
Exploring Volatile Precursors: Synthesis, Characterization and Thermal Properties of Thulium Fluoroacetylacetonate Complexes


Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of [Tm(tfaa)3(H2O)]2 (1)
2.2. Synthesis of [Tm(tfaa)3(H2O)2] (2a)
2.3. Synthesis of [Tm(tfaa)3(H2O)2] · Diglyme (2b)
2.4. Synthesis of [Tm(hfaa)3(H2O)2] (3) and [Tm(hfaa)2(tfa)(H2O)2]2 (4)
2.5. Synthesis of [Tm(tfaa)3(DME)] (5)
2.6. Synthesis of [TmCl2(tfaa)(diglyme)] (6)
2.7. Synthesis of [Tm(hfaa)3(DME)] (7), [Tm(hfaa)3(DME)(NaCl)]2 (8) and Na[Tm(hfaa)4] (9)
2.8. X-ray Crystal Structure Analyses
3. Results and Discussion
3.1. Syntheses
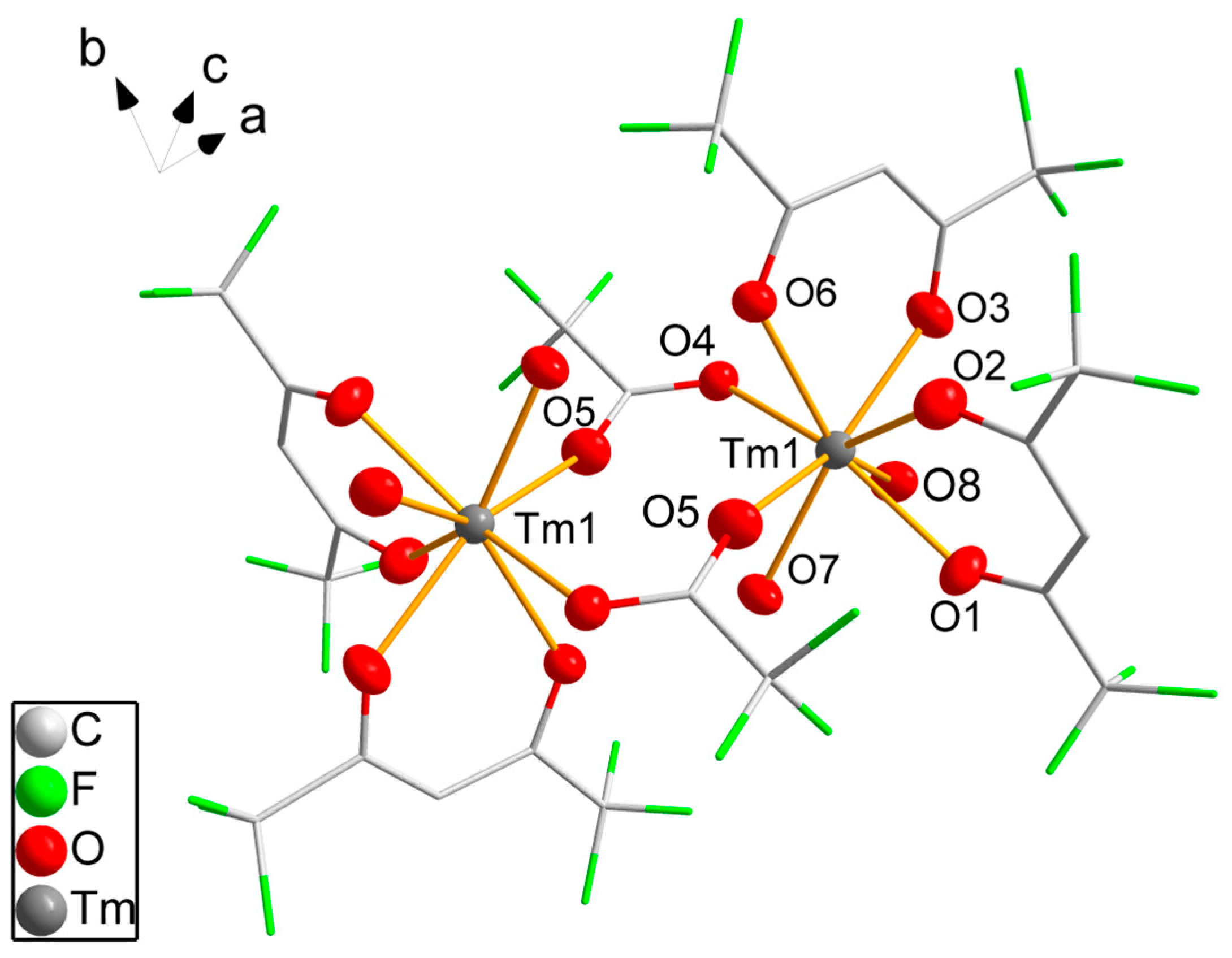
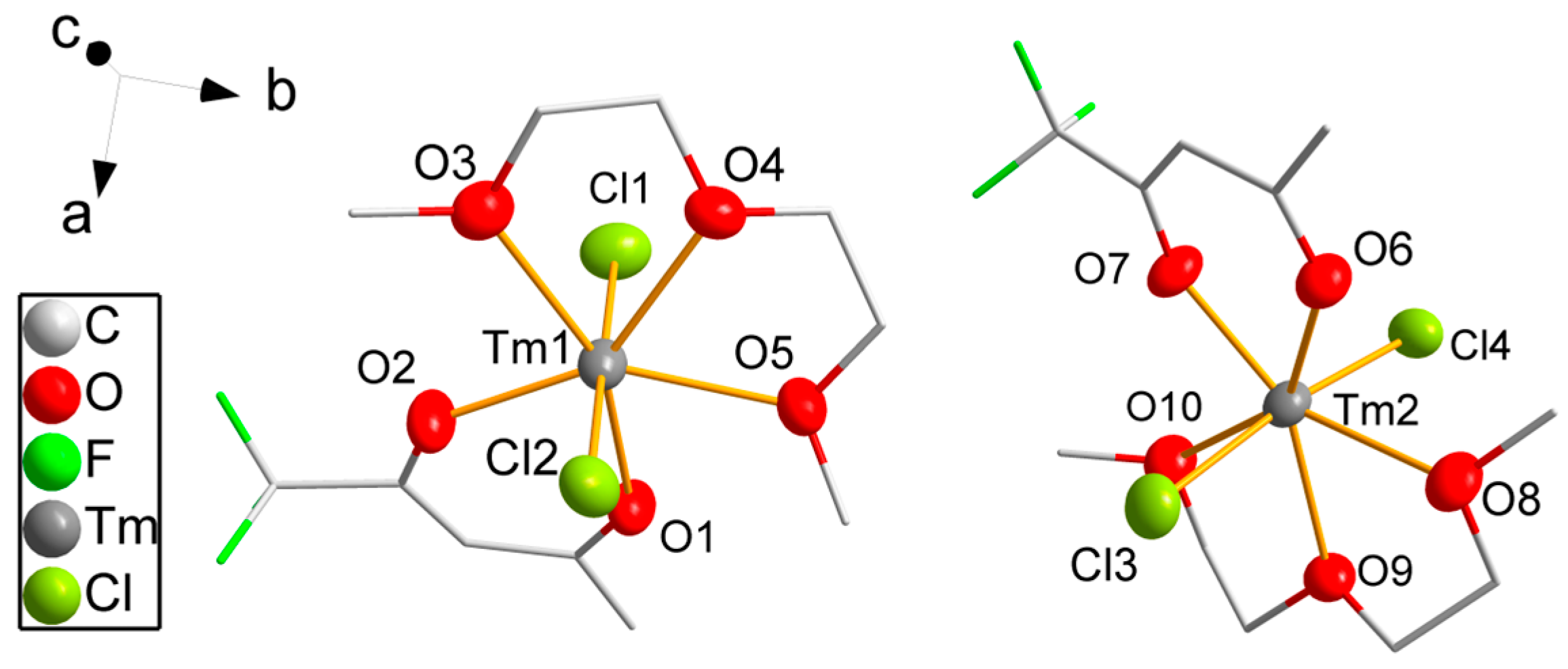
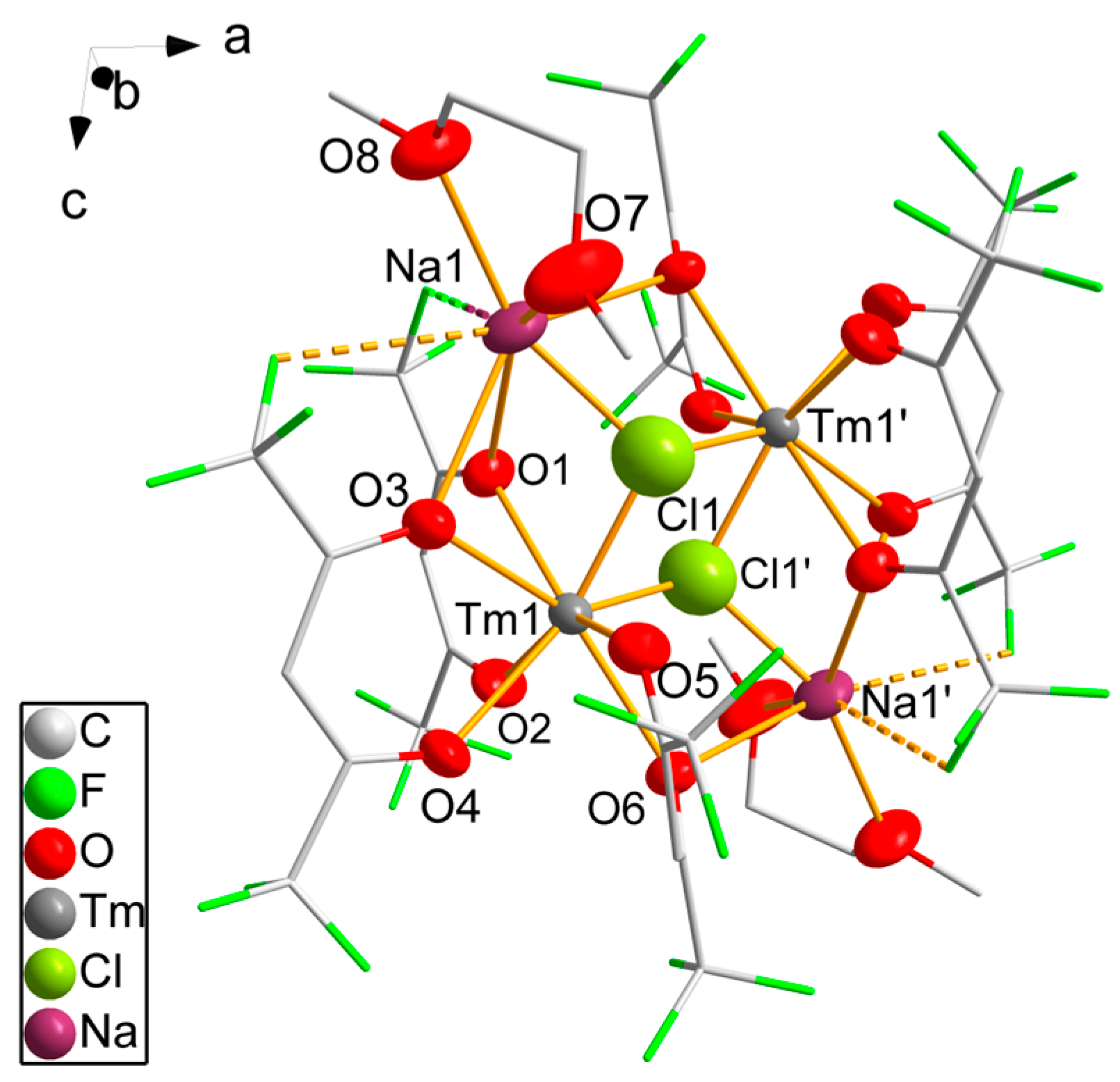
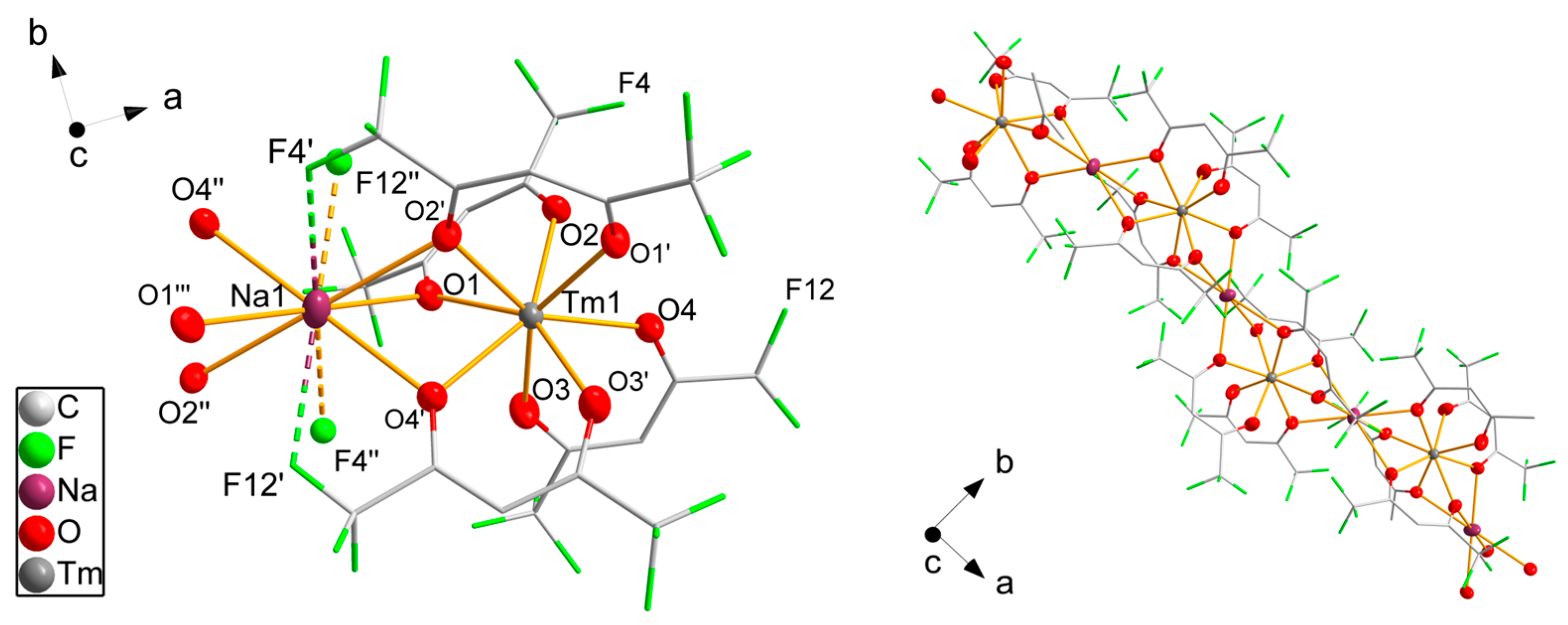
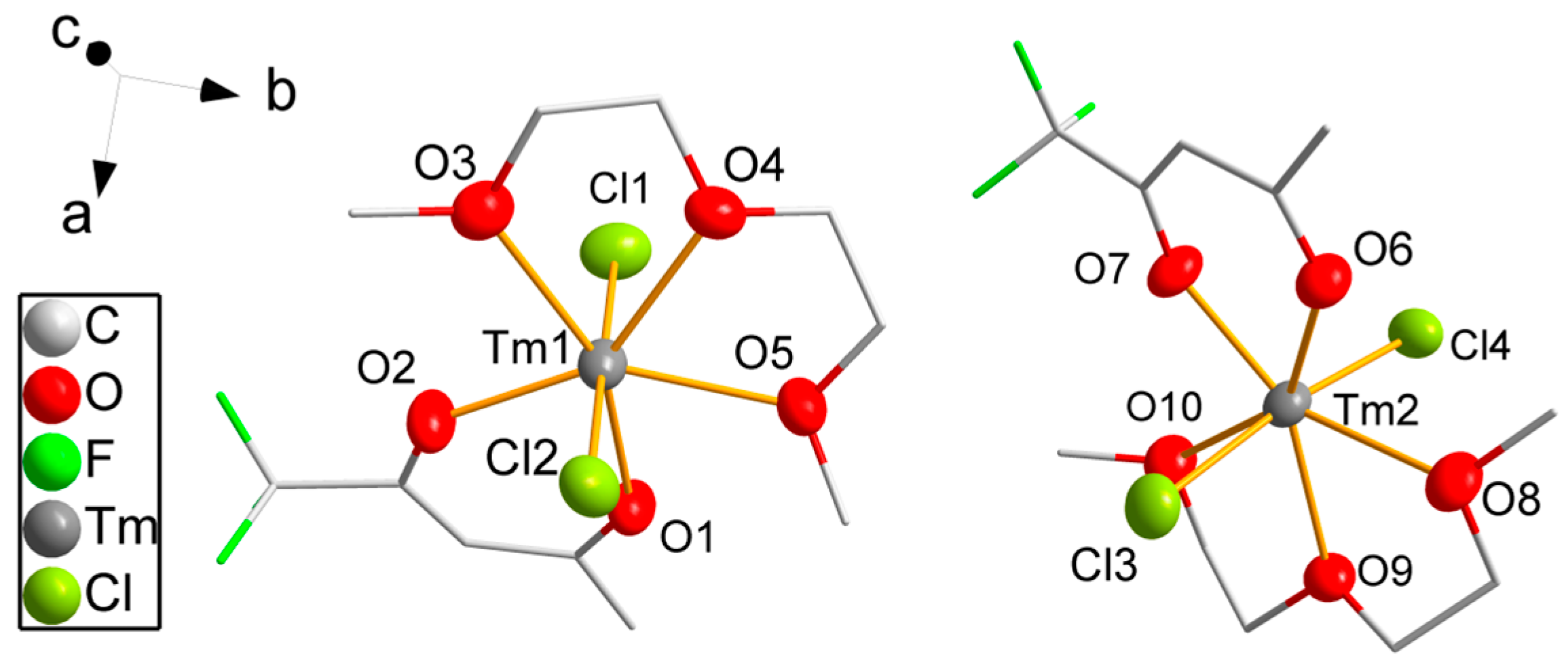
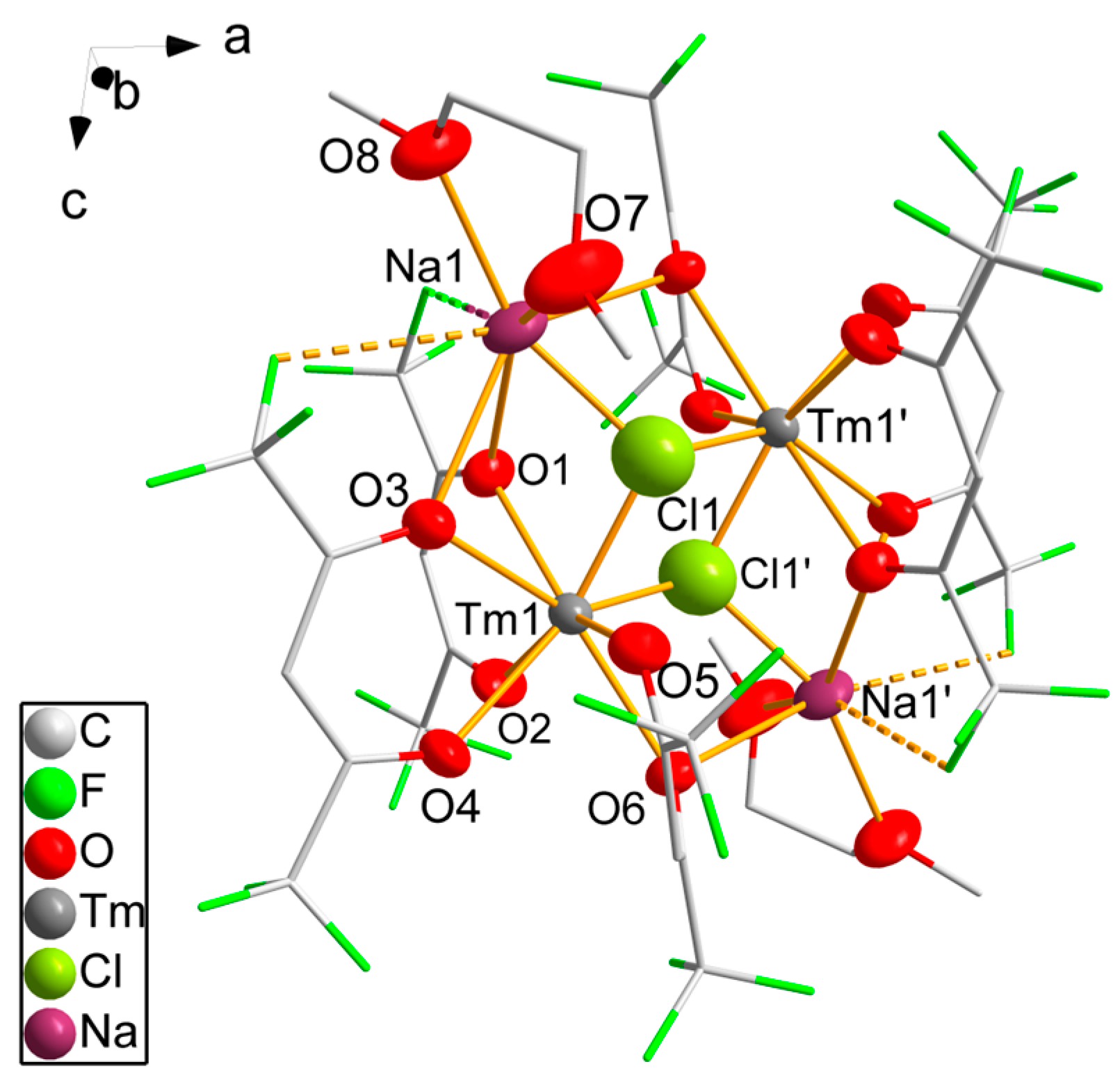
3.2. Single-Crystal Structure Analyses
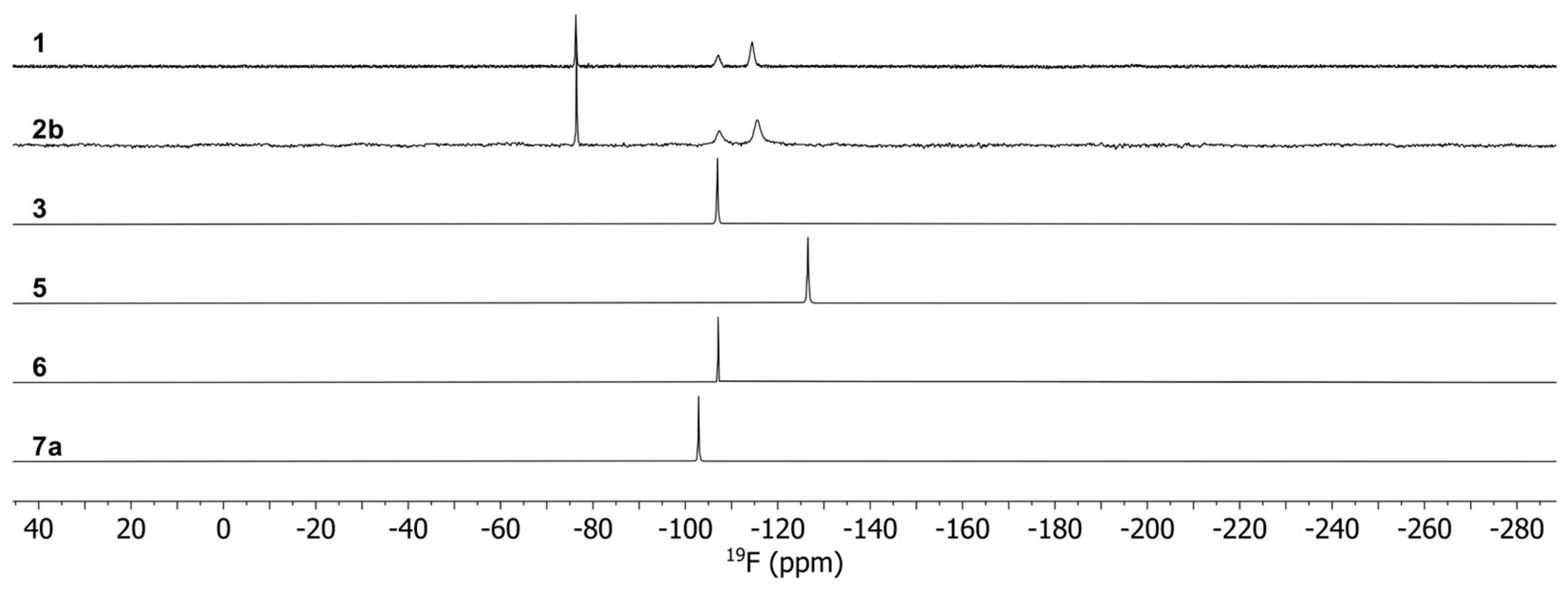
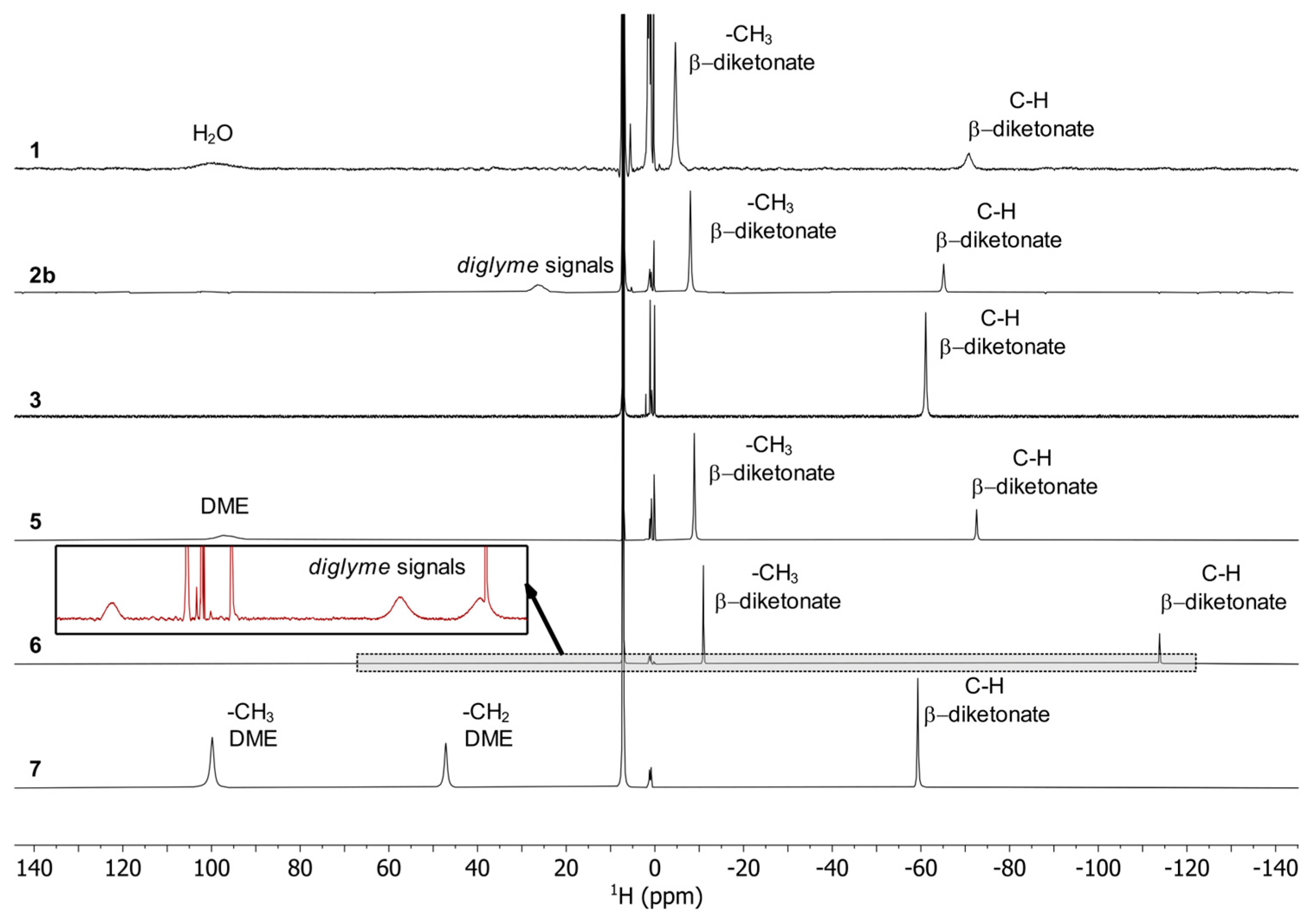
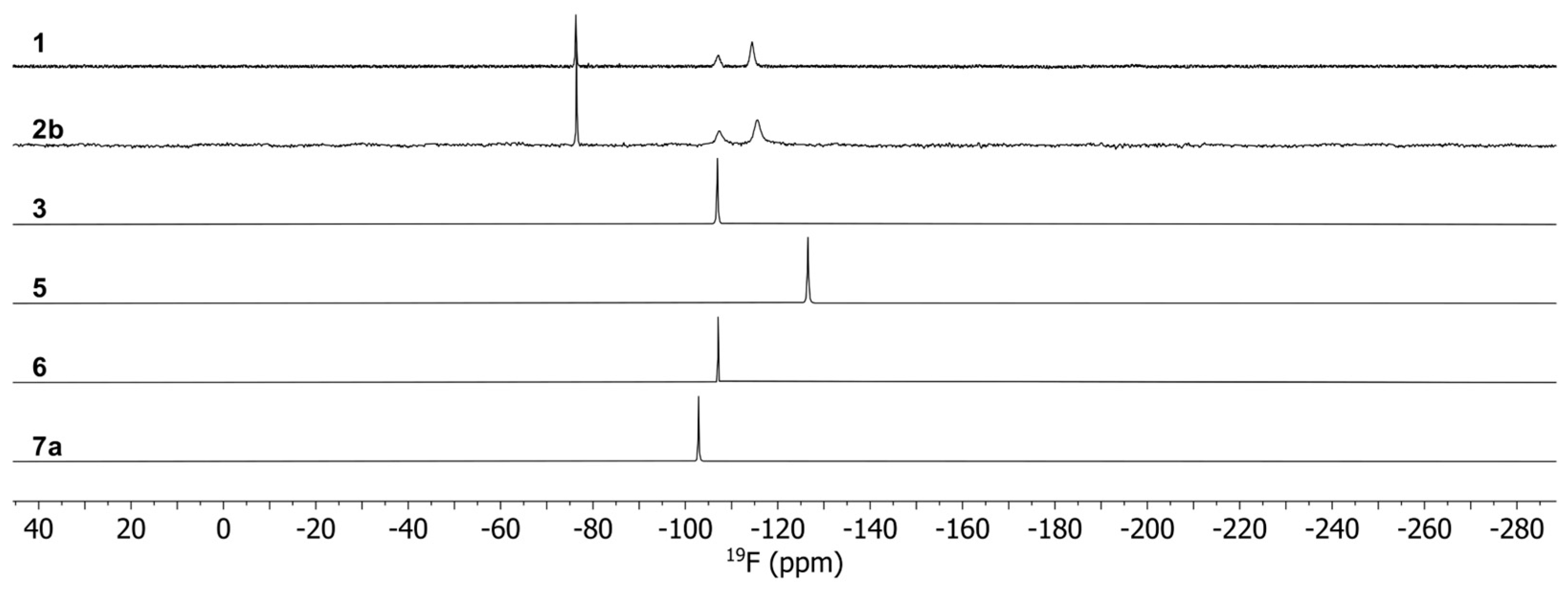
3.3. Spectroscopic Characterization of the Complexes
3.4. Thermal Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hebbink, G.A.; Grave, L.; Woldering, L.A.; Reinhoudt, D.N.; van Veggel, F.C.J.M. Unexpected Sensitization Efficiency of the Near-Infrared Nd3+, Er3+, and Yb3+ Emission by Fluorescein Compared to Eosin and Erythrosin. J. Phys. Chem. A 2003, 107, 2483–2491. [Google Scholar] [CrossRef]
- Armelao, L.; Quici, S.; Barigelletti, F.; Accorsi, G.; Bottaro, G.; Cavazzini, M.; Tondello, E. Design of luminescent lanthanide complexes: From molecules to highly efficient photo-emitting materials. Coord. Chem. Rev. 2010, 254, 487–505. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G.; Comby, S.; Chauvin, A.-S.; Vandevyver, C.D. New Opportunities for Lanthanide Luminescence. J. Rare Earth 2007, 25, 257–274. [Google Scholar] [CrossRef]
- Bettencourt-Dias, A. Lanthanide-based emitting materials in light-emitting diodes. Dalton Trans. 2007, 2007, 2229–2241. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.A.; Anwander, R.; Denk, M. Lanthanoiden-Komplexe, III. Flüchtige Neodym- und Yttrium-Alkoxide mit neuen sperrigen Chelatliganden. Chem. Berichte 1992, 125, 2399–2405. [Google Scholar] [CrossRef]
- Aspinall, H.C.; Bickley, J.F.; Gaskell, J.M.; Jones, A.C.; Labat, G.; Chalker, P.R.; Williams, P.A. Precursors for MOCVD and ALD of rare earth oxides-complexes of the early lanthanides with a donor-functionalized alkoxide ligand. Inorg. Chem. 2007, 46, 5852–5860. [Google Scholar] [CrossRef]
- Weber, A.; Suhr, H. Thin lanthanum oxide and rare-earth oxide films by PECVD of β-diketonate chelate complexes. Mod. Phys. Lett. B 1989, 3, 1001–1008. [Google Scholar] [CrossRef]
- Lo Nigro, R.; Toro, R.G.; Malandrino, G.; Raineri, V.; Fragalà, I.L. A Simple Route to the Synthesis of Pr2O3 High-k Thin Films. Adv. Mater. 2003, 15, 1071–1075. [Google Scholar] [CrossRef]
- Chevalier, S.; Bonnet, G.; Larpin, J.P. Metal-organic chemical vapor deposition of Cr2O3 and Nd2O3 coatings. Oxide growth kinetics and characterization. Appl. Surf. Sci. 2000, 167, 125–133. [Google Scholar] [CrossRef]
- Holzschuh, H.; Oehr, C.; Suhr, H.; Weber, A. Thin Films of Barium, Yttrium, Europium, Erbium, and Copper Oxides Prepared by Plasma-Enhanced CVD. Mod. Phys. Lett. B 1988, 2, 1253–1257. [Google Scholar] [CrossRef]
- Drake, S.R.; Lyons, A.; Otway, D.J.; Slawin, A.M.Z.; Williams, D.J. Lanthanide β-diketonate glyme complexes exhibiting unusual coordination modes. J. Chem. Soc. Dalton 1993, 1993, 2379–2386. [Google Scholar] [CrossRef]
- Xu, G.; Wang, Z.-M.; He, Z.; Lü, Z.; Liao, C.-S.; Yan, C.-H. Synthesis and structural characterization of nonanuclear lanthanide complexes. Inorg. Chem. 2002, 41, 6802–6807. [Google Scholar] [CrossRef] [PubMed]
- Malandrino, G.; Fragalà, I.L. Lanthanide “second-generation” precursors for MOCVD applications: Effects of the metal ionic radius and polyether length on coordination spheres and mass-transport properties. Coord. Chem. Rev. 2006, 250, 1605–1620. [Google Scholar] [CrossRef]
- Richardson, M.F.; Wagner, W.F.; Sands, D.E. Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 1968, 30, 1275–1289. [Google Scholar] [CrossRef]
- Malandrino, G.; Bettinelli, M.; Speghini, A.; Fragalà, I.L. Europium “Second Generation” Precursors for Metal-Organic Chemical Vapor Deposition: Characterization and Optical Spectroscopy. Eur. J. Inorg. Chem. 2001, 2001, 1039–1044. [Google Scholar] [CrossRef]
- Bradley, D.C.; Chudzynska, H.; Hursthouse, M.B.; Motevalli, M.; Wu, R. Volatile fluorinated tertiary alkoxides of some lanthanides, tris-hexafluoro-tertiary butoxides of lanthanum, praseodymium and europium. Polyhedron 1994, 13, 7–14. [Google Scholar] [CrossRef]
- Malandrino, G.; Licata, R.; Castelli, F.; Fragala, I.L.; Benelli, C. New Thermally Stable and Highly Volatile Precursors for Lanthanum MOCVD: Synthesis and Characterization of Lanthanum β-Diketonate Glyme Complexes. Inorg. Chem. 1995, 34, 6233–6234. [Google Scholar] [CrossRef]
- Pollard, K.D.; Jenkins, H.A.; Puddephatt, R.J. Chemical Vapor Deposition of Cerium Oxide Using the Precursors [Ce(hfac)3(glyme)]. Chem. Mater. 2000, 12, 701–710. [Google Scholar] [CrossRef]
- Katagiri, S.; Tsukahara, Y.; Hasegawa, Y.; Wada, Y. Energy-Transfer Mechanism in Photoluminescent Terbium(III) Complexes Causing Their Temperature-Dependence. Bull. Chem. Soc. Jpn. 2007, 80, 1492–1503. [Google Scholar] [CrossRef]
- Baxter, I.; Drake, S.R.; Hursthouse, M.B.; Abdul Malik, K.M.; McAleese, J.; Otway, D.J.; Plakatouras, J.C. Effect of Polyether Ligands on Stabilities and Mass Transport Properties of a Series of Gadolinium(III) β-Diketonate Complexes. Inorg. Chem. 1995, 34, 1384–1394. [Google Scholar] [CrossRef]
- Ye, H.-Q.; Peng, Y.; Li, Z.; Wang, C.-C.; Zheng, Y.-X.; Motevalli, M.; Wyatt, P.B.; Gillin, W.P.; Hernández, I. Effect of Fluorination on the Radiative Properties of Er3+ Organic Complexes: An Opto-Structural Correlation Study. J. Phys. Chem. C 2013, 117, 23970–23975. [Google Scholar] [CrossRef]
- Valore, A.; Cariati, E.; Righetto, S.; Roberto, D.; Tessore, F.; Ugo, R.; Fragalà, I.L.; Fragalà, M.E.; Malandrino, G.; Angelis, F.; et al. Fluorinated β-diketonate diglyme lanthanide complexes as new second-order nonlinear optical chromophores: The role of f electrons in the dipolar and octupolar contribution to quadratic hyperpolarizability. J. Am. Chem. Soc. 2010, 132, 4966–4970. [Google Scholar] [CrossRef]
- Pirri, A.; Maksimov, R.N.; Li, J.; Vannini, M.; Toci, G. Achievements and Future Perspectives of the Trivalent Thulium-Ion-Doped Mixed-Sesquioxide Ceramics for Laser Applications. Materials 2022, 15, 2084. [Google Scholar] [CrossRef] [PubMed]
- McComb, T.S.; Sims, R.A.; Willis, C.C.C.; Kadwani, P.; Sudesh, V.; Shah, L.; Richardson, M. High-power widely tunable thulium fiber lasers. Appl. Opt. 2010, 49, 6236–6242. [Google Scholar] [CrossRef] [PubMed]
- Neumeier, J.J.; Dalichaouch, Y.; Ferreira, J.M.; Hake, R.R.; Lee, B.W.; Maple, M.B.; Torikachvili, M.S.; Yang, K.N.; Zhou, H. Thulium barium copper oxide: A 90-K superconductor with a potential 1-MG upper critical field. Appl. Phys. Lett. 1987, 51, 371–373. [Google Scholar] [CrossRef]
- Prokić, M. Development of highly sensitive CaSO4:Dy/Tm and MgB4O7:Dy/Tm sintered thermoluminescent dosimeters. Nucl. Instrum. Methods 1980, 175, 83–86. [Google Scholar] [CrossRef]
- Hsu, C.-T. Growth of ZnS:Tm thin films by MOCVD. J. Cryst. Growth 2000, 208, 259–263. [Google Scholar] [CrossRef]
- Hara, K.; Tominaga, S.; Takano, A.; Dantani, K.; Yoshino, J.; Kukimoto, H. Preparation of ZnS:Tm Films by Metalorganic Chemical Vapor Deposition Using Thulium β-Diketonates as Dopants. Jpn. J. Appl. Phys. 1992, 31, L1661. [Google Scholar] [CrossRef]
- Forissier, S.; Roussel, H.; Jimenez, C.; Chaix, O.; Pereira, A.; Bensalah-Ledoux, A.; Deschanvres, J.-L.; Moine, B. Thulium and ytterbium-doped titania thin films deposited by MOCVD. Energy Procedia 2011, 10, 192–196. [Google Scholar] [CrossRef]
- Pellegrino, A.L.; Cortelletti, P.; Pedroni, M.; Speghini, A.; Malandrino, G. Nanostructured CaF2:Ln3+ (Ln3+ = Yb3+/Er3+, Yb3+/Tm3+) Thin Films: MOCVD Fabrication and Their Upconversion Properties. Adv. Mater. Interfaces 2017, 4, 1700245. [Google Scholar] [CrossRef]
- Pellegrino, A.L.; La Manna, S.; Bartasyte, A.; Cortelletti, P.; Lucchini, G.; Speghini, A.; Malandrino, G. Upconverting tri-doped calcium fluoride-based thin films: A comparison of the MOCVD and sol–gel preparation methods. J. Mater. Chem. C 2020, 8, 3865–3877. [Google Scholar] [CrossRef]
- Murthy, K.S.R.; Anjaneyulu, Y. Thermogravimetric and Gas Chromatographic Studies on Fluorinated β-diketone Chelates of Lanthanides. World App. Sci. J. 2014, 32, 939–944. [Google Scholar]
- Fatila, E.M.; Hetherington, E.E.; Jennings, M.; Lough, A.J.; Preuss, K.E. Syntheses and crystal structures of anhydrous Ln(hfac)3(monoglyme). Ln = La, Ce, Pr, Sm, Eu, Gd, Tb, Dy, Er, Tm. Dalton Trans. 2012, 41, 1352–1362. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, N.; Kuwamoto, T. Vapor pressures and enthalpies of sublimation and evaporation of trifluoroacetylacetonates in helium and helium containing the ligand vapor. Inorg. Chem. 1985, 24, 2697–2701. [Google Scholar] [CrossRef]
- Mullica, D.F.; Milligan, W.O.; Beall, G.W. Crystal structures of Pr(OH)3, Eu(OH)3 and Tm(OH)3. J. Inorg. Nucl. Chem. 1979, 41, 525–532. [Google Scholar] [CrossRef]
- Mestrelab Research SL. MestReNova Version 14.1.0-24037; Mestrelab Research SL: Santiago de Compostela, Spain, 2019. [Google Scholar]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- NETZSCH-Gerätebau GmbH. Proteus Analysis Version 5.0.1; NETZSCH-Gerätebau GmbH: Selb, Germany, 2009. [Google Scholar]
- STOE & Cie GmbH. X-Area Version 1.70; STOE & Cie GmbH: Darmstadt, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. Diamond Version 3.2k; Crystal Impact: Bonn, Germany, 2014. [Google Scholar]
- Bruker AXS. TOPAS Version 5; Bruker AXS: Karlsruhe, Germany, 2014. [Google Scholar]
- Malandrino, G.; Benelli, C.; Castelli, F.; Fragalà, I.L. Synthesis, Characterization, Crystal Structure and Mass Transport Properties of Lanthanum β-Diketonate Glyme Complexes, Volatile Precursors for Metal−Organic Chemical Vapor Deposition Applications. Chem. Mater. 1998, 10, 3434–3444. [Google Scholar] [CrossRef]
- Kang, S.-J.; Jung, Y.S.; Suh, I.-H. Synthesis and Characterization of Thermally Stable Ln(hfa)3(monoglyme) (Ln = Ho, Y, hfa = hexafluoroacetylacetone) Complexes. Bull. Korean Chem. Soc. 1999, 20, 95–98. [Google Scholar]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Van der Bondi, A. Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–664. [Google Scholar] [CrossRef]
- Tan, R.H.C.; Motevalli, M.; Abrahams, I.; Wyatt, P.B.; Gillin, W.P. Quenching of IR luminescence of erbium, neodymium, and ytterbium β-diketonate complexes by ligand C-H and C-D bonds. J. Phys. Chem. B 2006, 110, 24476–24479. [Google Scholar] [CrossRef] [PubMed]
- Pell, A.J.; Pintacuda, G.; Grey, C.P. Paramagnetic NMR in solution and the solid state. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 111, 1–271. [Google Scholar] [CrossRef]
- Meng, Q.; Witte, R.J.; May, P.S.; Berry, M.T. Photodissociation and Photoionization Mechanisms in Lanthanide-based Fluorinated β-Diketonate Metal−Organic Chemical-Vapor Deposition Precursors. Chem. Mater. 2009, 21, 5801–5808. [Google Scholar] [CrossRef]
- Talaga, D.S.; Hanna, S.D.; Zink, J.I. Luminescent Photofragments of (1,1,1,5,5,5-Hexafluoro-2,4-pentanedionato) Metal Complexes in the Gas Phase. Inorg. Chem. 1998, 37, 2880–2887. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2a | 2b * | 3 | 4 | |
|---|---|---|---|---|---|
| formula | C30H28F18O14Tm2 | C15H16F9O8Tm | C21H30F9O11Tm | C15H7F18O8Tm | C24H12F30O16Tm2 |
| molar mass M/g mol−1 | 1293.38 | 664.21 | 798.38 | 826.14 | 1464.20 |
| temperature/K | 200(2) | 180(2) | 180(2) | 120(2) | 180(2) |
| wavelength/pm | 71.073 | 154.186 | 154.186 | 154.186 | 154.186 |
| crystal color and shape | colorless needle | colorless needle | colorless needle | colorless block | colorless rod |
| crystal size/mm | 0.04·0.11·0.24 | 0.45·0.07·0.10 | 0.04·0.07·0.13 | 0.07·0.08·0.09 | 0.05·0.07·0.11 |
| crystal system | triclinic | triclinic | monoclinic | triclinic | triclinic |
| space group | P | P | Ic | P | P |
| a/pm | 815.99(8) | 1018.69(4) | 1339.28(4) | 991.1(2) | 894.14(2) |
| b/pm | 1102.04(8) | 1022.57(4) | 1444.32(3) | 1153.6(2) | 1066.73(2) |
| c/pm | 1213.9(1) | 1273.97(4) | 1645.70(5) | 1229.0(2) | 1221.65(3) |
| α/° | 65.335(6) | 87.962(3) | 90 | 67.931(9) | 69.838(2) |
| β/° | 72.933(8) | 74.854(3) | 108.453(2) | 73.717(9) | 88.985(2) |
| γ/° | 77.278(5) | 63.565(3) | 90 | 76.073(9) | 74.814(2) |
| V/106 pm3 | 1019.9(2) | 1142.02(8) | 3019.7(2) | 1235.4(3) | 1052.23(4) |
| Z | 1 | 2 | 4 | 2 | 1 |
| calc. density/g cm−3 | 2.10 | 1.92 | 1.76 | 2.22 | 2.31 |
| absorption coefficient µ/mm−1 | 4.47 | 8.33 | 6.49 | 8.43 | 9.56 |
| θ range/° | 2.6–26.0 | 3.6–70.0 | 4.1–70 | 3.9–70.5 | 3.9–63.3 |
| measured reflections | 7226 | 19,222 | 24,652 | 20,671 | 14,741 |
| independent reflections (Rint) | 3891 (0.0651) | 4245 (0.0296) | 4904 (0.0311) | 4584 (0.0290) | 3352 (0.0159) |
| observed reflections (I > 2σ(I)) | 3025 | 3566 | 4522 | 4016 | 3150 |
| parameters | 296 | 304 | 401 | 386 | 345 |
| R1 (observed reflections) | 0.0460 | 0.0496 | 0.0227 | 0.0429 | 0.0364 |
| wR2 (all data) | 0.1346 | 0.1353 | 0.0480 | 0.1170 | 0.0952 |
| max./min. residual e− density/10−6 pm−3 | 1.0 and −1.6 | 1.0 and −0.8 | 0.3 and −0.4 | 1.7 and −1.2 | 1.2 and −0.9 |
| CCDC no. | 2292075 | 2292076 | 2292077 | 2292079 | 2292080 |
| 5 | 6 | 8 * | 9 | |
|---|---|---|---|---|
| formula | C19H22F9O8Tm | C11H18Cl2F3O5Tm | C38H26Cl2F36Na2O16Tm2 | C20H4F24NaO8Tm |
| molar mass M/g mol−1 | 718.29 | 527.08 | 1877.33 | 1020.15 |
| temperature/K | 180(2) | 180(2) | 200(2) | 200(2) |
| wavelength/pm | 154.186 | 154.186 | 71.073 | 71.073 |
| crystal color and shape | colorless block | colorless block | colorless octahedron | colorless rod |
| crystal size/mm | 0.09·0.10·0.11 | 0.10·0.11·0.12 | 0.16·0.17·0.18 | 0.08·0.16·0.25 |
| crystal system | triclinic | monoclinic | monoclinic | monoclinic |
| space group | P | P21/c | P21/n | I2/c |
| a/pm | 779.94(3) | 1549.44(4) | 1257.94(6) | 1425.46(10) |
| b/pm | 1056.03(3) | 1564.79(3) | 1905.95(8) | 1207.07(10) |
| c/pm | 1706.90(5) | 1513.08(4) | 1283.96(6) | 1802.92(13) |
| α/° | 73.210(3) | 90 | 90 | 90 |
| β/° | 79.121(3) | 100.365(2) | 95.772(4) | 94.749(6) |
| γ/° | 76.174(3) | 90 | 90 | 90 |
| V/106 pm3 | 1296.06(8) | 3608.7(2) | 3062.8(2) | 3091.5(4) |
| Z | 2 | 8 | 2 | 4 |
| calc. density/g cm−3 | 1.84 | 1.94 | 2.04 | 2.19 |
| absorption coefficient µ/mm−1 | 7.40 | 12.37 | 3.15 | 3.07 |
| θ range/° | 4.4–70 | 2.9–70.5 | 2.3–25.8 | 2.2–27 |
| measured reflections | 21,933 | 33,844 | 28,017 | 9701 |
| independent reflections (Rint) | 4759 (0.0213) | 6798 (0.0311) | 28,017 (0.0508) | 3367 (0.0313) |
| observed reflections (I > 2σ(I)) | 4228 | 5344 | 19201 | 2838 |
| parameters | 337 | 401 | 426 | 244 |
| R1 (observed reflections) | 0.0295 | 0.0367 | 0.0558 | 0.0355 |
| wR2 (all data) | 0.0806 | 0.0898 | 0.1554 | 0.0896 |
| max./min. residual e− density/10−6 pm−3 | 1.0 and −0.9 | 1.2 and −0.5 | 1.5 and −1.6 | 1.1 and −1.0 |
| CCDC no. | 2292081 | 2292082 | 2292083 | 2292084 |
| 1 | 2a | 2b | 3 | 4 | 5 | 6 | 7 [33] | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Tm-O(keto) | 229.4(6) | 233.4(5) | 236.0(4) | 234.2(4) | 226.9(4) | 228.8(3) | 224.4(3) 1 | 228.5(5) | 238.0(6) | 231.1(3) |
| 222.9(7) | 230.2(5) | 231.8(3) | 226.7(4) | 230.2(4) | 228.8(3) | 220.4(3) 1 | 228.7(5) | 230.3(7) | 233.7(3) | |
| 234.4(6) | 233.8(5) | 230.7(4) | 231.5(4) | 228.3(4) | 229.3(3) | 224.7(3) 2 | 229.1(5) | 234.1(6) | 226.5(4) | |
| 227.0(6) | 225.9(5) | 227.4(4) | 231.9(4) | 237.2(4) | 228.9(3) | 220.6(3) 2 | 229.5(5) | 232.0(6) | 236.5(3) | |
| 243.2(9) * | 232.6(4) | 231.7(4) | 234.1(4) | 230.4(3) | 230.5(5) | 231.3(6) | ||||
| 247.8(5) * | 231.4(5) | 229.(4) | 234.2(4) | 226.8(2) | 230.7(5) | 232.0(6) | ||||
| 232.5(6) | ||||||||||
| Tm-O(H2O) | 232.6(5) | 235.7(4) | 233.6(4) | 233.7(4) | 235.2(4) | |||||
| 233.1(4) | 238.6(4) | 231.2(4) | 237.2(4) | |||||||
| Tm-O(DME) | 243.4(3) | 2.396(6) | ||||||||
| 246.9(3) | 2.431(6) | |||||||||
| Tm-O(tfa) | 226.9(4) | |||||||||
| 238.5(4) | ||||||||||
| Tm-O(diglyme) | 239.7(3) 1 | |||||||||
| 237.8(3) 1 | ||||||||||
| 239.8(4) 1 | ||||||||||
| 240.5(4) 2 | ||||||||||
| 238.8(3) 2 | ||||||||||
| 240.3(4) 2 | ||||||||||
| Tm-Cl | 256.0(2) 1 | 220.9(5) * | ||||||||
| 257.4(2) 1 | 221.9(5) * | |||||||||
| 255.9(2) 2 | ||||||||||
| 256.1(2) 2 | ||||||||||
| Na-O(keto) | 257.1(7) | 258.8(3) | ||||||||
| 260.7(8) | 255.1(3) | |||||||||
| 271.7(7) | 271.4(3) | |||||||||
| Na-O(DME) | 233.2(9) | |||||||||
| 233.0(8) | ||||||||||
| Na-Cl | 225.1(6) | |||||||||
| Na-F | 277.7(4) | |||||||||
| 294.5(4) |
| 1 [Tm(tfaa)3(H2O)]2 | 3 [Tm(hfaa)3(H2O)2] | 5 [Tm(tfaa)3(DME)] | 7 [Tm(hfaa)3(DME)] | |
|---|---|---|---|---|
| Isothermal thermogravimetric analysis at 100 °C | ||||
| Mass loss within 3 h/% | 0.28 | 1.37 | 2.80 | 4.4 |
| Calculated Tm transport rate/nmol cm−3 min−1 | 4 | 17 | - | 91 |
| Thermogravimetric analysis up to 600 °C | ||||
| Melting point/°C | 122 | 120 | 85 | 69 (lit.: 71–72) [33] |
| Total experimental mass loss/% | 58.0 | 89.5 | 65.3 | 97.5 |
| Calculated 1 experimental mass loss/% | 65.0 | 72.6 | 68.6 | 74.4 |
| Thermolysis product | TmF3 | TmF3 | TmF3 | TmF3 |
| Sublimation experiment at 100 °C in vacuo for 2 h | ||||
| Residue in starting zone | 1 with traces of decomposition products | 3 with traces of decomposition products | Decomposition products only | Pure 7 |
| Sublimation product | Pure 1 according to NMR spectra, but different crystal structure | Pure 3 | DME and decomposition products only | Pure 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuhrmann, D.; Kaden, K.; Kandler, M.; Icker, M.; Krautscheid, H. Exploring Volatile Precursors: Synthesis, Characterization and Thermal Properties of Thulium Fluoroacetylacetonate Complexes. Crystals 2023, 13, 1467. https://doi.org/10.3390/cryst13101467
Fuhrmann D, Kaden K, Kandler M, Icker M, Krautscheid H. Exploring Volatile Precursors: Synthesis, Characterization and Thermal Properties of Thulium Fluoroacetylacetonate Complexes. Crystals. 2023; 13(10):1467. https://doi.org/10.3390/cryst13101467
Chicago/Turabian StyleFuhrmann, Daniel, Kristin Kaden, Marika Kandler, Maik Icker, and Harald Krautscheid. 2023. "Exploring Volatile Precursors: Synthesis, Characterization and Thermal Properties of Thulium Fluoroacetylacetonate Complexes" Crystals 13, no. 10: 1467. https://doi.org/10.3390/cryst13101467
APA StyleFuhrmann, D., Kaden, K., Kandler, M., Icker, M., & Krautscheid, H. (2023). Exploring Volatile Precursors: Synthesis, Characterization and Thermal Properties of Thulium Fluoroacetylacetonate Complexes. Crystals, 13(10), 1467. https://doi.org/10.3390/cryst13101467