
Drug Repurposing for the Discovery of Potential Inhibitors Targeting DJ-1 (PARK7) Against Parkinson’s Disease

Abstract
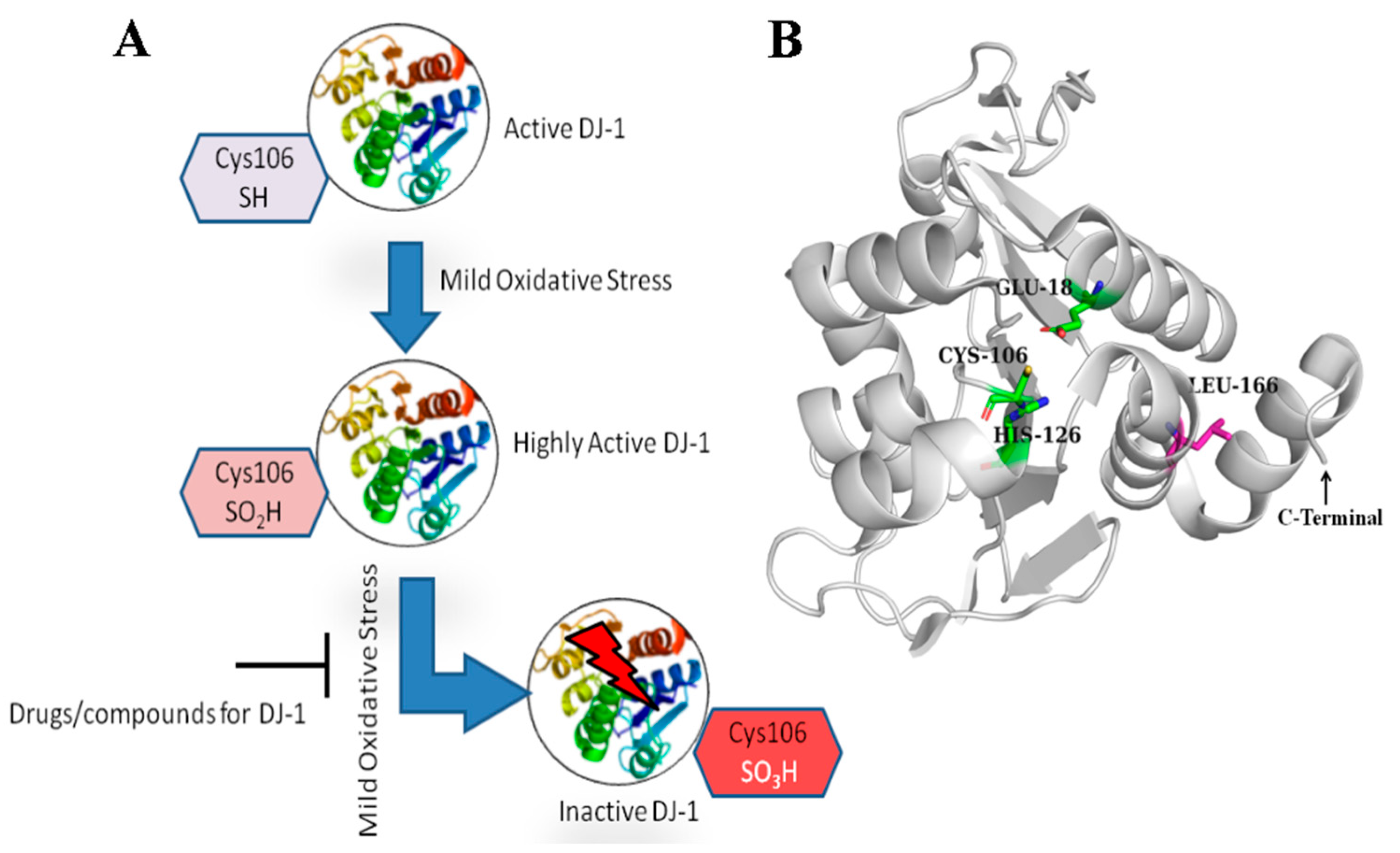
1. Introduction
2. Materials and Methods
2.1. Receptor Structure
2.2. Drug Library Preparation
2.3. Molecular Docking
2.4. Prediction of Activity Spectra for Substances (PASS)
2.5. Molecular Dynamics Simulations (MDSs)
2.6. Binding Energy (MMPBSA)
3. Results
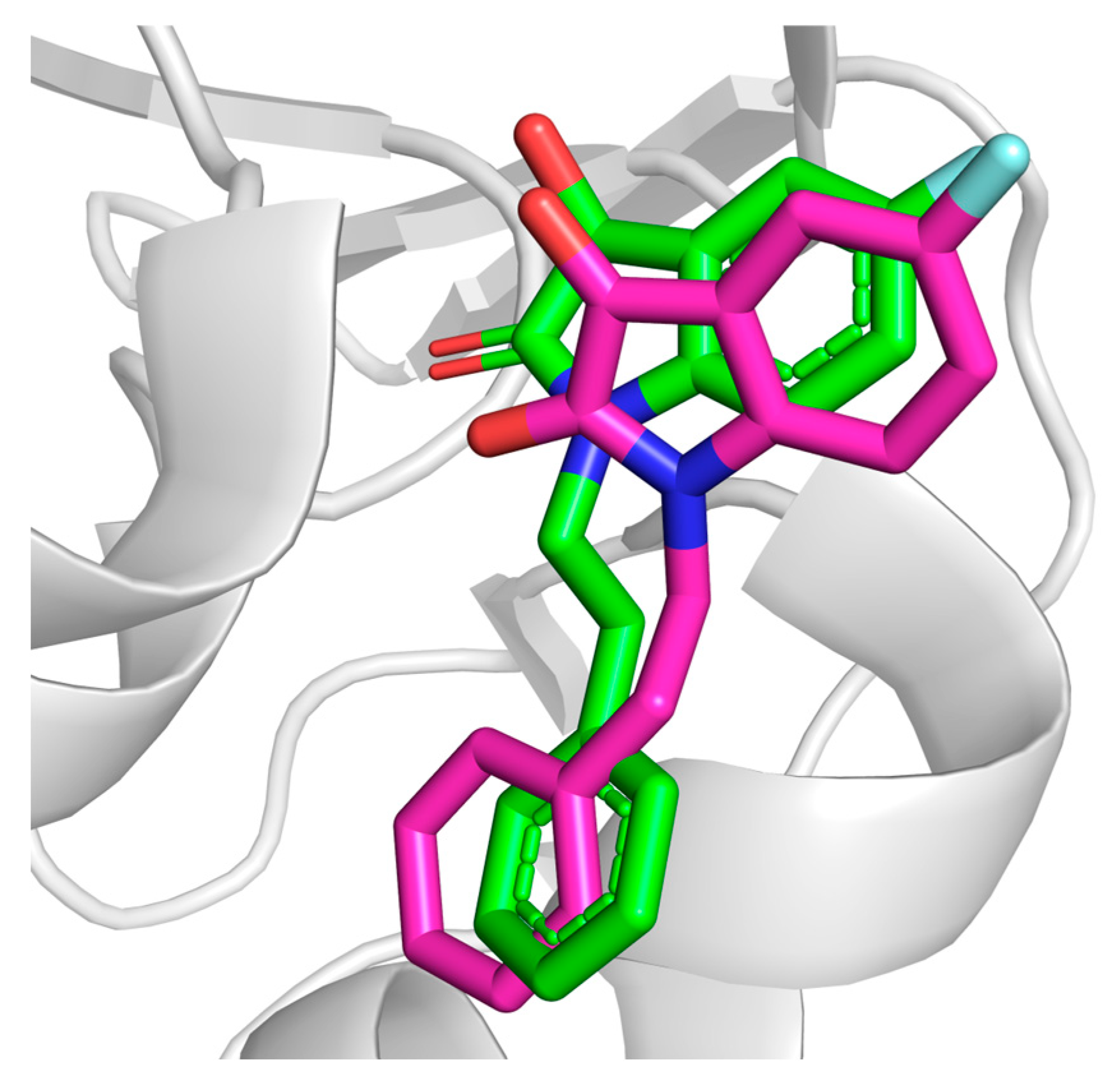
3.1. Docking Protocol Validation
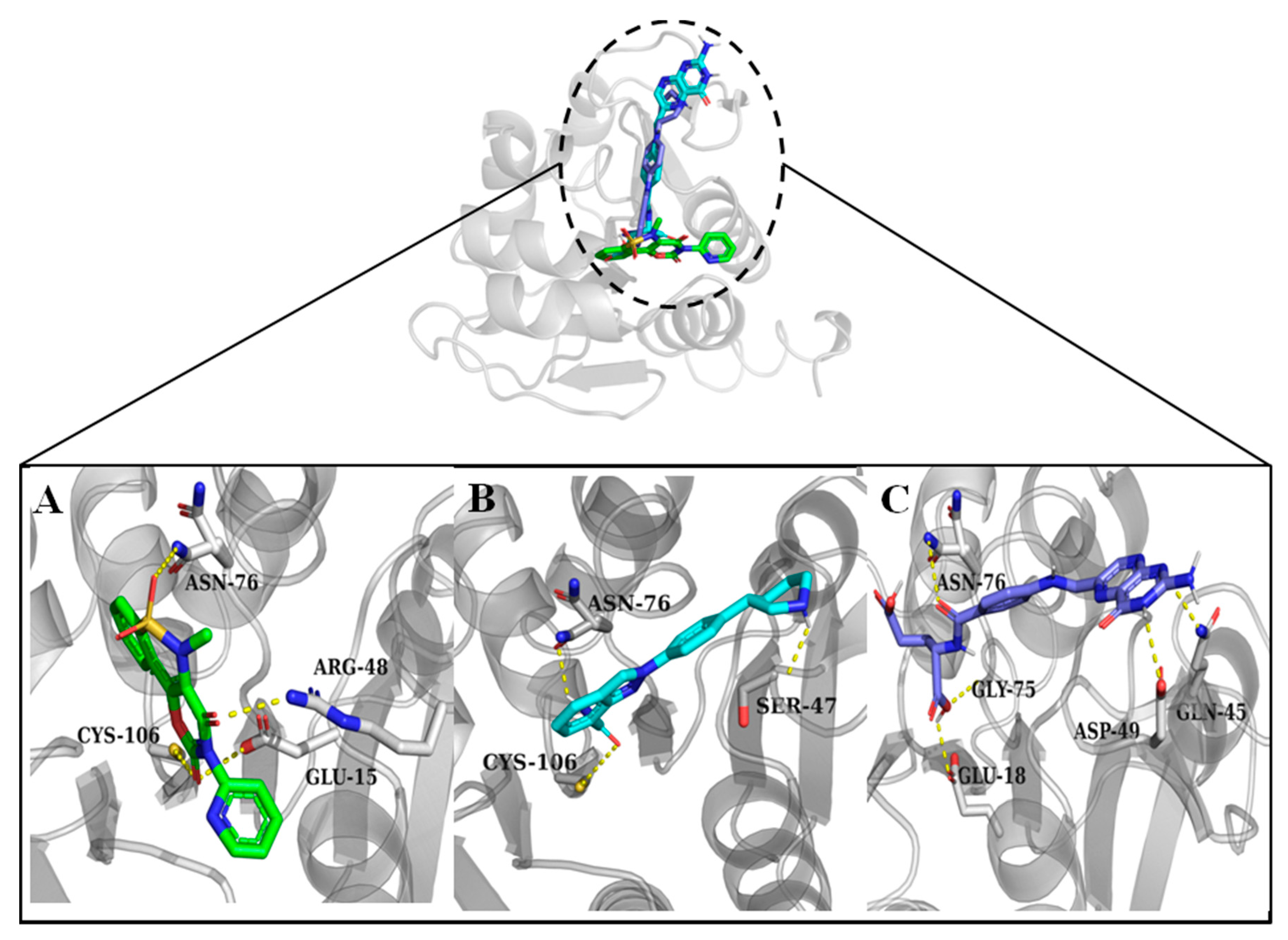
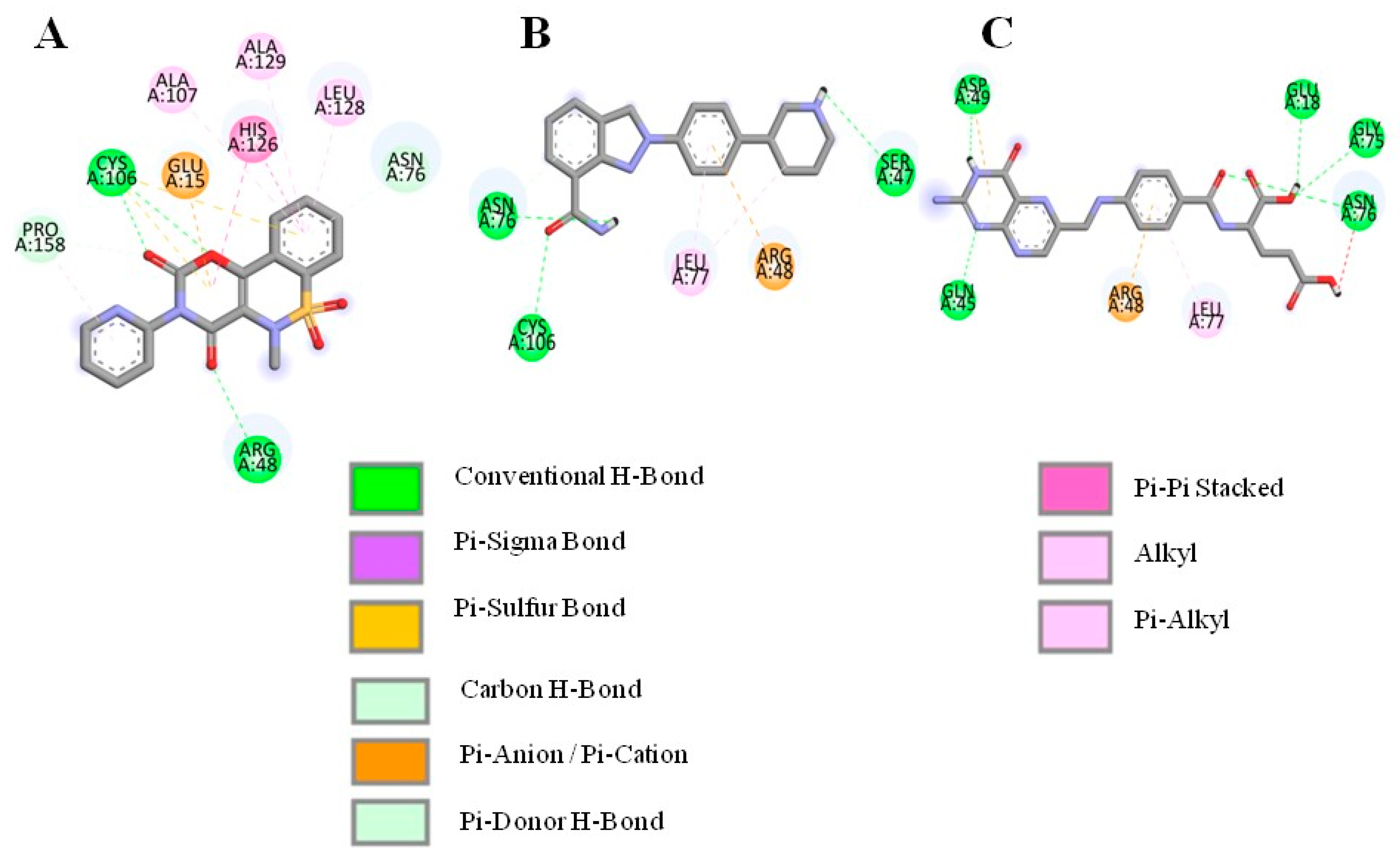
3.2. Molecular Docking and Interaction Analysis
3.3. PASS Online Analysis of Compounds Against Parkinson’s Disease
3.4. Molecular Dynamics Simulations (MDSs)
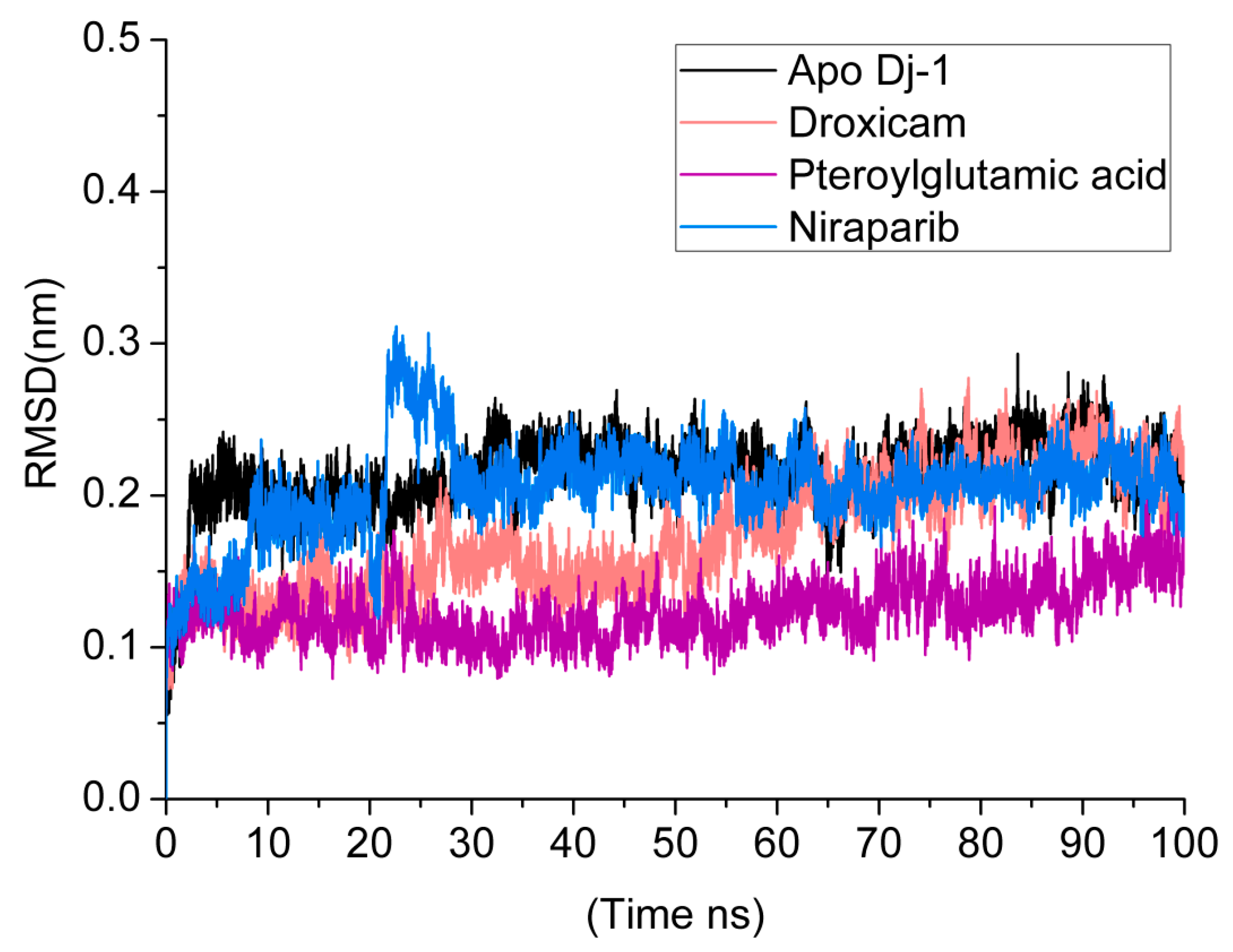
3.5. Root Mean Square Deviations (RMSDs)
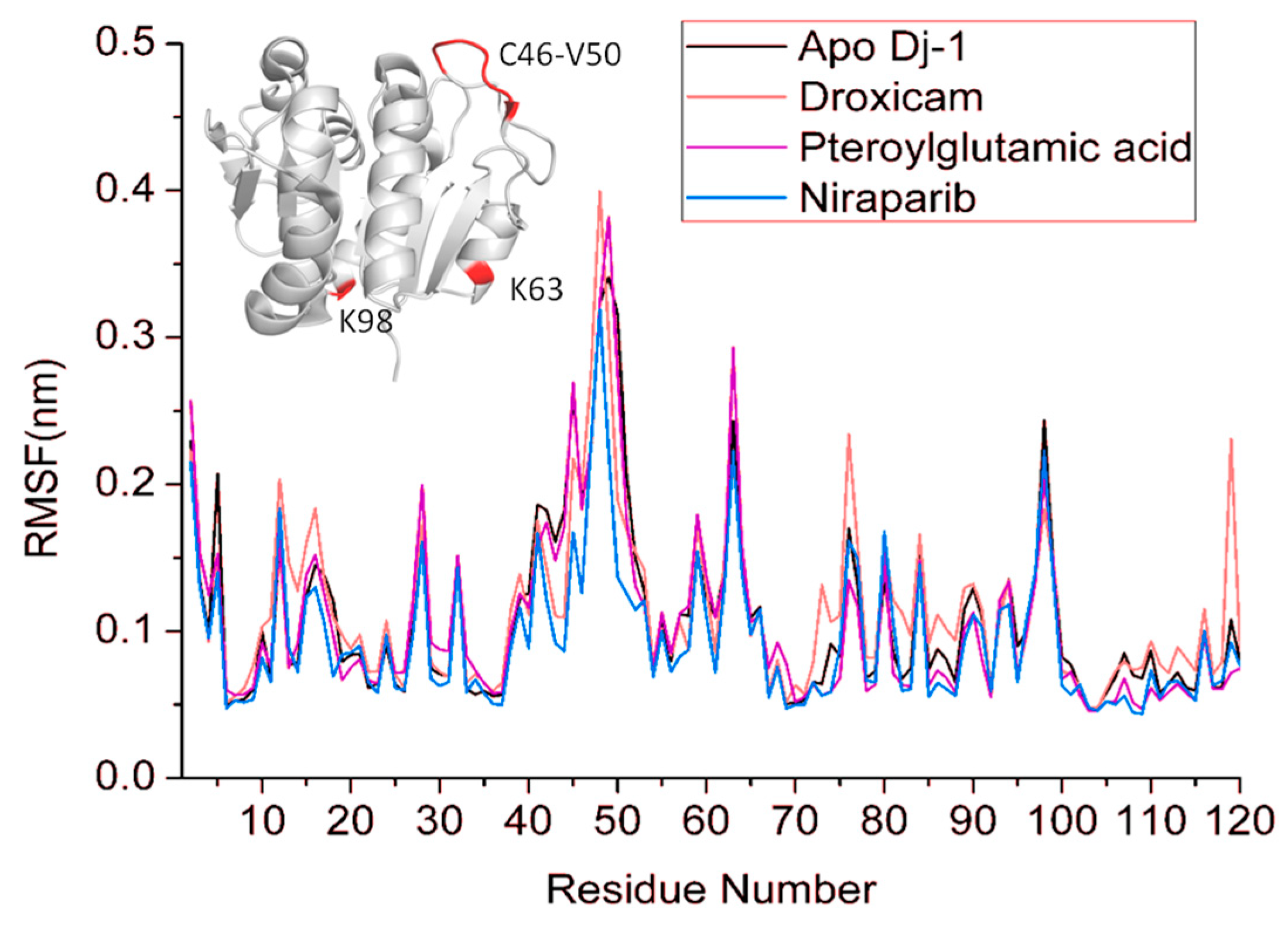
3.6. Root Mean Square Fluctuations (RMSF)
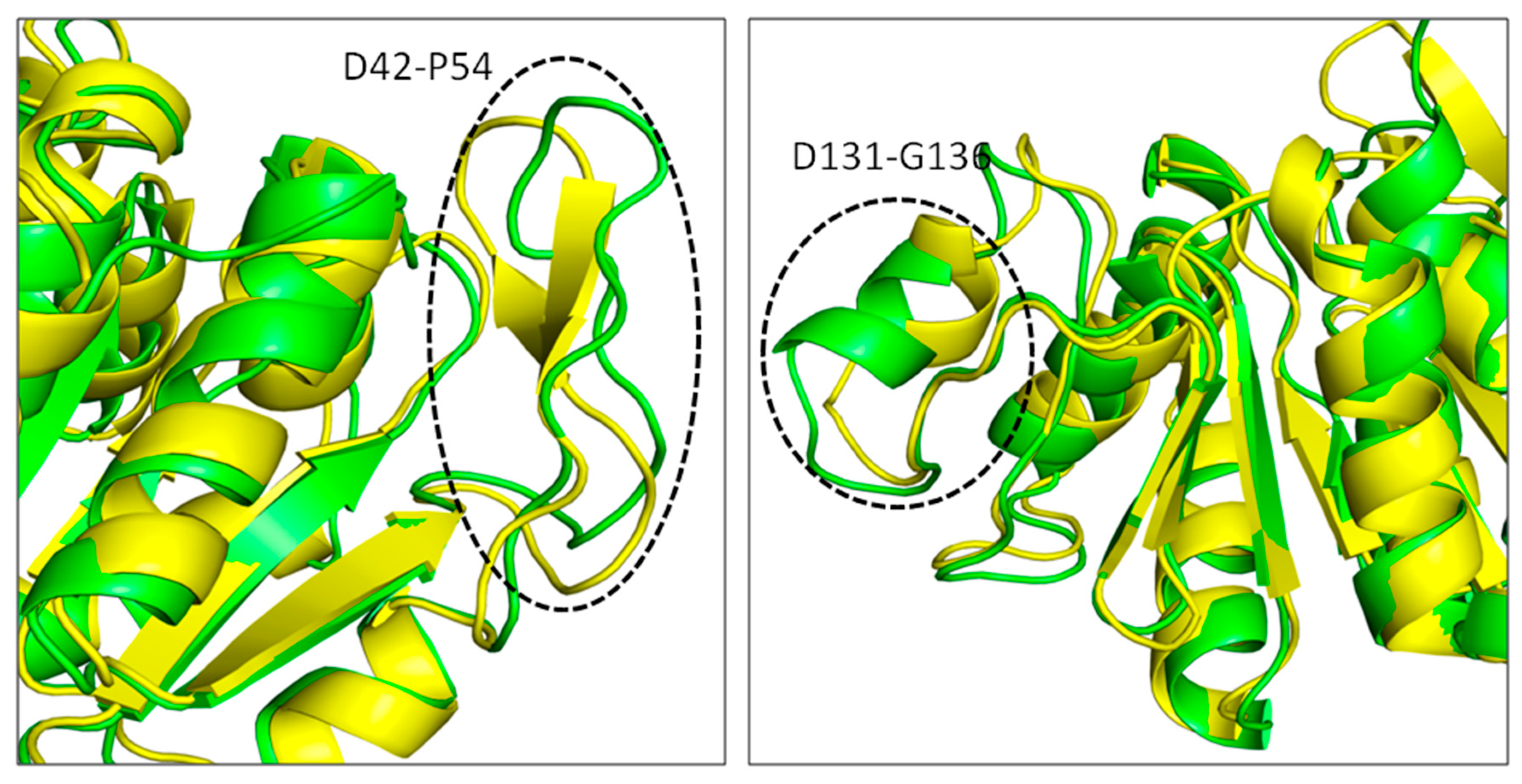
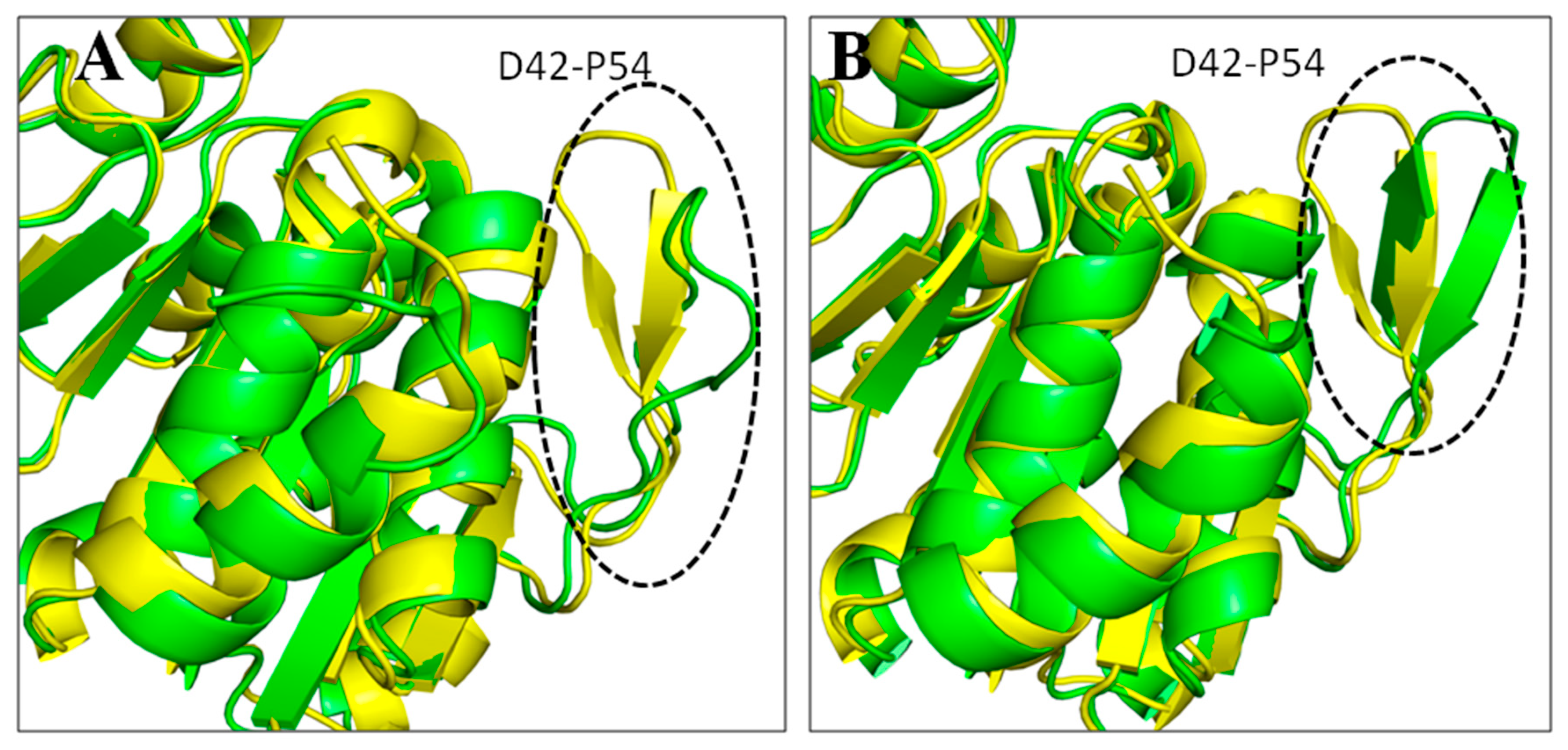
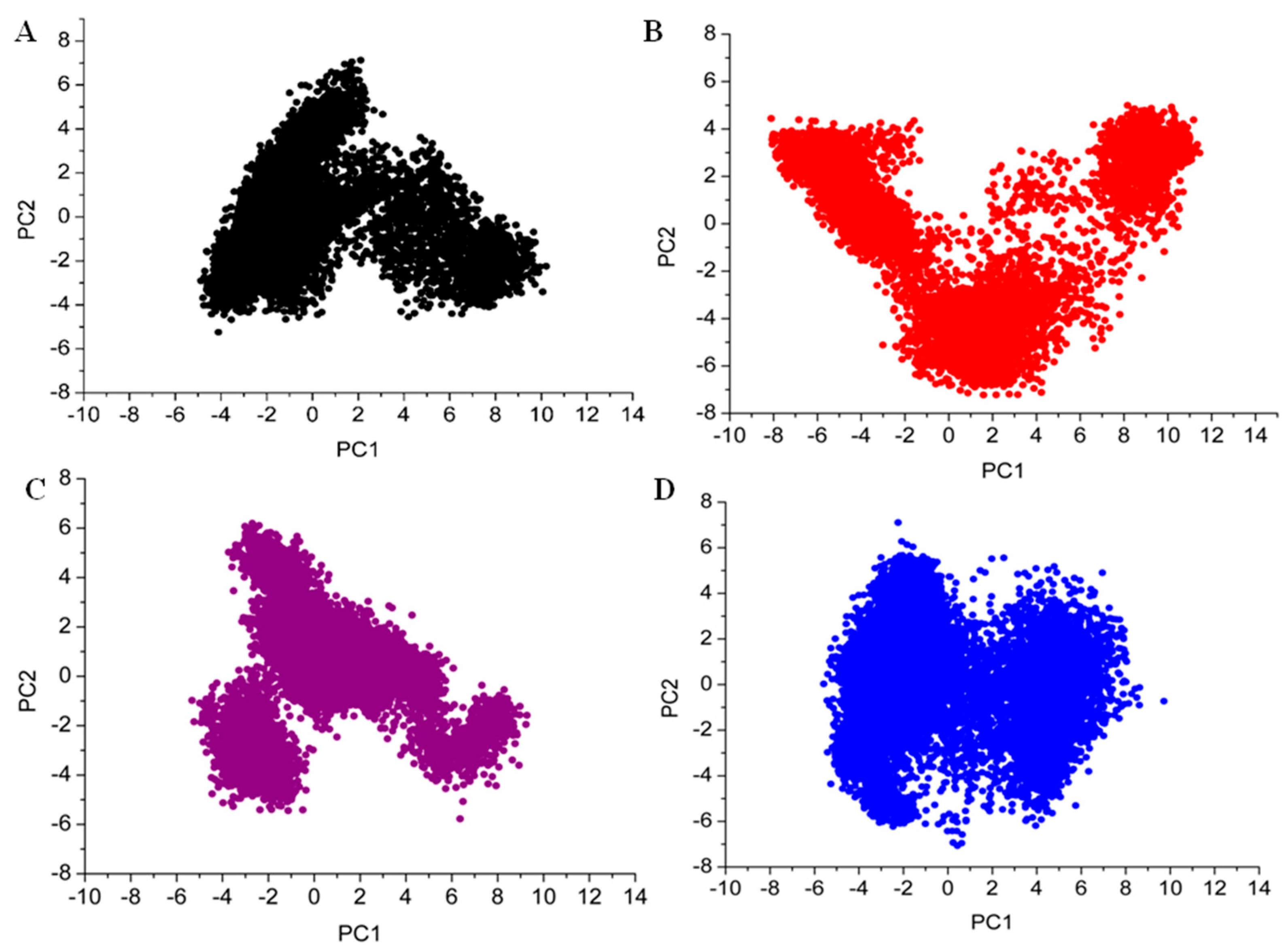
3.7. Principal Component Analysis
3.8. MMPBSA Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Dawson, V.L.; Dawson, T.M. Recent advances in the genetics of Parkinson’s disease. Annu. Rev. Genom. Hum. Genet. 2011, 12, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis; Codon Publications: Brisbane, Australia, 2018; pp. 3–26. [Google Scholar]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Hijioka, M.; Inden, M.; Yanagisawa, D.; Kitamura, Y. DJ-1/PARK7: A new therapeutic target for neurodegenerative disorders. Biol. Pharm. Bull. 2017, 40, 548–552. [Google Scholar] [CrossRef]
- Gao, H.; Yang, W.; Qi, Z.; Lu, L.; Duan, C.; Zhao, C.; Yang, H. DJ-1 protects dopaminergic neurons against rotenone-induced apoptosis by enhancing ERK-dependent mitophagy. J. Mol. Biol. 2012, 423, 232–248. [Google Scholar] [CrossRef]
- Honbou, K.; Suzuki, N.N.; Horiuchi, M.; Niki, T.; Taira, T.; Ariga, H.; Inagaki, F. The crystal structure of DJ-1, a protein related to male fertility and Parkinson’s disease. J. Biol. Chem. 2003, 278, 31380–31384. [Google Scholar] [CrossRef]
- Lee, S.J. Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J. Biol. Chem. 2003, 278, 44552–44559. [Google Scholar] [CrossRef]
- Tao, X.; Tong, L. Crystal structure of human DJ-1, a protein associated with early onset Parkinson’s disease. J. Biol. Chem. 2003, 278, 31372–31379. [Google Scholar] [CrossRef]
- Gautier, V.; Le, H.-T.; Malki, A.; Messaoudi, N.; Caldas, T.; Kthiri, F.; Landoulsi, A.; Richarme, G. YajL, the prokaryotic homolog of the Parkinsonism-associated protein DJ-1, protects cells against protein sulfenylation. J. Mol. Biol. 2012, 421, 662–670. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Brown, K.; Wilkinson, K.D.; Rees, H.D.; Huai, Q.; Ke, H.; Levey, A.I.; Li, L.; Chin, L.-S. Familial Parkinson’s disease-associated L166P mutation disrupts DJ-1 protein folding and function. J. Biol. Chem. 2004, 279, 8506–8515. [Google Scholar] [CrossRef]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s disease: Clinical insights and therapeutic perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Chen, S. DJ-1 in neurodegenerative diseases: Pathogenesis and clinical application. Prog. Neurobiol. 2021, 204, 102114. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Pacheco, A.B.; Hpc, L. Introduction to AutoDock and AutoDock Tools; Louisiana State University: Baton Rouge, LA, USA, 2012. [Google Scholar]
- Ravi, L.; Kannabiran, K. A handbook on protein-ligand docking tool: AutoDock 4. Innovare J. Med. Sci. 2016, 28–33. Available online: https://www.academia.edu/84591451/A_Handbook_on_Protein_Ligand_Docking_Tool_AutoDock_4 (accessed on 18 October 2024).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dong, R.; Huang, R.; Shi, X.; Xu, Z.; Mang, J. Exploration of the mechanism of luteolin against ischemic stroke based on network pharmacology, molecular docking and experimental verification. Bioengineered 2021, 12, 12274–12293. [Google Scholar] [CrossRef]
- Zhu, J.; Wei, J.; Lin, Y.; Tang, Y.; Su, Z.; Li, L.; Liu, B.; Cai, X. Inhibition of IL-17 signaling in macrophages underlies the anti-arthritic effects of halofuginone hydrobromide: Network pharmacology, molecular docking, and experimental validation. BMC Complement. Med. Ther. 2024, 24, 105. [Google Scholar] [CrossRef]
- Mellini, M.; Di Muzio, E.; D’Angelo, F.; Baldelli, V.; Ferrillo, S.; Visca, P.; Leoni, L.; Polticelli, F.; Rampioni, G. In silico selection and experimental validation of FDA-approved drugs as anti-quorum sensing agents. Front. Microbiol. 2019, 10, 2355. [Google Scholar] [CrossRef]
- Muneer, I.; ul Qamar, M.T.; Tusleem, K.; Rauf, S.A.; Hussain, H.M.; Siddiqi, A.R. Discovery of selective inhibitors for cyclic AMP response element-binding protein: A combined ligand and structure-based resources pipeline. Anti-Cancer Drugs 2019, 30, 363–373. [Google Scholar] [CrossRef]
- Lagunin, A.; Filimonov, D.; Poroikov, V. Multi-targeted natural products evaluation based on biological activity prediction with PASS. Curr. Pharm. Des. 2010, 16, 1703–1717. [Google Scholar] [CrossRef] [PubMed]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Vondrumen, R.v.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M. Gromacs-a parallel computer for molecular-dynamics simulations. In Proceedings of the 4th international conference on computational physics (PC 92), Prague, Czech Republic, 24–28 August 1992; pp. 252–256. [Google Scholar]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- AlRawashdeh, S.; Barakat, K.H. Applications of molecular dynamics simulations in drug discovery. Comput. Drug Discov. Des. 2023, 2714, 127–141. [Google Scholar]
- Bhat, S.S.; Sindhu, R.; Prasad, S.K. A Bioinformatics Approach Towards Plant-Based Anticancer Drug Discovery. In Computational Approaches in Biotechnology and Bioinformatics; CRC Press: Boca Raton, FL, USA, 2024; pp. 35–60. [Google Scholar]
- Wang, C.; Greene, D.A.; Xiao, L.; Qi, R.; Luo, R. Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef]
- Tashiro, S.; Caaveiro, J.M.; Nakakido, M.; Tanabe, A.; Nagatoishi, S.; Tamura, Y.; Matsuda, N.; Liu, D.; Hoang, Q.Q.; Tsumoto, K.J.A. Discovery and optimization of inhibitors of the Parkinson’s disease associated protein DJ-1. ACS Chem. Biol. 2018, 13, 2783–2793. [Google Scholar] [CrossRef]
- Rungrotmongkol, T.; Nunthaboot, N.; Malaisree, M.; Kaiyawet, N.; Yotmanee, P.; Meeprasert, A.; Hannongbua, S. Molecular insight into the specific binding of ADP-ribose to the nsP3 macro domains of chikungunya and Venezuelan equine encephalitis viruses: Molecular dynamics simulations and free energy calculations. J. Mol. Graph. Model. 2010, 29, 347–353. [Google Scholar] [CrossRef]
- Adelakun, N.; Obaseki, I.; Adeniyi, A.; Fapohunda, O.; Obaseki, E.; Omotuyi, O. Discovery of new promising USP14 inhibitors: Computational evaluation of the thumb-palm pocket. J. Biomol. Struct. Dyn. 2022, 40, 3060–3070. [Google Scholar] [CrossRef]
- Yau, M.Q.; Emtage, A.L.; Chan, N.J.; Doughty, S.W.; Loo, J.S. Evaluating the performance of MM/PBSA for binding affinity prediction using class A GPCR crystal structures. J. Comput.-Aided Mol. Des. 2019, 33, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bao, G.; Liu, D.; Yang, Y.; Li, X.; Cai, G.; Liu, Y.; Wu, Y. The association between folate and Alzheimer’s disease: A systematic review and meta-analysis. Front. Neurosci. 2021, 15, 661198. [Google Scholar] [CrossRef]
- Ma, F.; Wu, T.; Zhao, J.; Ji, L.; Song, A.; Zhang, M.; Huang, G. Plasma homocysteine and serum folate and vitamin B12 levels in mild cognitive impairment and Alzheimer’s disease: A case-control study. Nutrients 2017, 9, 725. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhou, X.; Li, Q.; Zhao, J.; Song, A.; An, P.; Du, Y.; Xu, W.; Huang, G. Effects of folic acid and vitamin B12, alone and in combination on cognitive function and inflammatory factors in the elderly with mild cognitive impairment: A single-blind experimental design. Curr. Alzheimer Res. 2019, 16, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Drug Bank Ids | Binding Score (Kcal/mol) | Hydrogen Bond Interactions | Other Interactions |
|---|---|---|---|---|
| Droxicam | DB09215 | −7.6 | Cys106, Arg48, Asn76, Glu15 | His126, Pro158, Leu128, Ala129, Ala107 |
| Niraparib | DB11793 | −7.6 | Ser47, Asn76, Cys106 | Leu77, Arg48 |
| Pteroylglutamic Acid | DB00158 | −7.5 | Gln45, Asp49, Glu18, Gly75, Asn76 | Arg48, Leu77 |
| Drug Name | Chemical Structure | MolWT (g/mol) | LogP | H-Bond Donors | H-Bond Acceptors |
|---|---|---|---|---|---|
| Reference |  | 269.27 | 3 | 0 | 3 |
| Droxicam | 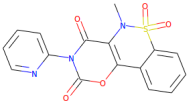 | 357.3 | 0.99 | 0 | 7 |
| Pteroylglutamic Acid | 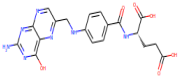 | 441.4 | 0.368 | 6 | 10 |
| Niraparib | 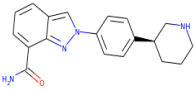 | 320.4 | 2.591 | 3 | 3 |
| Compound Name | Activity | Pa | Pi |
|---|---|---|---|
| Niraparib | Poly(ADP-ribose) polymerase inhibitor | 0.752 | 0.003 |
| Nootropic | 0.755 | 0.026 | |
| MAP kinase kinase 4 inhibitor | 0.390 | 0.006 | |
| Neurodegenerative diseases treatment | 0.381 | 0.081 | |
| Antiinflammatory, ophthalmic | |||
| Antineoplastic enhancer | 0.459 | 0.013 | |
| Anticonvulsant | 0.376 | 0.075 | |
| Antineurogenic pain | 0.238 | 0.144 | |
| Anti-neurotic | 0.303 | 0.244 | |
| Lysyl oxidase inhibitor | 0.226 | 0.204 | |
| Pteroylglutamic Acid | Gamma-glutamyl hydrolase inhibitor | 0.632 | 0.000 |
| Immunostimulant | 0.576 | 0.026 | |
| Autoimmune disorders treatment | 0.318 | 0.089 | |
| MAP3K5 inhibitor | 0.371 | 0.042 | |
| Thiol oxidase inhibitor | 0.575 | 0.014 | |
| Droxicam | Antiparkinsonian | 0.372 | 0.036 |
| Neurodegenerative diseases treatment | 0.490 | 0.034 | |
| Anxiolytic | 0.370 | 0.047 | |
| Nootropic | 0.497 | 0.131 | |
| Anti-inflammatory | 0.472 | 0.065 | |
| Anti-neurotic | 0.460 | 0.1294 | |
| Non-steroidal anti-inflammatory agent | 0.394 | 0.106 | |
| S)-6-hydroxynicotine oxidase inhibitor | 0.345 | 0.078 | |
| Chloride peroxidase inhibitor | 0.365 | 0.127 | |
| Hydroxylamine oxidase inhibitor | 0.439 | 0.119 |
| Energy Terms (Kcal/mol) | Droxicam | Pteroylglutamic Acid | Niraparib |
|---|---|---|---|
| ΔEvdw | −5.20 | −18.79 | −20.18 |
| ΔEelec | −3.74 | −5.06 | −52.51 |
| ΔESURF | −0.80 | −2.60 | −3.64 |
| ΔEGB | 6.57 | 15.04 | 62.84 |
| ΔGSOLV | 5.78 | 12.45 | 59.20 |
| ΔGGAS | −8.66 | −23.86 | −72.70 |
| ΔTOTAL | −2.88 | −11.41 | −13.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldakhil, T.; Altharawi, A. Drug Repurposing for the Discovery of Potential Inhibitors Targeting DJ-1 (PARK7) Against Parkinson’s Disease. Crystals 2025, 15, 239. https://doi.org/10.3390/cryst15030239
Aldakhil T, Altharawi A. Drug Repurposing for the Discovery of Potential Inhibitors Targeting DJ-1 (PARK7) Against Parkinson’s Disease. Crystals. 2025; 15(3):239. https://doi.org/10.3390/cryst15030239
Chicago/Turabian StyleAldakhil, Taibah, and Ali Altharawi. 2025. "Drug Repurposing for the Discovery of Potential Inhibitors Targeting DJ-1 (PARK7) Against Parkinson’s Disease" Crystals 15, no. 3: 239. https://doi.org/10.3390/cryst15030239
APA StyleAldakhil, T., & Altharawi, A. (2025). Drug Repurposing for the Discovery of Potential Inhibitors Targeting DJ-1 (PARK7) Against Parkinson’s Disease. Crystals, 15(3), 239. https://doi.org/10.3390/cryst15030239