C1–C2–C3 | 107.8(3) | *107.64 | C1–N1–S1 | 124.9(2) | *124.03 | |
| (IV) C19H20N2O3S | ||||||
| O2–S1–O1 | 121.46(9) | *122.66 | O2–S1–N1 | 104.85(8) | *104.53 | |
| O1–S1–N1 | 106.17(8) | *106.14 | O2–S1–C9 | 109.02(9) | *108.62 | |
| O1–S1–C9 | 108.96(9) | *108.80 | N1–S1–C9 | 105.11(8) | *104.57 | |
| C1–C2–C15 | 127.27(17) | *127.76 | C3–C2–C15 | 124.84(17) | *125.11 | |
| C2–C1–N1 | 109.91(16) | *109.87 | C1–C2–C3 | 107.50(16) | *107.12 | |
| C8-N1-S1 | 126.46(12) | *126.83 | C15-N2-C16 | 124.62(17) | *125.42 | |
| C1-N1-C8 | 108.16(15) | *108.57 | C1-N1-S1 | 122.87(12) | *121.94 | |
| Atoms | Torsions, ° | DFT, ° | Atoms | Torsions, ° | DFT, ° | |
| (I) C15H10N2O2S | ||||||
| O2–S1–N1–C1 | –158.34(18) | *–163.96 | O1–S1–N1–C1 | –28.9(2) | *–28.96 | |
| C9–S1–N1–C1 | 85.88(19) | *89.28 | O2–S1–N1–C8 | 35.5(2) | *38.43 | |
| O1–S1–N1–C8 | 164.89(19) | *169.60 | C9–S1–N1–C8 | –80.3(2) | *–76.31 | |
| N1–C1–C2–C15 | 179.6(2) | *178.54 | C4–C3–C2–C15 | 0.7(4) | *0.34 | |
| (II) C15H10N2O2S | ||||||
| O2–S1–N1–C8 | 38.8(2) | *20.81 | O1–S1–N1–C8 | 168.47(18) | *151.45 | |
| O2–S1–N1–C1 | –161.89(18) | *–171.16 | C9–S1–N1–C8 | –76.8(2) | *–93.75 | |
| O1–S1–N1–C1 | –32.2(2) | *–40.49 | C9–S1–N1–C1 | 82.6(2) | *74.28 | |
| S1–N1–C1–C2 | –165.63(19) | *–171.14 | C8–N1–C1–C2 | –3.2(13) | *–1.17 | |
| C8–N1–C1–C15 | 175.2(2) | *177.71 | S1–N1–C1–C15 | 12.7(3) | *7.73 | |
| C15–C1–C2–C3 | –176.3(2) | *–178.13 | N1–C1–C2–C3 | 2.0(3) | *0.72 | |
| (III) C16H9N3O2S | ||||||
| O1–S1–N1–C1 | –25.9(3) | *–40.02 | C9–S1–N1–C1 | 88.8(3) | *74.91 | |
| O2–S1–N1–C8 | 25.2(3) | *16.88 | O1–S1–N1–C8 | 155.0(3) | *147.35 | |
| C9–S1–N1–C8 | –90.3(3) | *–97.71 | C8–N1–C1–C2 | –1.7(4) | *–0.54 | |
| C15–C1–C3–C3 | –178.0(3) | *–177.96 | C16–C2–C3–C4 | –1.6(6) | *–0.20 | |
| N1–C1–C2–C16 | 179.8(3) | *179.59 | C15–C1–C2–C16 | 0.7(6) | *2.14 | |
| S1–N1–C1–C2 | 179.1(2) | *174.34 | N1–C1–C2–C3 | 1.4(4) | *0.31 | |
| (IV) C19H20N2O3S | ||||||
| O1–S1–N1–C1 | –155.47(15) | *–157.71 | O2–S1–N1–C1 | –25.74(17) | *–26.76 | |
| C9–S1–N1–C1 | 89.15(16) | *87.34 | O2–S1–N1–C8 | 174.42(16) | *173.89 | |
| C3–C2–C15–N2 | –154.56(19) | *–157.48 | C3–C2–C15–O3 | 28.5(3) | *23.44 | |
| O1–S1–N1–C8 | 44.69(18) | *42.94 | C9–S1–N1–C8 | –70.69(17) | *–72.02 | |
| C15–C2–C3–C8 | –173.38(18) | *–179.31 | N1–C1–C2–C15 | 174.10(18) | *178.79 | |
| CS–N1–C1–C2 | –164.64(14) | *–164.48 | C8–N1–C1–C2 | –1.6(2) | *–1.80 | |
DFT B3LYP 6-31 G(d) geometry optimization calculations for (I), (II), (III) and (IV) [13].
In (I) and (II) the cyano triple bond lengths are 1.131(4) Å and 1.150(4) Å, respectively. The longer bond in (II) may reflect electron donation from the indole nitrogen into the cyano π system. For comparison, 3-cyano-2-methyl-1-(4-methylphenyl)-5,6,7-trimethoxyindole has a C–N bond length of 1.142(2) Å [14], 5-azido-3-cyano-1-methylindole has a CN bond length of 1.149(2) Å [15], and 3-cyano-2,6-dimethyl-1-methoxyindole has a C–N bond length of 1.146(2) Å [16]. Similar comparisons with known 2-cyanoindoles could not be found. In dicyanoindole (III), the respective C–N bond lengths are identical, C–3 CN, 1.135(5) Å, and C–2 C–N, 1.132(5) Å. The C1–C2 indole double bond length in these three cyanoindoles is (I), 1.355(3) Å; (II), 1.337(4) Å; (III), 1.361(5) Å, which may reflect some well-known π-donation into the C–3 cyano group from the indole double bond which would lengthen C1–C2. Any π donation into the C–2 cyano group is much less significant. Accordingly, N1–C1 in (I) is shorter (1.374(3) Å) than N1–C1 in (II) (1.412(3) Å). In (III) this bond distance is 1.388(4) Å. The C15–C1–C2–C16 torsion angle is 0.7(6)°, the C15–C1–C2–C3 torsion angle is –178.0(3)°, and the N1–C1–C2–C16 torsion angle is 179.8(3)° indicating that the two cyano groups are coplanar and both lie in the plane of the indole ring. Likewise, in (I) the N1–C1–C2–C15 and C4–C3–C2–C15 torsion angles are 179.6(2)° and 0.7(4) °, respectively. In (II) the C3–C2–C1–C15 and S1–N1–C1–C15 torsion angles are –176.3(2)° and 12.7(3)°, respectively; the latter angle indicating that the N1–S1 bond is slightly out of the indole ring plane. In the crystal, weak C–H…O intermolecular interactions are observed in (I), (II), and (III) (Table 2). In addition, weak S–O…Cg (I) and C–H…Cg (II), (III), (IV) π-ring interactions and π–π stacking interactions in (II) and (III) are also present along with additional C–H…N interactions observed in (III) (Table 2). In (II) the π–π stacking interactions are observed between nearby phenyl rings (Cg2–Cg3), whereas in (III) these interactions exist on both the phenyl rings (Cg3–Cg3) as well as on the indole rings (Cg2–Cg1), forming a one-dimensional structure parallel to [111] and most likely as a result of the large difference in the indole-phenylsulfonyl dihedral angle observed between the actual and DFT calculated rings of 7.3(7)°. In (II) this difference was observed as 3.0(8)°. The influence of the additional C–H…N interaction in (III) appears to support this observation.
Table 2.
Hydrogen bond interactions for (I), (II), (III), and (IV) [Å and °].
| D–H...A | d(D–H) | d(H...A) | d(D...A) | <(DHA) |
|---|---|---|---|---|
| (I) | ||||
| C1–H1A...O1 #1 | 0.95 | 2.52 | 3.411(3) | 156 |
| S1–O2...Cg1 #2 | – | 3.09 | 3.8584(12) | 112 |
| S1–O4...Cg3 #2 | – | 3.22 | 3.9045(12) | 108 |
| (II) | ||||
| C2–H2A…O2 #3 | 0.95 | 2.50 | 3.435(3) | 168 |
| C5A–H5A…Cg3 #4 | – | 2.85 | 3.721(4) | 152 |
| Cg2…Cg2 #4 | – | – | 3.753(2) | – |
| (III) | ||||
| C5–H5A…N2 #5 | 0.95 | 2.61 | 3.540(5) | 172 |
| C13–H13A…O2 #6 | 0.95 | 2.52 | 3.257(5) | 134 |
| C4–H4A…Cg3 #7 | – | 2.61 | 3.499(4) | 156 |
| Cg2…Cg1 #7 | – | – | 3.797(2) | – |
| Cg3…Cg3 #8 | – | – | 3.809(2) | – |
| (IV) | ||||
| N2–H2N…O3 #9 | 0.85 | 2.12 | 2.967(2) | 178 |
| C11–H11A…O2 #10 | 0.95 | 2.56 | 3.457(3) | 157 |
| C17–H17E…O3 | 0.98 | 2.17 | 2.867(8) | 127 |
| C6–H6A…O1 #11 | 0.95 | 2.59 | 3.540(3) | 176 |
| C5–H5A…Cg1 #12 | – | 2.93 | 3.794(3) | 152 |
| C18–H18C…Cg1 #13 | – | 2.75 | 3.702(8) | 163 |
Symmetry codes: #1 x + 1/2, –y + 3/2, –z; #2 –1 + x, y, z; #3 x − 1/2, –y + 3/2, z − 1/2; #4 1 − x, 1 − y, 1 − z; #5 1 + x, 1 + y, z; #6 –1 + x, y, z; #7 1 − x, 2 − y, 1 − z; #8 –x, 1 − y, −z; #9 x, –y + 3/2, z − 1/2; #10 –x + 3/2, y, z + 1/2; #11 –x + 1;2, y, z + 1/2; #12 ½ − x, y, 1/2 + z; #13 x, 3/2 − y, –1/2 + z. In (I) Cg1 = N1/C1/C2/C3/C8 and Cg3 = C9/C10/C11/C12/C13/C14; In (II) Cg2 = C3/C4/C5/C6/C7C8, Cg3 = C9/C10/C11/C12/C13C14; In (III) Cg1 = N1/C1/C2/C3/C8, Cg2 = C3/C4/C5/C6/C7C8, Cg3 = C9/C10/C11/C12/C13/C14; In (IV) Cg1 = N1/C1/C2/C3/C8.
In (IV), the methyl and ethyl atoms of the tertiary butyl group are disordered over two sites in an occupancy ratio 0.544(10): 0.456(10). In the crystal, N–H…O and C–H…O classical hydrogen bonds are observed forming chains along [001] (Figure 4b, Table 3). Weak C–H…O and C–H…Cg π-ring interactions are also observed (Table 2) providing additional crystal stability. The amide functionality in (IV) is in the expected anti-periplanar conformation and, as revealed by the torsion angles C3–C2–C15–N2, –154.56(19)° and C3–C2–C15–O3, 28.5(3)°, is twisted out of conjugation with the indole double bond. The somewhat large C15–N2–C16 bond angle, 124.64(17)°, perhaps results from steric repulsion between the carbonyl group (C15–O3) and the C16 tertiary butyl group. Overall, bond lengths and bond angles are all within expected ranges [17], with small exceptions noted.
2.2. Theoretical Study of (I)
After a DFT geometry optimization calculation, the dihedral angle between the mean planes of the indole and phenylsulfonyl rings becomes 86.2(8)°, an increase of 0.8(6)°. Bond lengths and bond angles show only small changes with the exception of selected torsion angles consistent with the differences in the mean planes changes indicated above (Table 1). These changes suggest that the single weak C–H…O intermolecular interaction involving the indole ring and a sulfonyl oxygen atom plays only a small role in the crystal packing of the molecule (Table 2).
Calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals for (I) show three absorption band envelopes, exhibiting some blue shifts, which are consistent with the experimental data (Figure 6 and Table 3) with λmax values located at 292, 263, and 217 nm, respectively. The bands in the UV region 290–260 nm are assigned to cyano n → π* and π → π* transitions while the other band at 217 nm is assigned to aromatic π → π* transitions. In HOMO the electronic clouds are distributed primarily on the indole ring and cyano group. In HOMO–1 they are located only on the indole ring. In LUMO the electronic clouds are delocalized primarily on the phenyl ring while in LUMO+1 they are located on both the indole and phenyl rings, as well as on the cyano group. In LUMO+2 they are dispersed primarily on the indole ring. Therefore, the first absorption band envelope at 292 nm is assigned to contributions primarily from HOMO- > LUMO. The second absorption band at 263 nm is assigned to overlapping contributions from HOMO–1- > LUMO and HOMO- > LUMO+1. The third absorption band at 217 nm is assigned to overlapping contributions from HOMO–1- > LUMO+1, HOMO- > LUMO+2 and HOMO–1- > LUMO+2, respectively. It is evident that electron transitions among frontier molecular orbitals in (I) are corrsponding to n → π*and π → π* transitions.
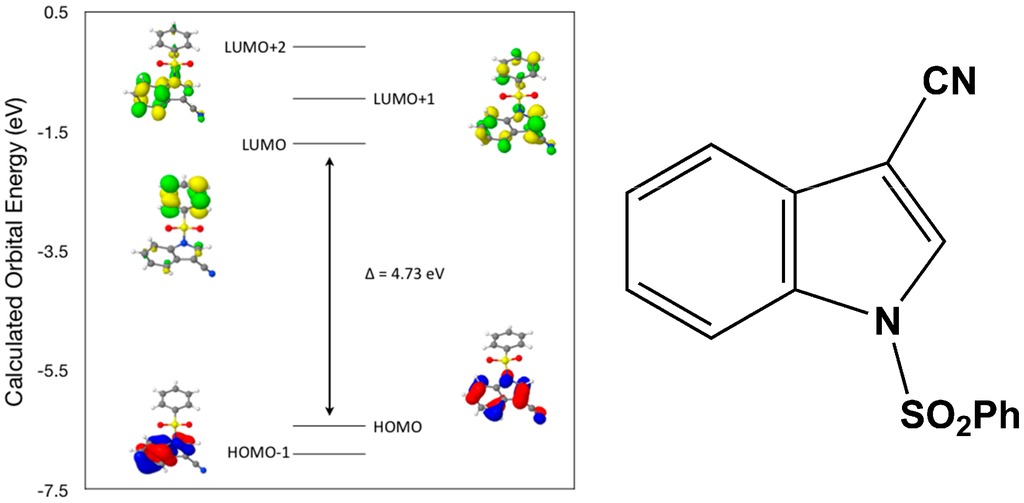
Figure 6.
Calculated frontier molecular orbitals for the C15H10N2O2S (I).
Table 3.
Experimental and calculated energy of molecular orbitals of (I) and associated transitions.
| Experimental | Calculated | ||
|---|---|---|---|
| λmax (nm/eV) | f | λmax (nm/eV) | MO Contributions |
| 292/4.25 | 0.07 | 262/4.73 | HOMO → LUMO |
| 263/4.71 | 0.13 | 238/5.20 | HOMO–1 → LUMO |
| 263/4.71 | 0.13 | 226/5.48 | HOMO → LUMO+1 |
| 217/5.71 | 0.25 | 208/5.95 | HOMO–1 → LUMO+1 |
| 217/5.71 | 0.25 | 195/6.35 | HOMO → LUMO+2 |
| 217/5.71 | 0.25 | 182/6.82 | HOMO–1 → LUMO+2 |
Ocsillator Strength, f = 4.32 × 10−9·εmax·Δω1/2.
2.3. Theoretical Study of (II)
After a DFT geometry optimization calculation, the dihedral angle between the mean planes of the indole and phenylsulfonyl rings becomes 84.1(9)°, a decrease of 3.0(8)°. Again, bond lengths and bond angles show only small changes with the exception of selected torsion angles consistent with the differences in the mean planes changes indicated above (Table 1). These changes also suggest that the single weak C–H…O intermolecular interaction involving the indole ring and a sulfonyl oxygen atom plays a small role in the crystal packing of the molecule (Table 2).
Calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals for (II) show three absorption band envelopes, exhibiting some blue shifts, which are consistent with the experimental data (Figure 7 and Table 4) with λmax values located at 310, 279 and 241 nm, respectively. The bands in the UV region 310–280 nm are assigned to cyano n → π* and π → π* transitions while the other band at 241 nm is assigned to aromatic π → π* transitions. In both HOMO and HOMO–1 the electronic clouds are distributed primarily on both the indole ring and cyano group. In LUMO the electronic clouds are delocalized primarily on the indole ring and cyano group while in LUMO+1 and LUMO+2 they are located only the phenyl ring. Electronic transitions are generally paired between the various molecular orbitals of the ground and excited states corresponding to these three band envelopes as indicated in Table 4. Therefore, the first absorption band envelope at 310 nm is assigned to contributions primarily from HOMO- > LUMO. The second absorption band envelope at 279 nm is assigned to overlapping contributions from HOMO–1- > LUMO and HOMO- > LUMO+1. The third absorption band at 241 nm is assigned to overlapping contributions from HOMO–1- > LUMO+1, HOMO- > LUMO+2 and HOMO–1- > LUMO+2, respectively. It is evident that electron transitions among frontier molecular orbitals in (II) are corrsponding to n → π*and π → π* transitions.
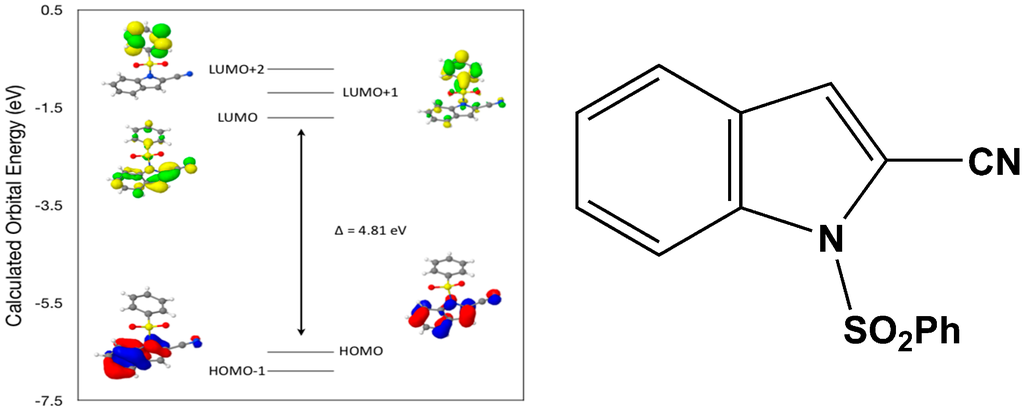
Figure 7.
Calculated frontier molecular orbitals for the C15H10N2O2S (II).
Table 4.
Experimental and calculated energy of molecular orbitals of (II) and associated transitions.
| Experimental | Calculated | ||
|---|---|---|---|
| λmax (nm/eV) | f | λmax (nm/eV) | MO Contributions |
| 310/4.00 | 0.02 | 258/4.81 | HOMO → LUMO |
| 279/4.44 | 0.09 | 238/5.20 | HOMO–1 → LUMO |
| 279/4.44 | 0.09 | 233/5.32 | HOMO → LUMO+1 |
| 241/5.14 | 0.01 | 217/5.71 | HOMO–1 → LUMO+1 |
| 241/5.14 | 0.01 | 214/5.81 | HOMO → LUMO+2 |
| 241/5.14 | 0.01 | 200/6.19 | HOMO–1 → LUMO+2 |
Oscillator Strength, f = 4.32 × 10−9·εmax·Δω1/2.
2.4. Theoretical Study of (III)
After a DFT geometry optimization calculation, the dihedral angle between the mean planes of the indole and phenylsulfonyl rings becomes 82.5(4) °, an increase of 7.3(7)°. Again, bond lengths and bond angles show only small changes with the exception of selected torsion angles consistent with the differences in the mean planes changes indicated above (Table 1). These changes suggest that the two weak intermolecular interactions involving the indole ring (C–H…O) with a sulfonyl oxygen atom and with a cyano group (C–H…N) nitrogen atom play significant roles in the crystal packing of the molecule (Table 2).
Calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals for (III) show two absorption band envelopes, exhibiting some blue shifts, which are consistent with the experimental data (Figure 8 and Table 5) with λmax values located at 298 and 229 nm, respectively. The band in the 300 nm UV region is assigned to cyano n → π* and π → π* transitions while the other band at 229 nm is assigned to aromatic π → π* transitions. In both HOMO and HOMO–1 the electronic clouds are distributed primarily on both the indole ring and cyano groups. In LUMO the electronic clouds are delocalized primarily on the indole ring and cyano group while in LUMO+1 and LUMO+2 they are located primarily on the phenyl ring. Electronic transitions are generally paired between the various molecular orbitals of the ground and excited states corresponding to these two band envelopes as indicated in Table 5. Therefore, the first absorption band envelope at 298 nm is assigned to overlapping contributions primarily from HOMO- > LUMO, HOMO–1- > LUMO and HOMO- > LUMO+1. The second absorption band at 229 nm is assigned to overlapping contributions from HOMO–1- > LUMO+1, HOMO- > LUMO+2 and HOMO–1- > LUMO+2, respectively. Again, it is evident that electron transitions among frontier molecular orbitals in (III) are corrsponding to n → π*and π → π* transitions.
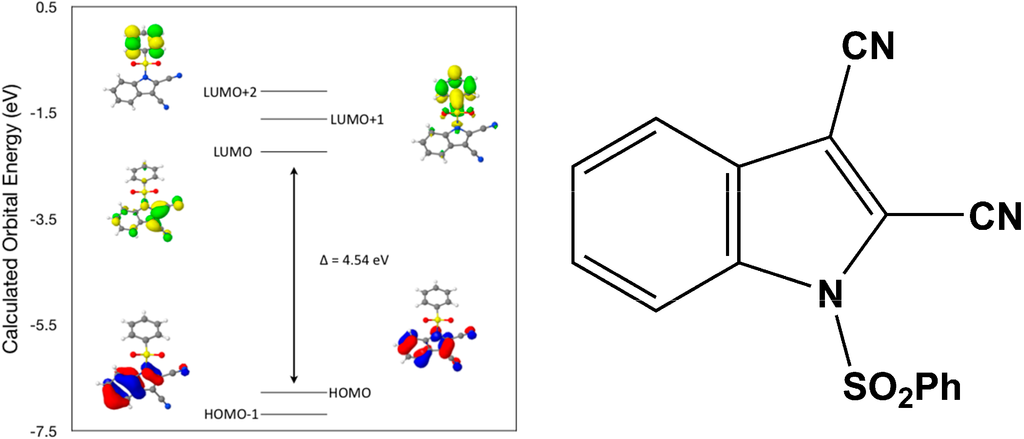
Figure 8.
Calculated frontier molecular orbitals for the C16H9N3O2S (III).
Table 5.
Experimental and calculated energy of molecular orbitals of (III) and associated transitions.
| Experimental | Calculated | ||
|---|---|---|---|
| λmax (nm/eV) | f | λmax (nm/eV) | MO Contributions |
| 298/4.16 | 0.2 | 273/4.54 | HOMO → LUMO |
| 298/4.16 | 0.2 | 250/4.95 | HOMO–1 → LUMO |
| 298/4.16 | 0.2 | 241/5.15 | HOMO → LUMO+1 |
| 229/5.41 | 0.19 | 223/5.57 | HOMO–1 → LUMO+1 |
| 229/5.41 | 0.19 | 218/5.68 | HOMO → LUMO+2 |
| 229/5.41 | 0.19 | 203/6.09 | HOMO–1 → LUMO+2 |
Oscillator Strength, f = 4.32 × 10−9·εmax·Δω1/2.
2.5. Theoretical Study of (IV)
After a DFT geometry optimization calculation, the dihedral angle between the mean planes of the indole and phenylsulfonyl rings becomes 89.5(3)°, an increase of 0.9(1)°. Again, bond lengths and bond angles show only small changes with the exception of selected torsion angles consistent with the differences in the mean planes changes indicated above (Table 1). These changes suggest that the hydrogen bonds involving the carboxamide ligand (C–H…O and N–H…O) in concert with weak C–H…O intermolecular interactions involving the indole and phenyl groups with the two sulfonyl oxygen atoms play only a small role in the crystal packing of the molecule (Table 2).
Calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals for (IV) show two absorption band envelopes, exhibiting some blue shifts, which are consistent with the experimental data (Figure 9 and Table 6) with λmax values located at 252 and 210 nm, respectively. Both bands in the 250 nm and 230 UV regions areassigned to aromatic π → π* transitions. In HOMO and HOMO–1 the electronic clouds are distributed primarily on the indole ring. In LUMO and LUMO+1 the electronic clouds are delocalized primarily on the phenyl ring while in LUMO+2 they are located only on the indole ring. Electronic transitions are generally paired between the various molecular orbitals of the ground and excited states corresponding to these two band envelopes as indicated in Table 6. Therefore, the first absorption band envelope at 252 nm is assigned to contributions primarily from HOMO- > LUMO and HOMO–1- > LUMO. The second absorption band at 210 nm is assigned to overlapping contributions from HOMO- > LUMO+1, HOMO- > LUMO+2, HOMO–1- > LUMO+1 and HOMO–1- > LUMO+2, respectively. Again, it is evident that electron transitions among frontier molecular orbitals in (IV) are corrsponding to n → π*and π → π* transitions.
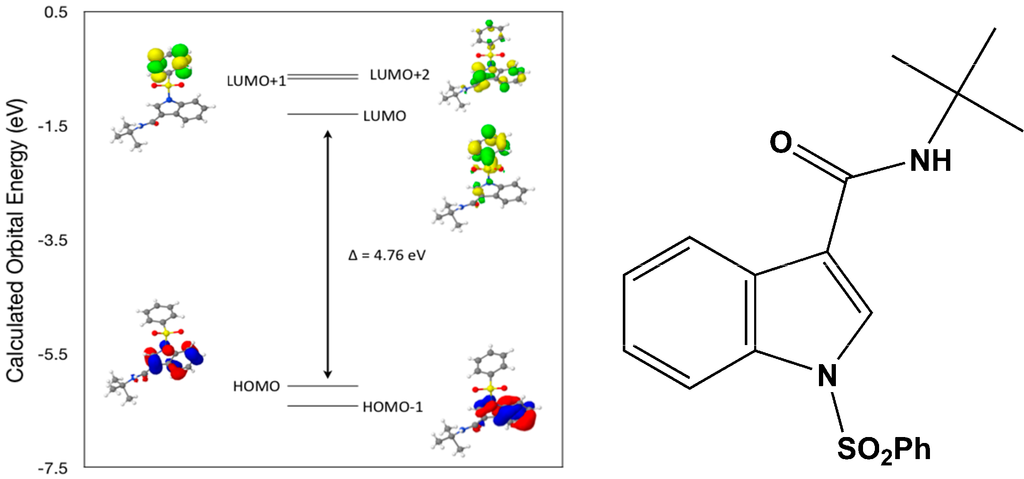
Figure 9.
Calculated frontier molecular orbitals for the C19H20N2O3S (IV).
Table 6.
Experimental and calculated energy of molecular orbitals of (IV) and associated transitions.
| Experimental | Calculated | ||
|---|---|---|---|
| λmax (nm/eV) | f | λmax (nm/eV) | MO Contributions |
| 252/4.92 | 0.24 | 260/4.76 | HOMO → LUMO |
| 252/4.92 | 0.24 | 242/5.11 | HOMO–1 → LUMO |
| 210/5.90 | 0.3 | 230/5.39 | HOMO → LUMO+1 |
| 210/5.90 | 0.3 | 227/5.45 | HOMO → LUMO+2 |
| 210/5.90 | 0.3 | 216/5.74 | HOMO–1 → LUMO+1 |
| 210/5.90 | 0.3 | 214/5.80 | HOMO–1 → LUMO+2 |
Oscillator Strength, f = 4.32 × 10−9·εmax·Δω1/2.
3. Experimental Procedures
3.1. Synthesis of 3-cyano-1-(phenylsulfonyl)indole (I)
A stirred solution of 1-(phenylsulfonyl)-N-(tert-butyl)indole-3-carboxamide (3.76 g, 10.5 mmol) in benzene (75 mL) under N2 was treated with phosphorus oxychloride (20 mL, 0.2 mol) (Scheme 1).
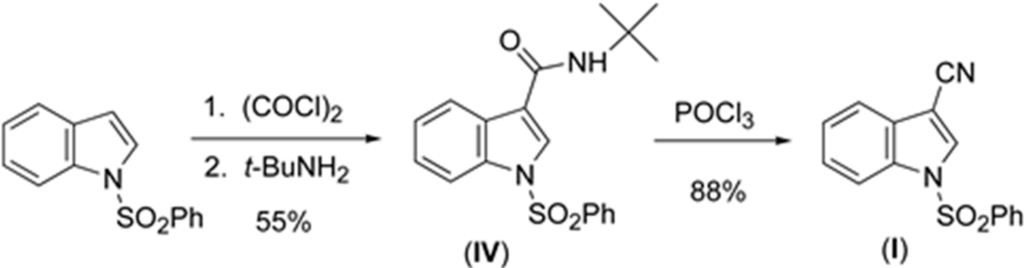
Scheme 1.
Synthesis of (I).
The mixture was refluxed overnight. The solution was quenched with aqueous saturated NaHCO3 (400 mL) and stirred until evolution of gas ceased. The solution was then extracted with methylene chloride (100 mL). The organic layer was washed with water, brine, dried over magnesium sulfate, filtered, and concentrated in vacuo to yield a white solid. The solid was purified using flash chromatography (100% CH2Cl2) to give the title compound as a white solid (2.61 g, 88%): mp 416–418 K (lit. mp [18] 424–425 K); 1H NMR (CDCl3) δ 8.11 (s, 1H) 7.99–8.02 (d, 1H) 7.94–7.97 (d, 2H) 7.69–7.71 (d, 1H) 7.61–7.64 (d, 1H) 7.50–7.55 (m, 2H) 7.42–7.48 (m, 1H) 7.36–7.41 (m, 1H); 13C NMR (CDCl3) δ 137.1, 134.9, 133.6, 133.1, 129.8, 128.3, 127.1, 126.6. 124.9, 120.3, 113.7, 113.4, 93.9. UV-vis data collected on a JASCO V-630 (JASCOINC, 28600 Mary’s Court, Easton, MD, USA) from 800–200 nm. Crystals suitable for X-ray analysis were grown from ethanol.
3.2. Synthesis of 2-cyano-1-(phenylsulfonyl)indole (II)
To a stirred solution of dry diisopropylamine (0.5 mL) in dry THF (10 mL) at 273 K under N2 was added n-butyllithium (2.5 M, 1.4 mL, 3.5 mmol). It was stirred at 273 K for 45 min. The freshly prepared LDA was then added to a stirred solution of 1-(phenylsulfonyl) indole (796 mg, 3.09 mmol) in dry THF (10 mL) at 195 K under N2 (Scheme 2).
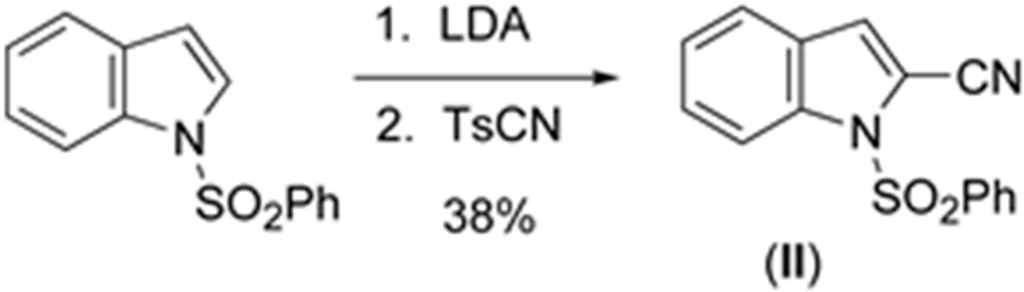
Scheme 2.
Synthesis of (II).
After stirring for 3 h, a suspension of p-toluenesulfonyl cyanide (840 mg, 4.6 mmol) in dry THF (5.0 mL) was added quickly. The reaction was allowed to slowly reach room temperature overnight. Thereafter, the mixture was quenched by the addition of aqueous saturated NH4Cl (100 mL) and stirred for 1 h. The mixture was extracted with methylene chloride (2 × 50 mL). The organic extracts were washed with water, brine, dried over magnesium sulfate, filtered, and concentrated in vacuo to afford a brown residue. This was subjected to flash chromatography [hexanes-CH2Cl2 (1:1)] to give the title compound as white needles (330 mg, 38%): mp 386–388 K (Lit. mp [19] 400.5–402 K) 1H NMR (CDCl3) δ 8.22–8.24 (d, 1H) 8.02–8.05 (d, 2H) 7.55–7.62 (m, 3H) 7.48–7.53 (m, 3H) 7.38 (s, 1H); 13C NMR (CDCl3) δ 136.9, 136.3, 134.6, 129.4, 128.5, 127.2, 126.8, 124.6, 123.1, 122.4, 114.3, 112.0, 108.7; IR (NaCl) 2231 (CN) cm−1. UV-VIS data collected on a JASCO V-630 from 800–200 nm. Crystals suitable for X-ray analysis were grown from ethanol.
3.3. Synthesis of 2,3-dicyano-1-(phenylsulfonyl)indole (III)
To a stirred solution of 3-cyano-1-(phenylsulfonyl)indole (438 mg, 1.55 mmol) in dry THF (16 mL) was added a solution of LDA in THF/heptane (2 M, 1 mL, 2 mmol) at 195 K under N2 (Scheme 3).
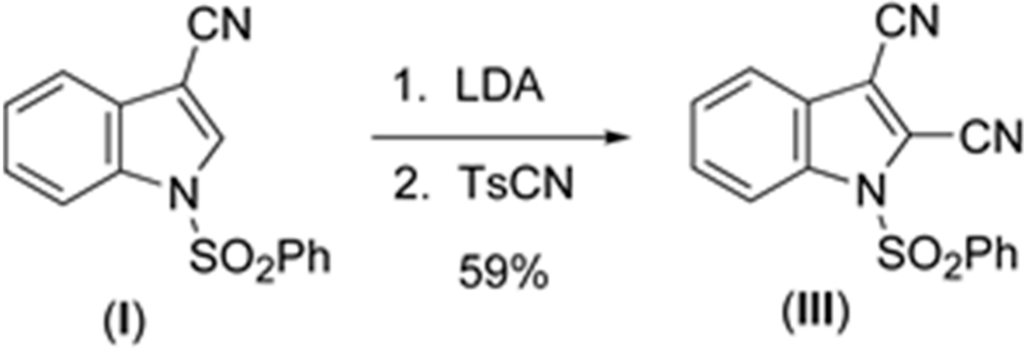
Scheme 3.
Synthesis of (III).
After stirring for 2 h, a suspension of p-toluenesulfonyl cyanide (434 mg, 2.4 mmol) in dry THF (2.0 mL) was added. The resulting mixture was allowed to slowly reach room temperature overnight. Thereafter, the mixture was quenched by the addition of aqueous saturated NH4Cl (50 mL) and stirred for 1 h. The mixture was extracted with methylene chloride (2 × 30 mL). The organic extracts were washed with water, brine, dried over magnesium sulfate, filtered, and concentrated in vacuo to afford a brown residue. This was subjected to flash chromatography [hexanes-CH2Cl2 (1:1)] to give the title compound as white needles (280 mg, 59%): mp 431–435 K (lit. mp [20] 440–442 K); 1H NMR (CDCl3) δ 8.25–8.28 (d, 1H) 8.07–8.10 (d, 2H) 7.64–7.75 (m, 3H) 7.55–7.60 (m, 2H) 7.47–7.52 (m, 1H); 13C NMR (CDCl3) δ 136.7, 136.1, 135.6, 130.5, 130.4, 127.7, 126.6, 126.4, 121.3, 115.0, 114.3, 110.9, 109.4, 105.8. UV-vis data collected on a JASCO V-630 from 800–200 nm. Crystals suitable for X-ray analysis were grown from dichloromethane.
3.4. Synthesis of 1-(phenylsulfonyl)-1-(1,1-dimethylethyl)indole-3-carboxamide (IV)
To a stirred solution of AlCl3 (80.0 g, 0.600 mol) in methylene chloride (600 mL) at 273 K under N2 was added dropwise oxalyl chloride (53.0 mL, 0.6 mol), resulting in a yellow solution. After 1 h at 273 K, 1-(phenylsulfonyl)indole (31.38 g, 0.122 mol) was added and the resulting red mixture was allowed to warm to room temperature. After 3 h, the reaction mixture was poured over ice (1200 mL) in a 5 L beaker. The aqueous layer was then extracted with methylene chloride (3 × 150 mL). The organic extracts were concentrated in vacuo to roughly half of the previous volume. The concentrated extracts were stirred overnight under N2 with excess tert-butylamine (60 mL, 0.60 mol). The reaction mixture was then washed sequentially with 10% aqueous HCl (600 mL), aqueous saturated NaHCO3 (600 mL), and brine (600 mL); the organic layer was dried with magnesium sulfate, filtered, and concentrated in vacuo, resulting in a dark brown solid. The crude product was recrystallized from ether to afford 22.60 g of the title compound as light tan needles (55%): mp 477–479 K (lit. mp [18] 482–483 K); 1H NMR (CDCl3) δ 8.05–8.40 (m, 1H) 7.93–7.96 (d, 1H) 7.84–7.86 (d, 1H) 7.48–7.54 (m, 1H) 7.36–7.42 (m, 2H) 7.28–7.34 (m, 2H) 6.00 (s, 1H) 1.49 (s, 9H); 13C NMR (CDCl3) δ 162.8, 137.6, 134.9, 134.3, 129.5, 127.9, 126.9, 126.9, 125.4, 124.2, 121.5, 118.9, 113.4, 51.8, 29.0. UV-VIS data collected on a JASCO V-630 from 800–200 nm. Crystals suitable for X-ray analysis were grown from ethanol.
3.5. X-ray Structure Analysis and Refinement
Individual crystals of compounds (I), (II), (III), and (IV) were mounted on a CryoLoop (Hampton Research, 34 Journey, Aliso Viejo, CA, USA) and placed in a –100 °C compressed air stream on an Agilent Gemini-EOS Single Crystal Autodiffractometer at Keene State College (Agilent Technologies, LTD, Yarnton, England,). Crystallographic data were collected using graphite monochromated 0.71073 Å Mo-Kα radiation and integrated and corrected for absorption using the CrysAlisRed (Oxford Diffraction, 2010 software package) [21]. The structures were solved using direct methods and refined using least-square methods on F-squared [22]. The hydrogen atoms were placed in their calculated positions and included in the refinement using the riding model. All other pertinent crystallographic details such as h, k, l ranges, 2θ ranges, and R-factors can be found in Table 1.
Table 7.
Crystal data and structure refinement for (I), (II), (III), and (IV).
| Identification Code | I | II | III | IV |
|---|---|---|---|---|
| Formula | C15H10N2O2S | C15H10N2O2S | C16H9N3O2S | C19H20N2O3S |
| Formula weight | 282.31 | 282.31 | 307.32 | 356.43 |
| Crystal color, habit | colorless, plate | colorless, block | colorless, block | colorless, block |
| Crystal size (mm) | 0.40 × 0.20 × 0.10 | 0.25 × 0.22 × 0.15 | 0.18 × 0.15 × 0.10 | 0.35 × 0.33 × 0.28 |
| Crystal system | orthorhombic | monoclinic | triclinic | orthorhombic |
| Space Group, Z | P 21 21 21, 4 | C 2/c, 8 | P –1, 2 | P ccn, 8 |
| Temperature, K | 173(2) | 173(2) | 173(2) | 173(2) |
| a (Å) | 4.9459(3) | 18.062(2) | 8.1986(8) | 13.7605(8) |
| b (Å) | 10.5401(7) | 11.293(2) | 9.6381(11) | 27.3177(14) |
| c (Å) | 25.0813(14) | 15.922(3) | 9.8113(5) | 9.7584(6) |
| α (°) | 90 | 90 | 95.053(6) | 90 |
| β (°) | 90 | 124.49(2) | 101.441(6) | 90 |
| γ (°) | 90 | 90 | 108.071(9) | 90 |
| Volume, Å3 | 1307.50(14) | 2676.7(7) | 713.02(11) | 3668.2(4) |
| F(000) | 584 | 1168 | 316 | 1504 |
| µ (mm−1) | 0.249 | 0.244 | 2.115 | 0.196 |
| Dcalc (Mg·m−3) | 1.434 | 1.401 | 1.431 | 1.291 |
| θmax/o with Mo/Cu Kα | 28.27 | 27.88 | 71.5 | 27.88 |
| Independent Reflections/Rint | 7904/0.0171 | 12107/0.0669 | 4435/0.0595 | 31435/0.018 |
| Reflections [I > 2σ (I)] | 3250 | 3188 | 2689 | - |
| R/Rw [I > 2σ (I)] | 0.0453/0.1107 | 0.0552/0.1457 | 0.0545/0.1697 | 0.0512/0.1164 |
| Collection range | - | - | - | - |
| h | –6 to 6 | –23 to 23 | –9 to 10 | –18 to 18 |
| k | –14 to 14 | –14 to 14 | –11 to 11 | –34 to 35 |
| l | –31 to 33 | –20 to 20 | –7 to 12 | –12 to 12 |
| GOF on F2 | 1.084 | 1.025 | 1.089 | 1.074 |
| (Δρ)max/min/e Å−3 | 0.24/–0.29 | 0.33/–0.31 | 0.38/–0.34 | 0.31/–0.32 |
| Measurement | GEMINI (Oxford Diffraction, 2007) | - | - | - |
| Program System | CrysAlisPro | - | - | - |
| Structure Determination | SHELXS97 | - | - | - |
3.6. Computational Details
A density functional theory (DFT) molecular orbital calculation (WebMo Pro [13] with the GAUSSIAN-03 program package [23] employing the B3LYP (Becke three parameter Lee-Yang-Parr exchange correlation functional), which combines the hybrid exchange functional of Becke [24,25] with the gradient correlation functional of Lee, Yang and Parr [23] and the 6-31 G(d) basis set [26] was performed on each of the four compounds. No solvent corrections were made with these calculations. Starting geometries were taken from X-ray refinement data. The optimized results in the free molecule state are, therefore, compared to those in the crystalline state. Experimentally determined oscillator strengths (f) were determined by use of the equation relating them to the molar decadic absorption coefficient (e) (f = 4.32 × 10−9·emax·Δω1/2) [27,28]. The molar decadic absorption coefficient measures the intensity of the optical absorption at a given wavelength. Deconvolution of the spectra to obtain the emax and Δω1/2 values was carried out by the IGOR program [29]. Discrepancies between the experimental and calculated band centers and band intensities exist. However, this does not prohibit us from making informed decisions on the observations since it is generally known that DFT often underestimates HOMO-LUMO gaps, thereby having a tendency to give excitations far too low in energy. All calculations were performed on a workstation PC using default convergence criteria.
3.7. Density Functional Theory (DFT) Calculations
A comparison of selected bond angles and bond distances in the crystal to that from the geometry optimized DFT calculations at the B3LYP 6-31G(d) level is given in Table 1. The differences between the two values are within normal ranges and generally consistent with bond lengths and angles for similar types of compounds. In addition, a comparison of the angles between mean planes of the indole and phenylsulfonyl rings in the crystal and with the DFT geometry optimized calculation in concert with strong and weak intermolecular hydrogen bond interactions has been included in a discussion of the structural aspects for each molecule. From a DFT molecular orbital calculation for each compound, surface plots for the two highest occupied molecular orbital (HOMO and HOMO–1) and three lowest unoccupied molecular orbitals (LUMO, LUMO+1, LUMO+2) are displayed to provide visual evidence of the molecular orbitals involved in the spectroscopic electronic energy transitions examined. Based on correlation of the energies of these HOMO-LUMO frontier surfaces to the UV-VIS absorption spectra, electronic excitation transition predications are suggested.
4. Summary and Conclusions
The crystal and molecular structure of three new cyano(phenylsulfonyl)indoles and a key synthetic precursor have been determined, along with the frontier molecular orbitals of each compound displayed through density function theory (DFT-B3LYP 6-31G(d)) geometry optimization and molecular orbital calculations. Correlation between the calculated molecular orbital energies (eV) for the surfaces of the frontier molecular orbitals to the electronic excitation transitions from the absorption spectrum of each compound has been determined. In each compound, the DFT molecular orbital calculation, supported by a geometry optimization calculation confirmed that the excitation energies of the surfaces of the frontier molecular orbitals from the HOMO–1 and HOMO to LUMO, LUMO+1, LUMO+2, and LUMO+3 electronic excitations closely match the λmax values of the absorption spectra in overlapping contributions from two, three or four of these excitations within each band envelope. In the crystal structures of three compounds, it has been determined that hydrogen bonds and/or weak C–H…O intermolecular interactions play a small role in the crystal packing of each molecule. In compound (III), the presence of a second cyano nitrogen atom plays a significant role in the observed intermolecular interactions and in the crystal packing. This is supported by changes in the mean planes between the rings within the asymmetric unit when a comparison is made between the crystal structures and density functional theory (DFT) geometry optimization calculations.
Acknowledgments
Jerry P. Jasinski acknowledges the NSF-MRI program (grant No.CHE-1039027) for funds to purchase the X-ray diffractometer, Justin M. Lopchuk acknowledges support from the Department of Education for a GAANN Fellowship, and Gordon W. Gribble acknowledges support from the Donors of the Petroleum Research Fund, administered by the American Chemical Society.
Author Contributions
William L. Montgomery and Justin M. Lopchuk designed the experiments and synthesized the compounds; Gordon W. Gribble contributed reagents/materials/analysis tools for the project, interpreted the NMR data and assisted with writing the paper; Jerry P. Jasinski collected the X-ray data, solved the X-ray structures, interpreted the crystallographic results, performed the DFT calculations and assisted with writing the paper.
Conflict of Interest
The authors declare no conflict of interest.
Appendix
CCDC 1412731 (I), 1412732 (II), 1412733 (III), 1412734 (IV) contains supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44-1223-336033. E-Mail: deposit@ccdc.cam.uk or at http://www.ccdc.cam.ac.uk).
References
- Gribble, G.W.; Saulnier, M.G.; Pelkey, E.T.; Kishbaugh, T.L.S.; Liu, Y.-B.; Jiang, J.; Trujillo, H.A.; Keavy, D.J.; Davis, D.A.; Conway, S.C.; et al. Novel indole chemistry in the synthesis of heterocycles. Curr. Org. Chem. 2005, 9, 1493–1519. [Google Scholar] [CrossRef]
- Okamoto, K.; Watanabe, M.; Murai, M.; Hatano, R.; Ohe, K. Practical synthesis of aromatic nitriles via gallium-catalysed electrophilic cyanation of aromatic C-H bonds. Chem. Commun. 2012, 48, 3127–3129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.; Chang, S. Copper-mediated selective cyanation of indoles and 2-phenylpyridines with ammonium iodide and DMF. Org. Lett. 2012, 14, 3924–3927. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Huang, X.; Hong, X.; Xu, B. Palladium-assisted regioselective C-H cyanation of heteroarenes using isonitrile as cyanide source. Org. Lett. 2012, 14, 4614–4617. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhao, J.; Hu, Z.; Liang, D.; Huang, J.; Zhu, Q. Palladium-catalyzed C(sp2) –H cyanation using tertiary amine derived isocyanide as a cyano source. Org. Lett. 2012, 14, 4966–4969. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, Z. Chemistry of the Cyano Group; Wiley: London, UK, 1970; pp. 121–312. [Google Scholar]
- Liskey, C.W.; Liao, X.; Hartwig, J.F. Cyanation of arenes via iridium-catalyzed borylation. J. Am. Chem. Soc. 2010, 132, 11389–11391. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Synthesis, Patents, Applications, 4th ed.; Thieme Medical: Leipzig, Germany, 2001. [Google Scholar]
- Sriram, D.; Yogeeswari, P. Medicinal Chemistry; Pearson Education: München, German, 2007; p. 35. [Google Scholar]
- Gomes, A.C.; Biswas, G.; Biswas, S.; Biswas, G.K.; Iitaka, Y.; Banerjee, A. Crystal structure of N-(4-nitrophenyl)-N-phenylsulfonamide. J. Crystallogr. Spectrosc. Res. 1986, 23, 513–517. [Google Scholar] [CrossRef]
- Jasinski, J.P.; Rinderspacher, A.; Gribble, G.W. Structures of three new (Phenylsulfonyl)indole derivatives. J. Chem. Crystallogr. 2010, 40, 40–47. [Google Scholar] [CrossRef]
- Beddoes, R.L.; Dalton, L.; Joule, J.A.; Mills, O.S.; Street, J.D.; Watt, C.I.F. The geometry at nitrogen in N-phenylsulphonyl-pyrroles and -indoles. The geometry of sulphonamides. J. Chem. Soc. Perkin Trans. 2 1986, 787–797. [Google Scholar] [CrossRef]
- WebMO Pro, version 8.0.01e; WebMO LLc: Holland, The Netherland, 2007.
- Du, Y.; Liu, R.; Linn, G.; Zhao, K. Synthesis of N-substituted indole derivatives via PIFA-mediated intramolecular cyclization. Org. Lett. 2006, 8, 5919–5922. [Google Scholar] [CrossRef] [PubMed]
- Pluta, K.; Andersen, K.V.; Jensen, F.; Becher, J. Nucleophilic tele-substitution in 2-Chloro-3-formylindoles via ring opening-ring closure. J. Chem. Soc. Chem. Commun. 1988, 1583–1584. [Google Scholar] [CrossRef]
- Du, Y.; Chang, J.; Reiner, J.; Zhao, K. Formation of N-alkoxyindole framework: Intramolecular heterocyclization of 3-alkoxyimino-2-arylalkylnitriles mediated by ferric chloride. J. Org. Chem. 2008, 73, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part I. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 2 1987, S1–S19. [Google Scholar] [CrossRef]
- Ketcha, D.M.; Gribble, G.W. A convenient synthesis of 3-acylindoles via a friedel-crafts acylation of 1-(Phenylsulfonyl)indole. A new route to pyridocarbazole-5,11-quinones and ellipticine. J. Org. Chem. 1985, 50, 5451–5457. [Google Scholar] [CrossRef]
- Gribble, G.W.; Barden, T.C.; Johnson, D.A. A directed metalation route to the zwitterionic indole alkaloids. Synthesis of sempervirine. Tetrahedron 1988, 44, 3195–3202. [Google Scholar] [CrossRef]
- Janosik, T.; Lipson, A.C.; Gribble, G.W. An efficient synthesis of 2,3-dicyanoindole. Org. Prep. Proc. Int. 2004, 36, 289–292. [Google Scholar] [CrossRef]
- CrysAlis PRO and CrysAlis RED, version 1.171.33.34d; Oxford Diffraction Ltd.: Abingdon, UK, 2010.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 03, version C.02; Gaussian Inc.: Wallingford, CT, USA, 2004.
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Random, L.; Schleyer, P.R.; Pople, J.A. Ab initio molecular orbital theory. Acc. Chem. Res. 1976, 9, 399–406. [Google Scholar] [CrossRef]
- Georgakopoulous, S.; Grondelle, R.V.; Zwan, G.V.D. Circular dichroism of carotenoides in bacterial light-harvesting complexes: Experimental and modeling. J. Biophys. 2005, 87, 3010–3022. [Google Scholar] [CrossRef] [PubMed]
- Guzin, A. Derivative spectrophotometric determination of caffeine in some beverages. Turk J. Chem. 2002, 26, 295–302. [Google Scholar]
- WaveMetrics, version 4.0; IGOR Pro Inc.: Lake Oswego, OR, USA, 1996.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).