Abstract
Montmorillonite clay (MMT) has been widely used in engineering and environmental applications as a landfill barrier and toxic waste repository due to its unique property as an expandable clay mineral that can absorb water easily. This absorption process rendered MMT to be highly exothermic due to electrostatic interactions among molecules and hydrogen bonds between surface atoms. A detailed study of a large supercell model of structural clay enables us to predict long-term nuclear waste storage. Herein, a large solvent MMT model with 4071 atoms is studied using ab initio density functional theory. The DFT calculation and analysis clarify the important issues, such as bond strength, solvation effect, elasticity, and seismic wave velocities. These results are compared to our previous study on crystalline MMT (dry). The solvated MMT has reduced shear modulus (G), bulk modulus (K), and Young’s modulus (E). We observe that the conduction band (CB) in the density of states (DOS) of solvated MMT model has a single, conspicuous peak at −8.5 eV. Moreover, the atom-resolved partial density of states (PDOS) summarizes the roles played by each atom in the DOS. These findings illuminate numerous potential sophisticated applications of MMT clay.
1. Introduction
Clay minerals have recently gained attention due to their numerous desirable properties, such as mechanical strength, optical absorption, and cation exchange capacity. Studying the structure and composition of clay minerals is expected to grow in the near future due to the discovery of new applications and refinement of existing ones [1,2]. This is particularly relevant for applications that require barriers for water movement, such as earthen dams, leaks in ponds, and toxic waste landfills [3]. Furthermore, there is a recent and fascinating application in the field of spectroscopy [4]. Clay minerals [5] are small crystalline granules that occur during the formation process of silicate-bearing rocks. They can make up to 40% of the minerals in sedimentary rocks and are a substantial part of soil. Additionally, they are commonly classified as hydrous layer aluminum phyllosilicate particles with size less than 2 µm [6,7]. Moreover, they have highly reactive surfaces capable of altering soil properties [8]. Clay minerals have always been of great interest to researchers, particularly in the interrelated disciplines of physics, chemistry, and earth sciences. A comprehensive understanding of their composition and physical characteristics is crucial to comprehend fundamental aspects, such as bond strength, solvation effect, spectral characterization, optical absorption, elasticity, and seismic wave velocities. This requires a thorough understanding of the atomistic composition, electronic structure, interatomic bonding, charge transfer, optical properties, and mechanical properties of these minerals. Even though clay minerals have been the subject of extensive experimental research, they still lack detailed understanding of their structure and characteristics, particularly when it comes to atomic bonding, density of states (DOS), and mechanical properties [9]. Ab initio computational modelling is an ideal method to gain this knowledge, as opposed to the expensive experimental trial-and-error approach in the laboratory. The ab initio approach is based on density functional theory (DFT) and is more accurate than the classical approach. The classical approach refers to methods that treat atoms as point particles with no internal structure and uses empirical potential energy functions to model their interactions. This approach is less accurate than ab initio methods since it does not account for the electronic degree of freedom to compute the forces and relies on assumptions and simplifications. In recent years, ab initio approaches have grown increasingly prominent in material research. They have led to a number of impressive discoveries [10,11] and successful applications of DFT to study the electronic structure and properties of atoms, molecules, and solids. For example, DFT calculations have been used to predict the stability and reactivity of transition metal catalysts, which are widely used in chemical reactions [12].
MMT clay is a type of smectite clay mineral. It is naturally abundant and can be chemically modified through the cation exchange [13,14,15]. Weathering of eruptive rock materials leads to the formation of MMT. MMT is considered to be a valuable model for studying the properties of smectite clay minerals, owing to its high swelling capacity and ability to produce stable suspensions [16]. Crystalline MMT has a layered structure that is characteristic of smectites, consisting of two tetrahedral sheets sandwiching a central octahedral sheet, as shown in Figure 1. Its layer charge and exchangeable cations are easily modifiable, making it a versatile material for investigating the properties of smectite clay minerals. Moreover, crystalline MMT has been widely studied in the literature, and its properties and behaviors are well documented, making it an excellent reference material for exploring other smectites. MMT is a class of materials that is hydrophilic and is capable of absorbing cations [17]. Therefore, it can be used in numerous fields, such as oil refining, pharmaceutical preparations, sugar refining, catalyst, and as a binder for insulating materials [18]. Moreover, MMT is used to design polymer–clay nanocomposites, which are capable of changing its strength and resistance [19]. Furthermore, it can form composites of biological entities, such as proteins and amino acids [20].
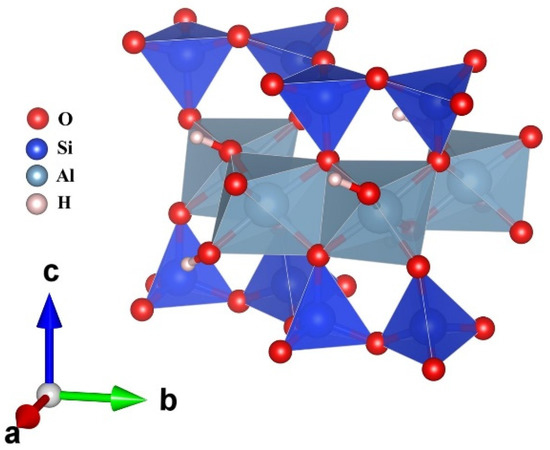
Figure 1.
Polyhedral structure for crystalline MMT model. SiO4 tetrahedra and AlO6 octahedra are shown in blue and light blue, respectively.
Computational modelling can provide valuable information to gain insight into the complex structures of clay minerals, including solvated montmorillonite (MMT). In comparison to crystalline MMT, the solvated MMT is far more challenging to study due to the presence of water [21,22] and other elements, such as Na, Mg, and Cl between these layers. Na+ has been identified as the main electric charge carrier [23] of the MMT layer. When a divalent metal ion takes the place of an aluminum ion in octahedral structure of MMT, it results in having a net negative charge. This layer attracts or binds to positively charged ions (also known as exchangeable cations), such as sodium, potassium, calcium, and magnesium, leading to the formation of an interlayer hydrated phase. These changes to the structure may have environmental impacts since they affect the mineral’s ability to adsorb and release ions and molecules, which can influence soil fertility, nutrient cycling, and water retention.
Understanding and predicting MMT clay’s interactions with water and other environmental fluids is a critical topic in geotechnical engineering, geo-environmental engineering, industrial applications, such as muds for oil well drilling, and water treatment. MMT clay possesses a remarkable capacity to absorb water into its interlayer spaces. This water absorption triggers a reaction within the clay, leading to its swelling. Due to the significant interest in understanding and predicting the swelling behavior of MMT clay, it becomes essential to develop and study a precise model that can effectively explain and forecast this phenomenon.
To gain a deeper understanding of various material properties, researchers can employ a range of approaches, such as theoretical simulations and experimental studies. These methods can establish meaningful correlations and comparisons between different properties, providing valuable insights and a wider perspective. Nevertheless, performing experimental research can be an expensive undertaking in terms of time and money due to the requirement for multiple physical fabrication attempts and practical limitations while computational simulations, such as the orthogonalized linear combination of atomic orbitals (OLCAO) [24] and molecular dynamic (MD), supported by robust algorithms and valid theoretical models, have emerged as a crucial element in contemporary research. These simulations serve as a bridge between theory and experiments [25]. OLCAO is a powerful computational tool for studying the electronic structure and properties of materials. This method can calculate complex systems with up to more than 4000 atoms, making it useful for investigating a wide range of materials, including complex minerals and alloys. By combining these approaches, researchers can gain a comprehensive understanding of the relationships between different material properties, paving the way for new discoveries and applications in materials science.
In this work, we performed detailed DFT calculations on the solvated MMT model. The model consists of 4071 atoms with metallic elements, such as Na, Mg, and Cl. Details of the solvated model are described in Section 2. We focus on the electronic structure, interatomic bonding, mechanical properties, and partial charge distributions of the solvated MMT. The electronic structure and properties of materials have been altered by the presence of both metallic elements and water molecules, leading to the possibility of unexpected applications. From the interatomic bonding, we have provided critical parameters, such as total and partial bond order density (TBOD and PBOD). The bond order can provide information about the internal cohesion of a material. Additionally, these parameters can be utilized to compare dissimilar materials and identify similarities and differences in their bonding behavior, thereby facilitating the design and optimization of materials with specific properties and functions.
2. Model Construction
The original model, created by N. Subramanian et al. [26] was constructed with five layers of MMT clay stacked on top of each other. This unit cell is then replicated in the a-b plane, followed by isomorphic substitution of Mg2+ for Al3+ in the octahedral sheet to produce a MMT layer with hydroxyl groups in places compatible with experimental results for Wyoming-MMT with a charge of −0.57e− per unit cell (O20(OH)4 [27]. Na+ was added to neutralize the net structural charge of the MMT layer. N. Subramanian et al., created the molecular structure of MMT using HRTEM lattice parameters as a monoclinic unit cell in the C 121 space group; the lattice constants a = 5.25 Å, b = 9.03 Å, c = 10.07 Å, and β = 99.5° from Douillard et al. [28]. The solvated MMT layer used in this work was extracted from the model of N. Subramanian et al., as illustrated in Figure 2. The solvated MMT model of N. Subramanian et al. mainly has one layer, that is replicated five times to form a large model. We chose to extract only one layer since we have a periodic boundary condition in our model. One of the major difficulties we encountered during the extraction for the solvated MMT layer was the cell size, which needs to be optimized. This has been successfully resolved by optimization via VASP code [29,30]. The optimized lattice parameters are a = 51.29 Å, b = 49.12 Å, and c = 18.27 Å. The final large supercell model has 4071 atoms including 1057 water molecules.
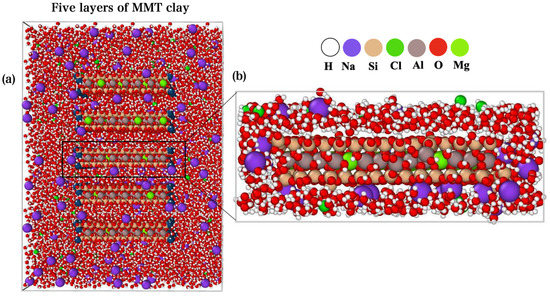
Figure 2.
Two-dimensional (2D) structure sketch of (a) N. Subramanian et al. model with five layers of MMT. The (b) extracted layer for solvated MMT model in ball and stick form.
3. Methods
In this study, we have used two DFT-based packages—Vienna ab initio simulation package (VASP) [31] and the orthogonalized linear combination of atomic orbitals (OLCAO) [24,32]. VASP is utilized for the optimization of the structures and for elastic and mechanical properties. In VASP calculation, projector augmented wave (PAW) potential [33] with a generalized gradient approximation (GGA) Perdew–Burke–Ernzerhof (PBE) [34] was used for the exchange and correlation part of the DFT potential. We used an energy cut-off of 600 eV, with electronic force convergence at 10–5 eV, and single KPOINTS for the large solvated MMT model. The optimized structure obtained from VASP is used as input in the OLCAO calculation for electronic structure and interatomic bonding. OLCAO method uses atomic orbitals in the basis expansion [32]. The combination of OLCAO method with VASP has been very successful in electronic property calculations, especially for large and complex systems. This success is due to the flexibility and economic use of the basis set [35,36,37]. We have used a minimal basis (MB) for the self-consistent potential, and the total density of states (TDOS), effective charges , and bond order (BO) calculations. The effective charges and bond order (BO) values were calculated based on Mulliken scheme [38,39].
In Equations (1) and (2), the overlap integrals are between the orbital in the atom and orbital in the atom. are the eigenvector coefficients of the band, orbital in the atom [24].
The partial charge (PC) for each atom is calculated from the effective charge using the following equation [24]:
where is the charge of the neutral atom. The BO shown in Equation (2) quantifies the relative strength of the bonds. By normalizing the total bond order of all the atomic pairs with the cell volume, total bond order density (TBOD) is obtained. TBOD is a single metric which evaluates the internal cohesion of the system and can be further resolved into the partial bond order density (PBOD).
The stress (σj) vs. strain () response analysis scheme [40,41] is applied to the fully optimized structure to obtain the elastic coefficients Cij (i, j = 1, 2, 3, 4, 5, 6). From and the calculated σj, Cij is calculated via solving the following linear equation:
From the elastic coefficients Cij, mechanical parameters, such as bulk modulus (K), shear modulus (G), Young’s modulus (E), Poisson’s ratio (ɳ), and Pugh’s modulus ratio (G/K) are calculated using Voight–Reuss–Hill (VRH) polycrystals approximation [42,43]. The Voight approximation assumes uniform strain distribution in the structure, which results in an upper limit of the polycrystalline bulk moduli. Whereas the Reuss approximation [42] assumes a uniform stress distribution resulting in the lower limits. The average of these two limits gives the so-called Hill approximation. Hill approximation is considered more realistic and can be compared with measured elastic parameters.
4. Results and Discussion
4.1. Electronic Structure and Interatomic Bonding
Total density of states (TDOS) and atom-resolved partial DOS (PDOS) are the most effective ways to summarize the electronic structures of a material. In this investigation, the DOS computation was carried out using minimal basis (MB). These results for solvated MMT are compared with crystalline MMT [44] and shown in Figure 3. We also calculated the TDOS and PDOS for both models within an energy range from −20.21 eV to 15.88 eV. In the crystalline MMT, the valance band (VB) DOS has the highest peak at −1.5 eV, while the highest peak in solvated MMT is at −8.5 eV. In crystalline MMT, the DOS in the conduction band (CB) has multiple peaks ranging from 6 eV to 15.88 eV, which mostly originates from O, Si, and Al atoms. Whereas in solvated MMT, there is a single prominent peak at 8.5 eV, which originates from O. The atom-resolved PDOS summarizes the contributions from each atom to the VB and CB. In both cases, it shows the lower part of the VB DOS mainly originating from the O atoms. There is a clear HOMO–LUMO band gap (Egap) of 5.11 eV in the crystalline model, but no band gap in the solvated model due to the presence of water molecules. Both oxygen and hydrogen atoms are filling the gap with further interaction to Si and Al atoms. It is important to note that the location of 0 eV is not the same. The 0 eV is at the highest occupied state at HOMO–LUMO. In the solvated model, the water molecules occupied those states. Furthermore, solvation refers to the process of dissolving a material in a solvent, which can result in the formation of a solvated shell around the material. The solvation shell can interact with MMT’s electronic orbital, leading to changes in their energy levels or charge distribution. As a result, we can claim that when the size of the alkali ions increases, the band gap decreases as in the hydrated MMT.
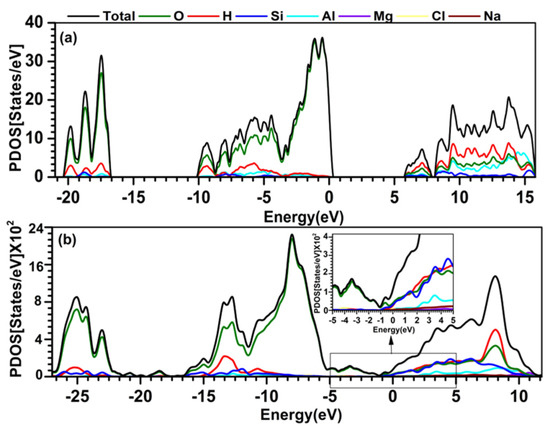
Figure 3.
Calculated TDOS and PDOS of the (a) crystalline MMT model, (b) solvated MMT model with inset focused on −5 eV to 5 eV. The atom-resolved PDOS is shown in color. O: green; H: red; Si: blue; Al: light blue; Mg: purple; Cl: yellow; Na: brown.
The most critical component of the electronic structure is interatomic bonding. Crystalline MMT has only four types of bonds: O-H, Si-O, Al-O, and O⋯H, which are also in the solvated model. The solvated MMT is signficantly more complicated. It has a total of fifteen types of bonds: Al-H, Al-O, Cl-H, Cl-Na, Cl-O, H-H, H-Mg, H-Na, O-H, O⋯H, H-Si, Mg-O, Na-O, O-O, and Si-O. In crystalline MMT, the O-H bond exhibits a strong bond with a bond order (BO) value of approximately 0.29 e−, as depicted in Figure 4a. Additionally, there are some hydrogen bonds (HB) O⋯H with very low BO and larger bond length (BL) between 2.47 and 3.49 Å. The Si-O and higher BO followed by Al-O bonds and both bonds act as a unit in the crystalline MMT. In solvated MMT, there is a wide range of BO for O-H, Si-O, and Al-O bonds. It is worth noting that the bond length has lower variation, while the bond order fluctuates significantly. This fluctuation in bond order could be explained by the presence of water molecules. Additionally, there is a notable presence of weak O⋯H bonds ranging from 1.50 Å to 3.49 Å. In solvated MMT, O-O bond has the strongest BO value of around 0.51 e−, followed by the H-H bond with a BO value of approximately 0.47 e−, as shown in Figure 4b. Interestingly, there are short H-H bonds with higher BO in the solvated MMT. These short H-H bonds are from two different H2O molecules. Mg-O and Na-O have bonds with noticeable BO. Cl-H has higher BO of around 0.28 e− in comparison to Al-H, H-Mg, H-Na, and H-Si bonds. Al-H, H-Mg, H-Si, and H-Na have lower BO at higher BL and are overlapped by other data in Figure 4b. There is one Cl-Na bond with BO of 0.06 e− at 2.69 Å. These bonds may not have a strong effect in the solvated MMT.
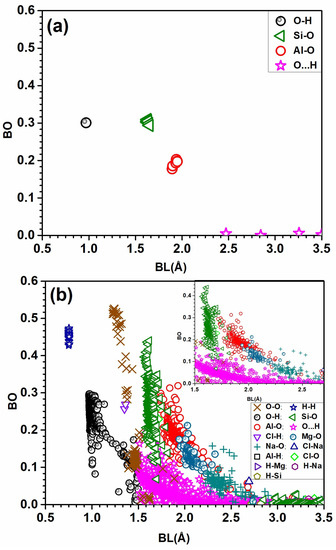
Figure 4.
(a) BO vs. BL distributions for all atomic pairs in the (a) crystalline MMT model and (b) solvated MMT with inset focused on BL from 1.5 Å to 2.7 Å.
TBOD is a useful metric to study the crystal cohesiveness. The TBOD for the crystalline MMT is 0.033 e−/Å3, whereas for the solvated MMT, it is 0.022 e−/Å3. To identify the reason behind lower TBOD in solvated MMT, we further analyzed the percentage contribution from the PBOD for each type of bond as shown in Figure 5. Si-O bonds with 62.29% contribution in TBOD of crystalline MMT is significantly higher. Si-O bonds form a strong three-dimensional network of tetrahedral sheets, providing structural stability and cohesiveness. On the other hand, solvated MMT contains water molecules and exchangeable cations between the layers, which create weaker electrostatic interactions with the mineral, reducing its overall cohesiveness. Although the Si-O bonds in solvated MMT are also covalent in nature, they contribute less to the stability than in crystalline MMT. Therefore, understanding the difference in the contribution of Si-O bonds between the two types of MMT is essential for understanding their distinct physical and chemical properties. We observe that the Al-O bonds also make higher contribution of 29.67% in crystalline MMT in comparison to solvated MMT with modest contribution of 12.34%.
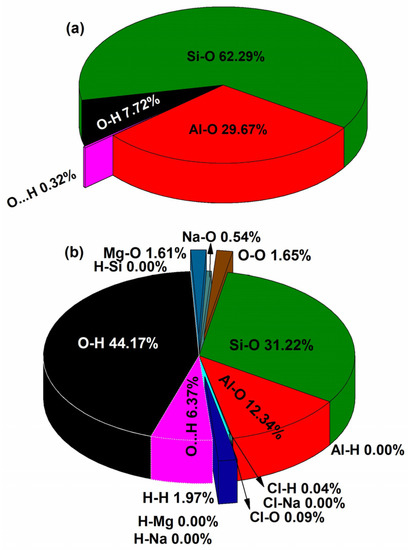
Figure 5.
Pie chart for the pair contributions of: (a) Crystalline MMT model and (b) solvated MMT model.
The presence of water in solvated MMT leads to the formation of a strong hydrogen [45] bonding network between the absorbed water in the interlayer, which is energetically favorable and exothermic. These findings have significant implications for applications, such as nuclear waste storage. In addition, the strong O-H bonds found in water molecules as well as in the hydroxyl groups have key contribution of 44.17% in the solvated MMT. Whereas the O-H bonds have only 7.72% contribution in crystalline MMT. Following a similar pattern, the solvated MMT has higher O⋯H contribution in comparison to crystalline MMT. Higher O-H and O⋯H contribution in the solvated MMT is clearly due to water molecules, since MMT is known to be hydrophilic and this notable characteristic differentiates it from crystalline MMT and other clay minerals. Furthermore, there is a very low contribution of Mg-O, O-O, and H-H bonds as shown in Figure 5b. The lower BO contribution of Al-H, Cl-Na, H-Na, H-Mg, and H-Si bonds leads to negligible percentage contribution in TBOD. Solvated MMT leads to a significantly more complicated bonding arrangement and a wide range of BO values. A comprehensive explanation of interatomic bonding in a complex system, as this large model shows, is a challenging endeavor.
4.2. Mechanical Properties
The study of mechanical properties of clay minerals is critical, particularly for engineering [46,47]. However, due to their tiny particle size the mechanical properties of clay minerals are difficult to evaluate directly. It is hard to isolate a large enough individual crystal of clay and impossible to directly measure their acoustic properties. However, we employed a computational approach using VASP to calculate the elastic coefficients for both crystalline and solvated MMT. Table 1 shows the calculated mechanical properties of crystalline and solvated MMT. The calculated mechanical properties are bulk modulus (K), shear modulus (G), Young’s modulus (E), Poisson’s ratio (η), Pugh’s modulus ratio (k = G/K), and Vicker’s hardness (HV). Vicker’s hardness is calculated using Tian et al. [48] equation HV = 0.92k1.137G0.708. Shear modulus (G) measures how resistant a material is to shear deformation. On the other hand, bulk modulus (K) measures a material’s resistance to volume compression. The compressibility of a material increases as its K decreases. Additionally, the sound velocity is closely related to the G and K, making it a useful parameter for analyzing the seismic and sonic log data [49]. Finally, Young’s modulus (E) measures how stiff a material is or how well it can resist deforming in the direction of an applied force. This study compares the mechanical properties of the solvated model to the crystalline model and reveals a significant decrease in the G, K, and E of solvated MMT. This implies that water solvation in MMT reduces the mechanical strength. However, the Poisson’s ratio of both models remains similar, exhibiting only slight variations in their values. This means that water has no effect on the material’s ability to contract laterally while being stretched longitudinally. It is also interesting to note that Pugh’s modulus ratio and Vicker’s hardness are slightly lower for solvated MMT. This indicates that solvated MMT is less stiff and less resistant to deformation than crystalline MMT. We were unable to find any experimental data on the mechanical properties of solvated MMT for compositions analogous to those used in this paper. In order to visualize the anisotropy of the Young’s and shear moduli, we have plotted the three-dimensional (3D) anisotropic surface figures using ELATE [50] software for both crystalline and solvated MMT in Figure 6. The surfaces of Young’s modulus are shown in green color in both crystalline and solvated MMT, indicating the positive values.

Table 1.
The calculated bulk modulus (K), shear modulus (G), Young’s modulus (E), Poisson’s ratio (η), Pugh’s modulus ratio (k = G/K), and Vicker’s hardness (HV) for crystalline and solvated models.
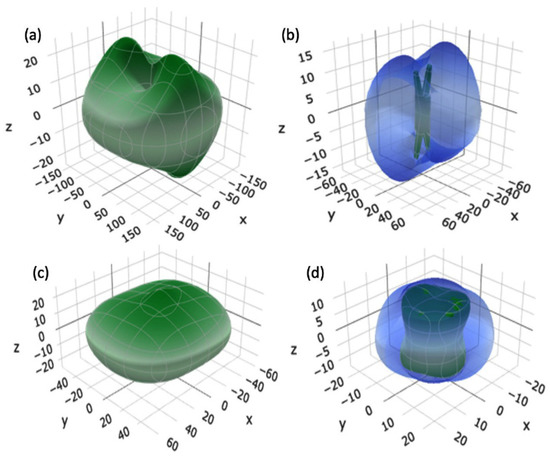
Figure 6.
A 3D representation of (a) Young’s modulus and (b) shear modulus for crystalline MMT; (c) Young’s modulus and (d) shear modulus for solvated MMT model.
The degree of deviation of the surfaces in Young’s and shear moduli from the spherical shape determines the anisotropy. Figure 6a,b shows that the degree of elastic anisotropy in Young’s and shear moduli is higher in crystalline MMT compared to solvated MMT, as shown in Figure 6c,d. This can be attributed to the fact that crystalline MMT has a more spherical shape in all three planes, which leads to a more isotropic distribution [51]. In solvated MMT, the presence of absorbed water molecules in the interlayer region causes the clay to swell, resulting in a more elongated shape and a higher degree of elastic anisotropy. In contrast, crystalline MMT has a more compact and ordered structure, which leads to a lower degree of anisotropy in its elastic properties.
It is crucial to determine the sound velocities once we have the elastic tensors for the solvated MMT. The following equations can be used to obtain the transverse sound velocity and longitudinal sound velocity [52].
where represents the density. In Table 2, we compare the calculated densities, and , of crystalline and solvated MMT. In the solvated MMT, and are both larger than in the crystalline case. These results may have applications in seismology and geosciences. The values of and can be used to determine the elastic properties of a material and to infer its geological structure. Compared with other clay minerals, such as illite, it has been found that the elastic properties of MMT are highly dependent on the layer charge density and the nature of the interlayer cations. This makes them a potentially useful tool for exploring geological structures and characterizing subsurface environments.
Table 2.
Density () and values of sound velocities ( and ).
In particular, the high swelling capacity of MMT and its ability to form stable suspensions make it a promising candidate for use in drilling fluids, where it can help in stabilizing boreholes and prevent blowouts.
5. Conclusions
In this article, we explore the computational simulation of montmorillonite (MMT) in both crystalline and solvated forms. Our study presents a comprehensive analysis of the electronic structure, interatomic bonding, and mechanical properties for solvated MMT clay mineral. This is the first study on solvated MMT using large-scale supercell modelling based on density functional theory (DFT). Our meticulous analysis involves bonds between each pair of atoms in the solvated MMT model, which reveals the complexity of covalent and hydrogen bonds. One of the key findings is that the presence of water in this model leads to a strong hydrogen bonding network with adsorbed water molecules in the interlayer. The O-H, Si-O, Al-O, and O⋯H are important components of interatomic bonding, which is found in both crystalline and solvated MMT. We can see that O-H and O⋯H bonds have a higher contribution in the solvated MMT compared to crystalline MMT due to water molecules. Conversely, the Si-O and Al-O bonds have less contribution in the solvated MMT compared to crystalline MMT. Moreover, our analysis of the electronic structure revealed a single prominent peak in the DOS in the conduction band (CB) located at −8.5 eV in the solvated MMT model. In addition, we analyzed the atom-resolved partial density of states (PDOS) to determine the contributions made by each atom in the VB and CB. We found that the majority of the lower part of the VB DOS originates from the O atom, while the CB DOS is contributed by H atoms. One of the most significant findings is that the solvation of montmorillonite (MMT) leads to a significant decrease in its mechanical strength, as evidenced by reduced values of G, K, and E. The presence of water does not affect the material’s lateral contraction ability while being longitudinally stretched, as indicated by similar Poisson’s ratios for solvated and crystalline MMT. These results from mechanical properties also reveal important insights into sound velocities. Comparing the densities, and , we observe that both values of and are higher in solvated MMT compared to crystalline MMT, while the converse is true in densities. Our findings have implications for the potential application of MMT clay in environmental applications as a landfill barrier and toxic waste repository. In summary, our study sheds light on the electronic structure and bonding network of solvated MMT clay, which can guide future research into its potential applications.
Author Contributions
W.-Y.C. conceived the project; L.S. and S.S. performed the calculations; L.S. made most of the figures; L.S., S.S., P.A. and W.-Y.C. drafted the paper. All authors participated in the discussion and interpretation of the results. All authors edited and proofread the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data can be provided on request to the corresponding author.
Acknowledgments
This research used the resources of the National Energy Research Scientific Computing Centre supported by DOE under Contract No. DE-AC03-76SF00098 and the Research Computing Support Services (RCSS) of the University of Missouri System. L.S. thanks Al-Jouf University and SACM sponsorship for giving her a full scholarship to pursue her Ph.D. study and work on this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Murray, H.H. Overview—Clay mineral applications. Appl. Clay Sci. 1991, 5, 379–395. [Google Scholar] [CrossRef]
- Sarkar, B.; Rusmin, R.; Ugochukwu, U.C.; Mukhopadhyay, R.; Manjaiah, K.M. Modified clay minerals for environmental applications. In Modified Clay and Zeolite Nanocomposite Materials; Elsevier: Amsterdam, The Netherlands, 2019; pp. 113–127. [Google Scholar]
- Keith, K.; Murray, H. Clay liners and barriers; Society for Mining, Metallurgy and Exploration: Littleton, CO, USA, 1994; pp. 249–255. [Google Scholar]
- Giovannini, G.; Garoli, D.; Rupper, P.; Neels, A.; Rossi, R.M.; Boesel, L.F. Metal-modified montmorillonite as plasmonic microstructure for direct protein detection. Sensors 2021, 21, 2655. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Mohan, C. Basics of Clay Minerals and Their Characteristic Properties; IntechOpen: London, UK, 2021; Volume 24, pp. 1–29. [Google Scholar]
- Huggett, J. Glauconites. In Encyclopedia of Geology; Selley, R.C., Cocks, L.R.M., Plimer, I.R., Eds.; Elsevier: Oxford, UK, 2005; pp. 542–548. [Google Scholar]
- Brindley, G.W. CLAYS, CLAY MINERALSClays, clay minerals. In Mineralogy; Springer: Boston, MA, USA, 1983; pp. 69–80. [Google Scholar]
- Essington, M.E. Soil and Water Chemistry: An Integrative Approach; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Sparks, D.L. Kinetics of ionic reactions in clay minerals and soils. Adv. Agron. 1986, 38, 231–266. [Google Scholar]
- Young, D. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Parr, R.G. Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry; Springer: Berlin/Heidelberg, Germany, 1980; pp. 5–15. [Google Scholar]
- Lee, Y.-K. Density Functional Theory (DFT) Calculations and Catalysis. Catalysts 2021, 11, 454. [Google Scholar] [CrossRef]
- Lorenz, P.; Meier, L.; Kahr, G. Determination of the cation exchange capacity (CEC) of clay minerals using the complexes of copper (II) ion with triethylenetetramine and tetraethylenepentamine. Clays Clay Miner. 1999, 47, 386–388. [Google Scholar]
- Marchuk, S.; Rengasamy, P.; Churchman, J. Cation exchange as influenced by the type of cations in different clay minerals. In Proceedings of the Australian Regolith and Clays Conference, Mildura, Australia, 7–10 February 2012. [Google Scholar]
- Fernandes, M.M.; Baeyens, B. Cation exchange and surface complexation of lead on montmorillonite and illite including competitive adsorption effects. Appl. Geochem. 2019, 100, 190–202. [Google Scholar] [CrossRef]
- Luckham, P.F.; Rossi, S. The colloidal and rheological properties of bentonite suspensions. Adv. Colloid Interface Sci. 1999, 82, 43–92. [Google Scholar] [CrossRef]
- Al-saeedi, A.A. Clay basics and their physical and chemical properties. Iaetsd J. Adv. Res. Appl. Sci. 2022, 10, 13–18. [Google Scholar]
- Hartwell, J. The diverse uses of montmorillonite. Clay Miner. 1965, 6, 111–118. [Google Scholar] [CrossRef]
- Toth, R.; Voorn, D.-J.; Handgraaf, J.-W.; Fraaije, J.G.; Fermeglia, M.; Pricl, S.; Posocco, P. Multiscale computer simulation studies of water-based montmorillonite/poly (ethylene oxide) nanocomposites. Macromolecules 2009, 42, 8260–8270. [Google Scholar] [CrossRef]
- Katti, D.R.; Ghosh, P.; Schmidt, S.; Katti, K.S. Mechanical properties of the sodium montmorillonite interlayer intercalated with amino acids. Biomacromolecules 2005, 6, 3276–3282. [Google Scholar] [CrossRef] [PubMed]
- Honorio, T.; Brochard, L.; Vandamme, M. Hydration phase diagram of clay particles from molecular simulations. Langmuir 2017, 33, 12766–12776. [Google Scholar] [CrossRef]
- Teich-McGoldrick, S.L.; Greathouse, J.A.; Jove-Colon, C.F.; Cygan, R.T. Swelling properties of montmorillonite and beidellite clay minerals from molecular simulation: Comparison of temperature, interlayer cation, and charge location effects. J. Phys. Chem. C 2015, 119, 20880–20891. [Google Scholar] [CrossRef]
- Takata, M.; Tomozawa, M.; Watson, E. Effect of Water Content on Transport in Na2O·3SiO2 Glass. J. Am. Ceram. Soc. 1982, 65, 91–93. [Google Scholar] [CrossRef]
- Ching, W. Theoretical studies of the electronic properties of ceramic materials. J. Am. Ceram. Soc. 1990, 73, 3135–3160. [Google Scholar] [CrossRef]
- Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.-h.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036, 012010. [Google Scholar] [CrossRef]
- Subramanian, N.; Whittaker, M.L.; Ophus, C.; Lammers, L.N. Structural Implications of Interfacial Hydrogen Bonding in Hydrated Wyoming-Montmorillonite Clay. J. Phys. Chem. C 2020, 124, 8697–8705. [Google Scholar] [CrossRef]
- Caruba, R.; Baumer, A.; Ganteaume, M.; Iacconi, P. An experimental study of hydroxyl groups and water in synthetic and natural zircons: A model of the metamict state. Am. Mineral. 1985, 70, 1224–1231. [Google Scholar]
- Douillard, J.-M.; Lantenois, S.; Prelot, B.; Zajac, J.; Henry, M. Study of the influence of location of substitutions on the surface energy of dioctahedral smectites. J. Colloid Interface Sci. 2008, 325, 275–281. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Vienna Ab-Initio Simulation Package (VASP); Vienna University: Vienna, Austria, 2001. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Sun, G.; Kürti, J.; Rajczy, P.; Kertesz, M.; Hafner, J.; Kresse, G. Performance of the Vienna ab initio simulation package (VASP) in chemical applications. J. Mol. Struct. THEOCHEM 2003, 624, 37–45. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Ching, W.-Y.; San, S.; Zhou, C.; Sakidja, R. Ab Initio Simulation of Structure and Properties in Ni-Based Superalloys: Haynes282 and Inconel740. Materials 2023, 16, 887. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, P.; Jawad, B.; Rao, P.; Podgornik, R.; Ching, W.-Y. Delta variant with P681R critical mutation revealed by ultra-large atomic-scale ab initio simulation: Implications for the fundamentals of biomolecular interactions. Viruses 2022, 14, 465. [Google Scholar] [CrossRef] [PubMed]
- Khaoulaf, R.; Adhikari, P.; Harcharras, M.; Brouzi, K.; Ez-Zahraouy, H.; Ching, W.-Y. Atomic-scale understanding of structure and properties of complex pyrophosphate crystals by first-principles calculations. Appl. Sci. 2019, 9, 840. [Google Scholar] [CrossRef]
- Mulliken, R. Electronic population analysis on LCAO-MO molecular wave functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Mulliken, R. Overlap populations, bond orders and covalent bond energies. J. Chem. Phys 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Nielsen, O.; Martin, R.M. First-principles calculation of stress. Phys. Rev. Lett. 1983, 50, 697. [Google Scholar] [CrossRef]
- Yao, H.; Ouyang, L.; Ching, W.Y. Ab initio calculation of elastic constants of ceramic crystals. J. Am. Ceram. Soc. 2007, 90, 3194–3204. [Google Scholar] [CrossRef]
- Reuß, A. Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle. ZAMM-J. Appl. Math. Mech. Z. Für Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Shafei, L.; Adhikari, P.; Ching, W.-Y. DFT Study of Electronic Structure and Optical Properties of Kaolinite, Muscovite, and Montmorillonite. Crystals 2021, 11, 618. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, Y.; Guo, X.; Xia, T.; Wang, T.; Jia, H.; Zhu, L. Charge mediated interaction of polystyrene nanoplastic (PSNP) with minerals in aqueous phase. Water Res. 2020, 178, 115861. [Google Scholar] [CrossRef] [PubMed]
- Pusch, R. Mechanical properties of clays and clay minerals. Dev. Clay Sci. 2006, 1, 247–260. [Google Scholar]
- Mhamdi, M.; Gasmi, N.; Elaloui, E.; Kbir-Ariguib, N.; Trabelsi-Ayadi, M. Study of the mechanical properties of clay containing smectite and carbonate. IOP Conf. Ser. Mater. Sci. Eng. 2010, 13, 012027. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Met. Hard Mater. 2012, 33, 93–106. [Google Scholar] [CrossRef]
- Mavko, G.; Mukerji, T.; Dvorkin, J. The Rock Physics Handbook; Cambridge University Press: Cambridge, UK, 2020. [Google Scholar]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Škundrić, T.; Matović, B.; Zarubica, A.; Zagorac, J.; Tatarko, P.; Zagorac, D. Structure prediction and mechanical properties of silicon hexaboride on ab initio level. Materials 2021, 14, 7887. [Google Scholar] [CrossRef]
- Korabel’nikov, D.; Zhuravlev, Y.N. Ab initio investigations of the elastic properties of chlorates and perchlorates. Phys. Solid State 2016, 58, 1166–1171. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).