
LaZn1−xBi2 as a Candidate for Dirac Nodal-Line Intermetallic Systems



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
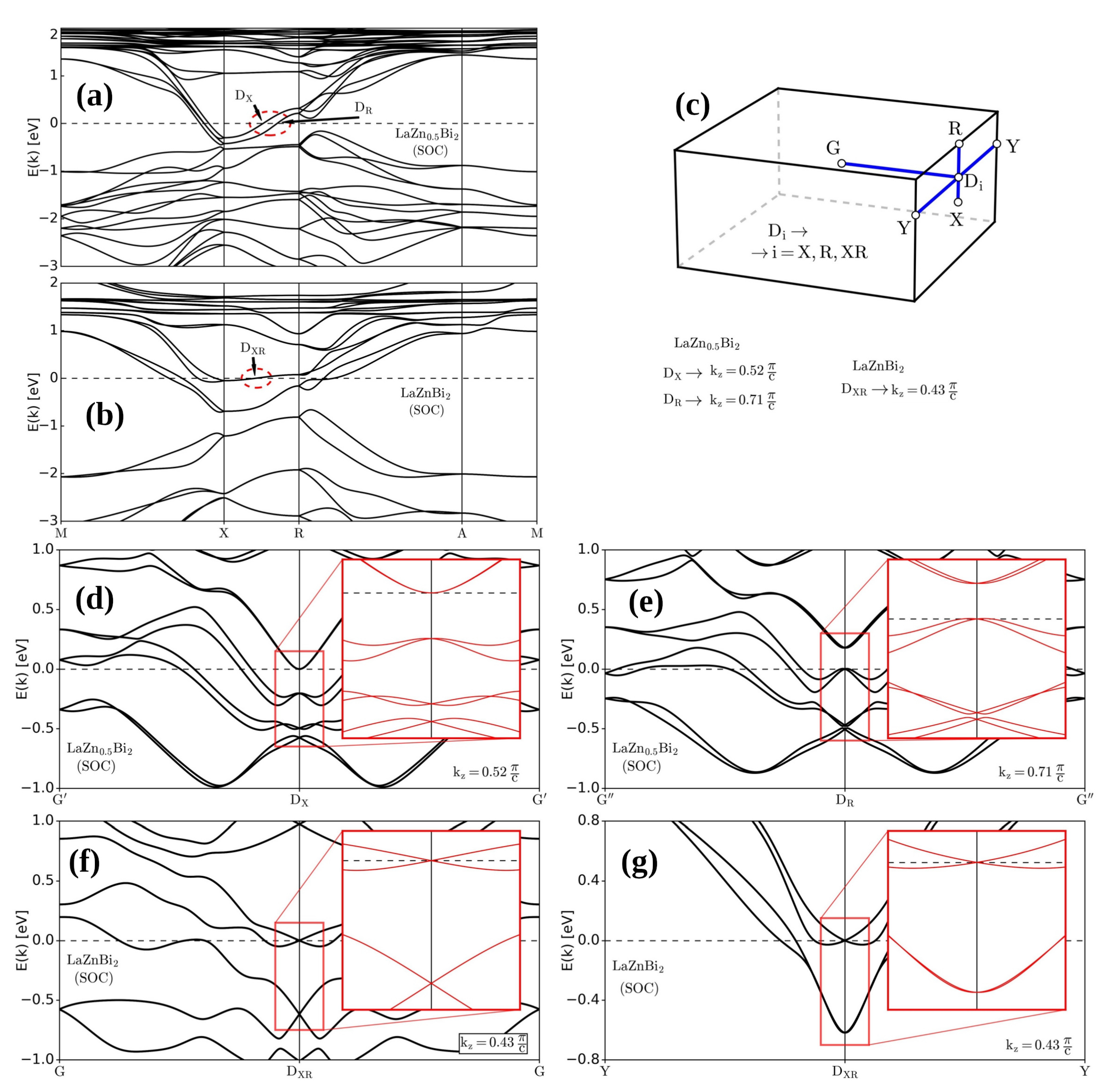
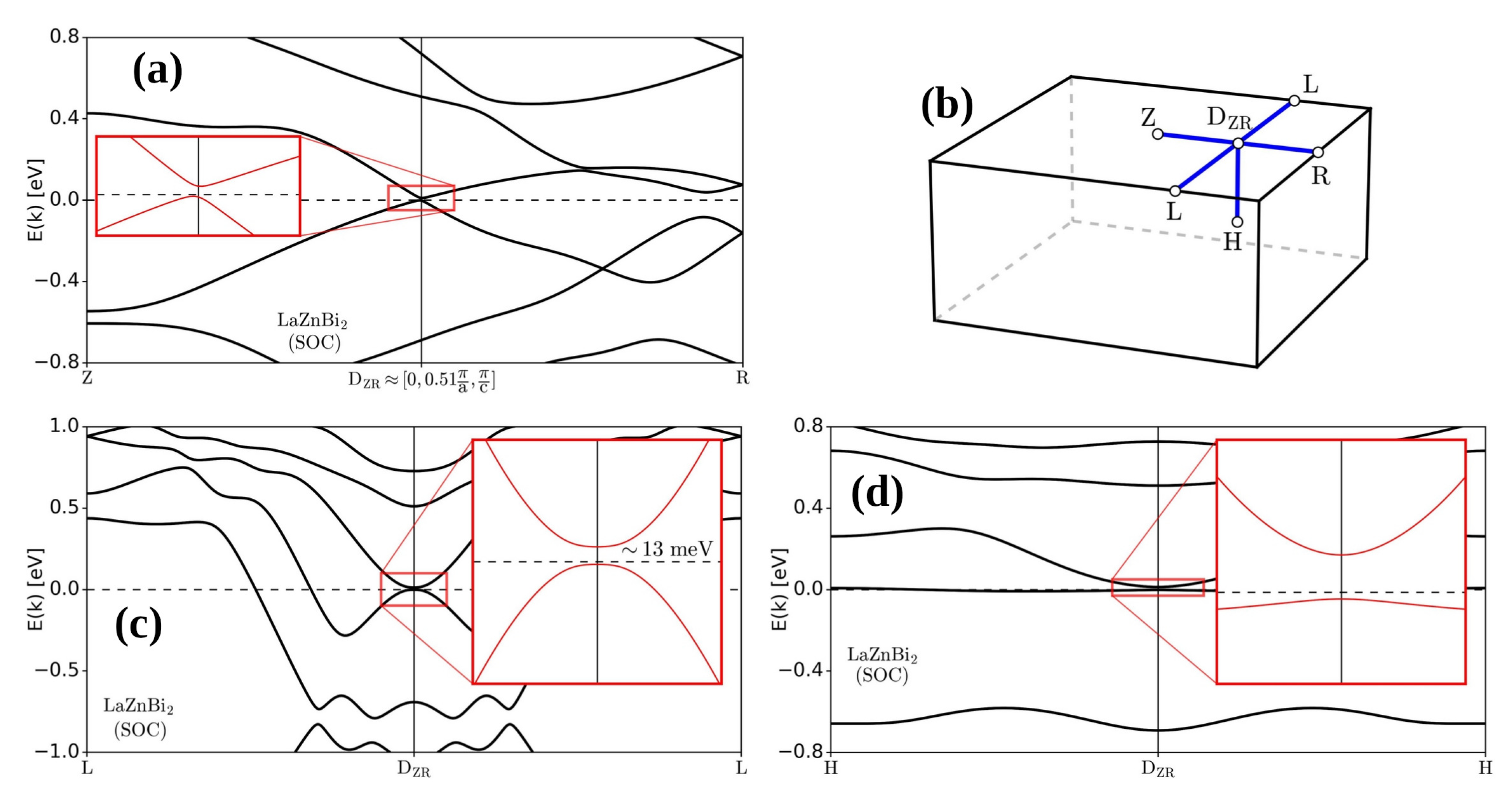
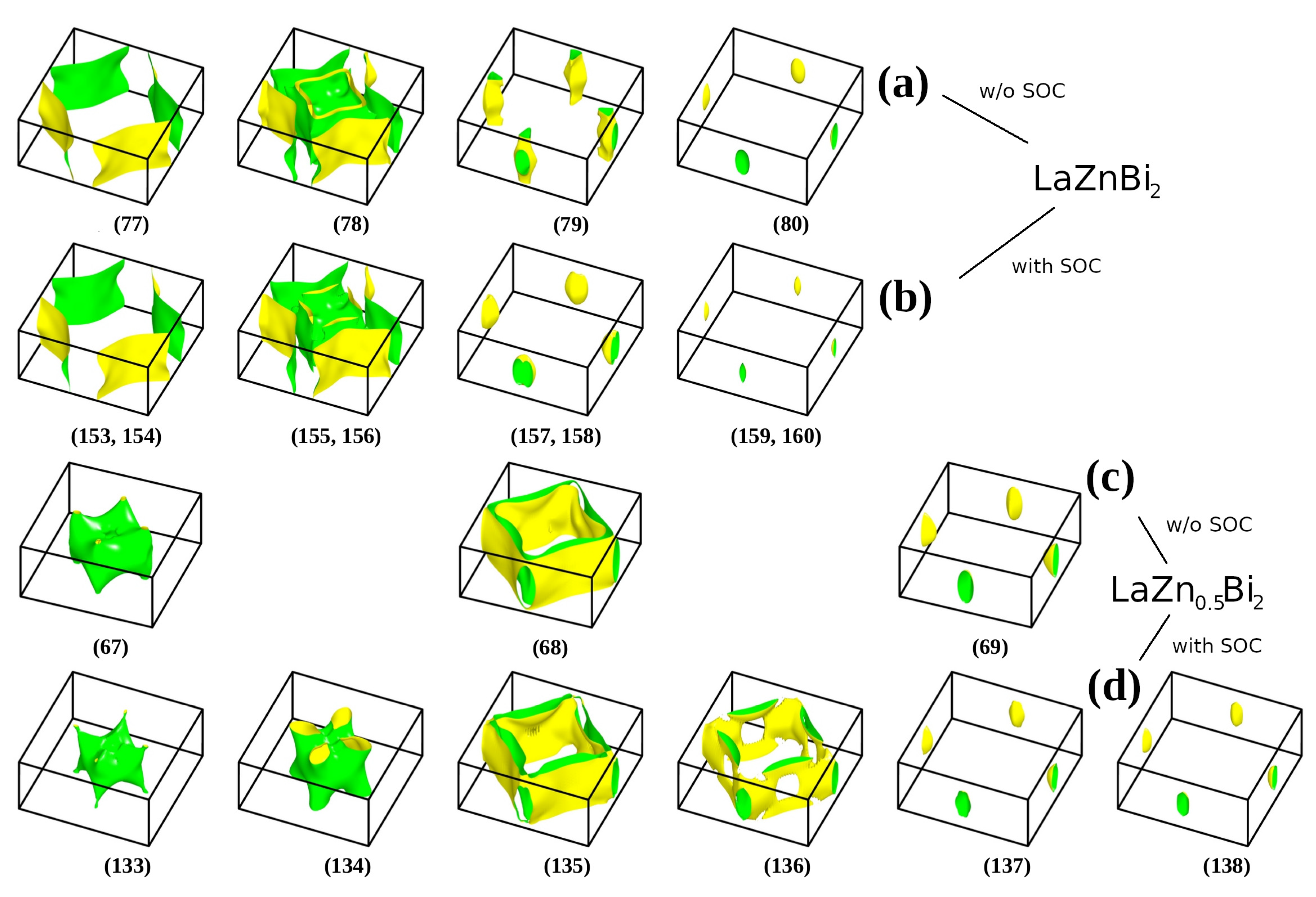
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Armitage, N.P.; Mele, E.J.; Vishwanath, A. Weyl and Dirac semimetals in three-dimensional solids. Rev. Mod. Phys. 2018, 90, 015001. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Matsuishi, S.; Hirano, M.; Tochibana, M.; Muromachi, E.T.; Kawaji, H.; Hosono, H. Coexistence of Light and Heavy Carriers Associated with Superconductivity and Antiferromagnetism in CeNi0.8Bi2 with a Bi Square Net. Phys. Rev. Lett. 2011, 106, 057002. [Google Scholar] [CrossRef]
- Han, F.; Malliakas, C.D.; Stoumpos, C.C.; Sturza, M.; Claus, H.; Chung, D.Y.; Kanatzidis, M.G. Superconductivity and strong intrinsic defects in LaPd1−xBi2. Phys. Rev. B 2013, 88, 144511. [Google Scholar] [CrossRef]
- Muro, Y.; Takeda, N.; Ishikawa, M. Magnetic and transport properties of dense Kondo systems, CeTSb2 (T = Ni, Cu, Pd and Ag). J. Alloys Compd. 1997, 257, 23–29. [Google Scholar] [CrossRef]
- Koyama, T.; Matsumoto, M.; Wada, S.; Muro, Y.; Ishikawa, M. Combination of Dense Kondo Effect and RKKY Interactions in CeCuSb2, Investigated by NMR. J. Phys. Soc. Jpn. 2001, 70, 3667–3672. [Google Scholar] [CrossRef]
- Inada, Y.; Tamizabel, A.; Sawai, Y.; Ikeda, S.; Shishido, H.; Okubo, T.; Yamada, M.; Onuki, Y.; Ebihara, T. Quasi-2D Fermi Surfaces of the Magnetic Compound CeAgSb2. Acta Phys. Pol. B 2003, 34, 1141–1144. Available online: http://www.actaphys.uj.edu.pl/R/34/2/1141/pdf (accessed on 31 January 2024).
- Zou, Y.; Logg, P.; Barber, M.; Feng, Z.; Ebihara, T.; Grosche, F.M. Thermodynamics of field-tuned quantum critical point in CeAgSb2. Phys. Status Solidi B 2013, 250, 529–532. [Google Scholar] [CrossRef]
- Chen, R.Y.; Liu, J.L.; Shi, L.Y.; Wang, C.N.; Zhang, S.J.; Wang, N.L. Possible orbital crossover in the ferromagnetic Kondo lattice compound CeAgSb2. Phys. Rev. B 2019, 99, 205107. [Google Scholar] [CrossRef]
- Adriano, C.; Rosa, P.F.S.; Jesus, C.B.R.; Mardegan, J.R.L.; Garitezi, T.M.; Grant, T.; Fisk, Z.; Garcia, D.J.; Reyes, A.P.; Kuhns, P.L.; et al. Physical properties and magnetic structure of the intermetallic CeCuBi2 compound. Phys. Rev. B 2014, 90, 235120. [Google Scholar] [CrossRef]
- Masuda, H.; Sakai, H.; Tokunaga, M.; Yamasaki, Y.; Miyake, A.; Shiogai, J.; Nakamura, S.; Awaji, S. Quantum Hall effect in a bulk antiferromagnet EuMnBi2 with magnetically confined two-dimensional Dirac fermions. Sci. Adv. 2016, 2, e1501117. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Graf, D.; Petrovic, C. Quasi-two-dimensional Dirac fermions and quantum magnetoresistance in LaAgBi2. Phys. Rev. B 2013, 87, 235101. [Google Scholar] [CrossRef]
- Shi, X.; Richard, P.; Wang, K.; Liu, M.; Matt, C.E.; Xu, N.; Dhaka, R.S.; Ristic, Z.; Qian, T.; Yang, Y.-F.; et al. Observation of Dirac-like band dispersion in LaAgSb2. Phys. Rev. B 2016, 93, 081105. [Google Scholar] [CrossRef]
- Wang, K.; Petrovic, C. Multiband effects and possible Dirac states in LaAgSb2. Phys. Rev. B 2012, 86, 155213. [Google Scholar] [CrossRef]
- Borisenko, S.; Evtushinsky, D.; Gibson, Q.; Yaresko, A.; Koepernik, K.; Kim, T.; van den Brink, M.A.J.; Hoesch, M.; Fedorov, A.; Haubold, E.; et al. Time-reversal symmetry breaking type-II Weyl state in YbMnBi2. Nat. Commun. 2019, 10, 3424. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Cheng, B.; Yaresko, A.; Gibson, Q.D.; Cava, R.J.; Armitage, N.P. Optical investigation of the strong spin-orbit-coupled magnetic semimetal YbMnBi2. Phys. Rev. B 2017, 96, 075151. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Xu, S.; Sun, L.-L.; Xia, T.-L. Quantum oscillations and coherent interlayer transport in a new topological Dirac semimetal candidate YbMnSb2. Phys. Rev. Mater. 2018, 2, 021201. [Google Scholar] [CrossRef]
- Ray, S.J.; Alff, L. Superconductivity and Dirac fermions in 112-phase pnictides. Phys. Status Solidi B 2017, 254, 1600163. [Google Scholar] [CrossRef]
- Klemenz, S.; Lei, S.; Schoop, L.M. Topological Semimetals in Square-Net Materials. Annu. Rev. Mater. Res. 2019, 49, 185–206. [Google Scholar] [CrossRef]
- Tamura, S.; Katayama, N.; Yamada, Y.; Sugiyama, Y.; Sugawara, K.; Sawa, H. Various Arsenic Network Structures in 112-Type Ca1–xLaxFe1–yPdyAs2 Revealed by Synchrotron X-ray Diffraction Experiments. Inorg. Chem. 2017, 56, 3030–3035. [Google Scholar] [CrossRef] [PubMed]
- Klemenz, S.; Hay, A.K.; Teicher, S.M.L.; Topp, A.; Cano, J.; Schoop, L.M. The Role of Delocalized Chemical Bonding in Square-Net-Based Topological Semimetals. J. Am. Chem. Soc. 2020, 142, 6350–6359. [Google Scholar] [CrossRef] [PubMed]
- Zelinska, O.Y.; Mar, A. Structure and electrical resistivity of rare-earth zinc bismuthides REZn1−xBi2 (RE = La, Ce, Pr). J. Alloys Compd. 2008, 451, 606–609. [Google Scholar] [CrossRef]
- Lakshmi, K.V.; Menon, L.; Nigam, A.K.; Das, A.; Malik, S.K. Magneto-resistance studies on RTSb2 compounds (R = La, Ce and T = Ni, Cu). Phys. B Condens. Matter 1996, 223–224, 289–291. [Google Scholar] [CrossRef]
- Kodama, K.; Wakimoto, S.; Igawa, N.; Shamoto, S.; Mizoguchi, H.; Hosono, H. Crystal and magnetic structures of the superconductor CeNi0.8Bi2. Phys. Rev. B 2011, 83, 214512. [Google Scholar] [CrossRef]
- Retzlaff, R.; Buckow, A.; Komissinskiy, P.; Ray, S.; Schmidt, S.; Mühlig, H.; Schmidl, F.; Seidel, P.; Kurian, J.; Alff, L. Superconductivity and role of pnictogen and Fe substitution in 112-LaPdxPn2 (Pn = Sb, Bi). Phys. Rev. B 2015, 91, 104519. [Google Scholar] [CrossRef]
- Ramachandran, K.K.; Genet, C.; Mar, A. Quaternary rare-earth arsenides REAg1−xZnyAs2 (RE = La–Nd, Sm, Gd–Dy) with tetragonal SrZnBi2- and HfCuSi2-type structures. J. Solid State Chem. 2015, 231, 204–211. [Google Scholar] [CrossRef]
- Malick, S.; Ghosh, A.; Barman, C.K.; Alam, A.; Hossain, Z.; Mandal, P.; Nayak, J. Weak antilocalization effect and triply degenerate state in Cu-doped CaAuAs. Phys. Rev. B 2022, 105, 165105. [Google Scholar] [CrossRef]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743–1757. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Eschrig, H.; Richter, M.; Opahle, I. Relativistic solid state calculations. In Relativistic Electronic Structure Theory, Part 2. Applications (Theoretical and Computational Chemistry); Schwerdtfeger, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 14, pp. 723–776. [Google Scholar]
- Schoop, L.M.; Ali, M.N.; Straßer, C.; Topp, A.; Varykhalov, A.; Marchenko, D.; Duppel, V.; Parkin, S.S.P.; Lotsch, B.V.; Ast, C.R. Dirac cone protected by non-symmorphic symmetry and three-dimensional Dirac line node in ZrSiS. Nat. Commun. 2016, 7, 11696. [Google Scholar] [CrossRef]
- Brylak, M.; Möller, M.H.; Jeitschko, W. Ternary Arsenides ACuAs2 and Ternary Antimonides AAgSb2 (A = Rare-Earth Elements and Uranium) with HfCuSi2-Type Structure. J. Solid State Chem. 1995, 115, 305–308. [Google Scholar] [CrossRef]
- Stoyko, S.S.; Mar, A. Ternary rare-earth zinc arsenides REZn1–xAs2 (RE = La–Nd, Sm). J. Solid State Chem. 2011, 184, 2360–2367. [Google Scholar] [CrossRef]
- Salamakha, L.P.; Mudryi, S.I. Crystal structure of the RZn1−xSb2 compounds (R = La, Ce). J. Alloys Compd. 2003, 359, 139–142. [Google Scholar] [CrossRef]
- Zelinska, O.Y.; Mar, A. Structure and physical properties of rare-earth zinc antimonides REZn1–xSb2 (RE = La, Ce, Pr, Nd, Sm, Gd, Tb). J. Solid State Chem. 2006, 179, 3776–3783. [Google Scholar] [CrossRef]
- Lin, X.; Stoyko, S.S.; Mar, A. Ternary rare-earth zinc arsenides REZn2As3 (RE = La–Pr) containing defect fluorite-type slabs. J. Solid State Chem. 2013, 199, 189–195. [Google Scholar] [CrossRef]
- Seibel, E.M.; Schoop, L.M.; Xie, W.; Gibson, Q.D.; Webb, J.B.; Fuccillo, M.K.; Krizan, J.W.; Cava, R.J. Gold-Gold Bonding: The Key to Stabilizing the 19-Electron Ternary Phases LnAuSb (Ln = La–Nd and Sm). J. Am. Chem. Soc. 2015, 137, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Ruszała, P.; Winiarski, M.J.; Samsel-Czekała, M. Dirac-like band structure of LaTESb2 (TE = Ni, Cu, and Pd) superconductors by DFT calculations. Comput. Mater. Sci. 2018, 154, 106–110. [Google Scholar] [CrossRef]
- Ren, W.; Wang, A.; Graf, D.; Liu, Y.; Zhang, Z.; Yin, W.-G.; Petrovic, C. Absence of Dirac states in BaZnBi2 induced by spin-orbit coupling. Phys. Rev. B 2018, 97, 035147. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Guo, P.-J.; Yu, Q.-H.; Xu, S.; Liu, K.; Xia, T.-L. Magneto-transport and electronic structures of BaZnBi2. New J. Phys. 2017, 19, 123044. [Google Scholar] [CrossRef]
- Ruszała, P.; Winiarski, M.J.; Samsel-Czekała, M. Dirac-Like Electronic-Band Dispersion of LaSb2 Superconductor and its Counterpart LaAgSb2. Acta Phys. Pol. A 2020, 138, 748–751. [Google Scholar] [CrossRef]
- Chamorro, J.R.; Topp, A.; Fang, Y.; Winiarski, M.J.; Ast, C.R.; Krivenkov, M.; Varykhalov, A.; Ramshaw, B.J.; Schoop, L.M.; McQueen, T.M. Dirac fermions and possible weak antilocalization in LaCuSb2. APL Mater. 2019, 7, 121108. [Google Scholar] [CrossRef]
- Seibel, E.M.; Xie, W.; Gibson, Q.D.; Cava, R.J. Structure and magnetic properties of the REAuBi2 (RE = La–Nd, Sm) phases. J. Solid State Chem. 2015, 230, 318–324. [Google Scholar] [CrossRef]
- Petrovic, C.; Bud’ko, S.L.; Strand, J.D.; Canfield, P.C. Anisotropic properties of rare earth silver dibismites. J. Magn. Magn. Mater. 2003, 261, 210–221. [Google Scholar] [CrossRef]
- Akiba, K.; Umeshita, N.; Kobayashi, T.C. Observation of superconductivity and its enhancement at the charge density wave critical point in LaAgSb2. Phys. Rev. B 2022, 106, L161113. [Google Scholar] [CrossRef]
- Uykur, E.; Maulana, L.Z.; Schoop, L.M.; Lotsch, B.V.; Dressel, M.; Pronin, A.V. Magneto-optical probe of the fully gapped Dirac band in ZrSiS. Phys. Rev. Res. 2019, 1, 032015. [Google Scholar] [CrossRef]
- Topp, A.; Queiroz, R.; Grüneis, A.; Müchler, L.; Rost, A.W.; Varykhalov, A.; Marchenko, D.; Krivenkov, M.; Rodolakis, F.; McChesney, J.L.; et al. Surface Floating 2D Bands in Layered Nonsymmorphic Semimetals: ZrSiS and Related Compounds. Phys. Rev. X 2017, 7, 041073. [Google Scholar] [CrossRef]
- Lee, G.; Farhan, M.A.; Kim, J.S.; Shim, J.H. Anisotropic Dirac electronic structures of AMnBi2 (A = Sr,Ca). Phys. Rev. B 2013, 87, 245104. [Google Scholar] [CrossRef]
- Klemenz, S.; Schoop, L.; Cano, J. Systematic study of stacked square nets: From Dirac fermions to material realizations. Phys. Rev. B 2020, 101, 165121. [Google Scholar] [CrossRef]
- Young, S.M.; Kane, C.L. Dirac Semimetals in Two Dimensions. Phys. Rev. Lett. 2015, 115, 126803. [Google Scholar] [CrossRef]
- Shao, D.-F.; Zhang, S.-H.; Dang, X.; Tsymbal, E.Y. Tunable two-dimensional Dirac nodal nets. Phys. Rev. B 2018, 98, 161104. [Google Scholar] [CrossRef]
- Fu, B.-B.; Yi, C.-J.; Zhang, T.-T.; Caputo, M.; Ma, J.-Z.; Gao, X.; Lv, B.Q.; Kong, L.-Y.; Huang, Y.-B.; Richard, P.; et al. Dirac nodal surfaces and nodal lines in ZrSiS. Sci. Adv. 2019, 5, eaau6459. [Google Scholar] [CrossRef]
- Rosmus, M.; Olszowska, N.; Bukowski, Z.; Starowicz, P.; Piekarz, P.; Ptok, A. Electronic Band Structure and Surface States in Dirac Semimetal LaAgSb2. Materials 2022, 15, 7168. [Google Scholar] [CrossRef]
- Murase, M.; Sasagawa, T. Large Magnetoresistance and Dirac Line Node in LaAgBi2. J. Phys. Soc. Jpn. 2020, 89, 055003. [Google Scholar] [CrossRef]
- Ekahana, S.A.; Wu, S.-C.; Jiang, J.; Okawa, K.; Prabhakaran, D.; Hwang, C.-C.; Mo, S.-K.; Sasagawa, T.; Felser, C.; Yan, B.; et al. Observation of nodal line in non-symmorphic topological semimetal InBi. New J. Phys. 2017, 19, 065007. [Google Scholar] [CrossRef]
- Regmi, S.; Dhakal, G.; Kabeer, F.C.; Harrison, N.; Kabir, F.; Sakhya, A.P.; Gofryk, K.; Kaczorowski, D.; Oppeneer, P.M.; Neupane, M. Observation of multiple nodal lines in SmSbTe. Phys. Rev. Mater. 2022, 6, L031201. [Google Scholar] [CrossRef]
- Feng, Y.; Jiang, Q.; Feng, B.; Yang, M.; Xu, T.; Liu, W.; Yang, X.; Arita, M.; Schwier, E.F.; Shimada, K.; et al. Rashba-like spin splitting along three momentum directions in trigonal layered PtBi2. Nat. Commun. 2019, 10, 4765. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.M.; Fazzio, A.; Dalpian, G.M. Zeeman-type spin splitting in nonmagnetic three-dimensional compounds. NPJ Quantum Mater. 2019, 4, 41. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Z.; Chen, C.; Shi, Y.; Xie, Z.; Yi, H.; Liang, A.; He, S.; He, J.; Peng, Y.; et al. Strong Anisotropy of Dirac Cones in SrMnBi2 and CaMnBi2 Revealed by Angle-Resolved Photoemission Spectroscopy. Sci. Rep. 2014, 4, 5385. [Google Scholar] [CrossRef] [PubMed]
- Bosak, A.; Souliou, S.-M.; Faugeras, C.; Heid, R.; Molas, M.R.; Chen, R.-Y.; Wang, N.-L.; Potemski, M.; Tacon, M.L. Evidence for nesting-driven charge density wave instabilities in the quasi-two-dimensional material LaAgSb2. Phys. Rev. Res. 2021, 3, 033020. [Google Scholar] [CrossRef]
- Thirupathaiah, S.; Efremov, D.; Kushnirenko, Y.; Haubold, E.; Kim, T.K.; Pienning, B.R.; Morozov, I.; Aswartham, S.; Buchner, B.; Borisenko, S.V. Absence of Dirac fermions in layered BaZnBi2. Phys. Rev. Mater. 2019, 3, 024202. [Google Scholar] [CrossRef]
- Ryu, H.; Park, S.Y.; Li, L.; Ren, W.; Neaton, J.B.; Petrovic, C.; Hwang, C.; Mo, S.-K. Anisotropic Dirac Fermions in BaMnBi2 and BaZnBi2. Sci. Rep. 2018, 8, 15322. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruszała, P.; Winiarski, M.J.; Samsel-Czekała, M. LaZn1−xBi2 as a Candidate for Dirac Nodal-Line Intermetallic Systems. Crystals 2024, 14, 209. https://doi.org/10.3390/cryst14030209
Ruszała P, Winiarski MJ, Samsel-Czekała M. LaZn1−xBi2 as a Candidate for Dirac Nodal-Line Intermetallic Systems. Crystals. 2024; 14(3):209. https://doi.org/10.3390/cryst14030209
Chicago/Turabian StyleRuszała, Piotr, Maciej J. Winiarski, and Małgorzata Samsel-Czekała. 2024. "LaZn1−xBi2 as a Candidate for Dirac Nodal-Line Intermetallic Systems" Crystals 14, no. 3: 209. https://doi.org/10.3390/cryst14030209
APA StyleRuszała, P., Winiarski, M. J., & Samsel-Czekała, M. (2024). LaZn1−xBi2 as a Candidate for Dirac Nodal-Line Intermetallic Systems. Crystals, 14(3), 209. https://doi.org/10.3390/cryst14030209