
First-Principles Calculations of the Phonon, Mechanical and Thermoelectric Properties of Half-Heusler Alloy VIrSi Alloys

 , ,
, , 
Abstract
1. Introduction
2. Computational Procedure
3. Results and Discussion
3.1. Structural and Electronic Properties
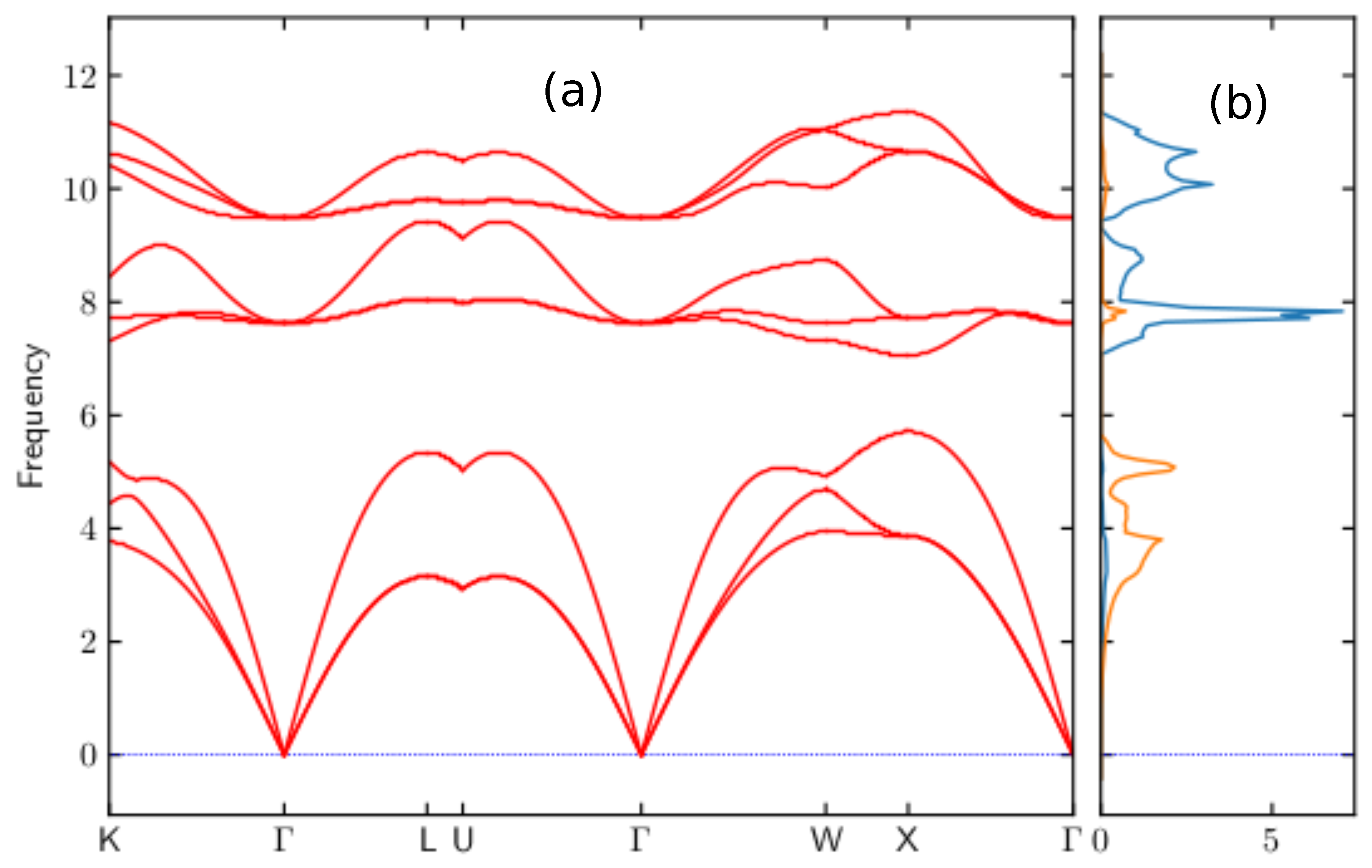
3.2. Phonon Dispersion Curve of VIrSi Half-Heusler Alloys
3.3. Mechanical Properties
3.4. Thermoelectric Properties of VIrSi Half-Heusler Alloys
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowe, D.M. CRC Handbook of Thermoelectrics; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar] [CrossRef]
- Kanatzidis, M.G. Nanostructured thermoelectrics: The new paradigm. Chem. Mater. 2010, 22, 648–659. [Google Scholar] [CrossRef]
- Zheng, X.F.; Liu, C.X.; Yan, Y.Y.; Wang, Q. A review of thermoelectrics research – Recent developments and potentials for sustainable and renewable energy applications. Renew. Sustain. Energy Rev. 2014, 32, 486. [Google Scholar] [CrossRef]
- Nolas, G.S.; Poon, J.; Kanatzidis, M. Recent Developments in Bulk Thermoelectric Materials. MRS Bull. 2006, 31, 199. [Google Scholar] [CrossRef]
- Zhao, L.D.; Dravid, V.P.; Kanatzidis, M.G. The panoscopic approach to high performance thermoelectrics. Energy Environ. Sci. 2014, 7, 251–268. [Google Scholar] [CrossRef]
- Uto, O.T.; Adebambo, P.O.; Akinlami, J.O.; Kenmoe, S.; Adebayo, G.A. Electronic, Structural, Mechanical, and Thermodynamic Properties of CoYSb (Y = Cr, Mo, W) Half-Heusler Compounds as Potential Spintronic Materials. Solids 2022, 3, 22–33. [Google Scholar] [CrossRef]
- Shutoh, N.; Sakurada, S. Thermoelectric properties of the TiX(Zr0.5Hf0.5)1-xNiSn half-Heusler compounds. J. Alloys Compd. 2005, 389, 204. [Google Scholar] [CrossRef]
- Culp, S.R.; Poon, S.J.; Hickman, N.; Tritt, T.M.; Blumm, J. Effect of substitutions on the thermoelectric figure of merit of half-Heusler phases at 800 °C. Appl. Phys. Lett. 2006, 88, 042106. [Google Scholar] [CrossRef]
- Kim, S.W.; Kimura, Y.; Mishima, Y. High temperature thermoelectric properties of TiNiSn-based half-Heusler compounds. Intermetallics 2007, 15, 349. [Google Scholar] [CrossRef]
- Katsuyama, S.; Matsuo, R.; Ito, M. Thermoelectric properties of half-Heusler alloys Zr1-xYxNiSn1-ySby. J. Alloys Compd. 2007, 428, 262. [Google Scholar] [CrossRef]
- Sakurada, S.; Shutoh, N. Effect of Ti substitution on the thermoelectric properties of (Zr,Hf)NiSn half-Heusler compounds. Appl. Phys. Lett. 2005, 86, 3159. [Google Scholar] [CrossRef]
- Yu, C.; Zhu, T.J.; Shi, R.Z.; Zhang, Y.; Zhao, X.B.; He, J. High-performance half-Heusler thermoelectric materials Hf1−xZrxNiSn1−ySby prepared by levitation melting and spark plasma sintering. Acta Mater. 2009, 57, 2757–2764. [Google Scholar] [CrossRef]
- Shen, Q.; Chen, L.; Goto, T.; Hirai, T.; Yang, J.; Meisner, G.P.; Uher, C. Effects of partial substitution of Ni by Pd on the thermoelectric properties of ZrNiSn-based half-Heusler compounds. Appl. Phys. Lett. 2001, 79, 4165–4167. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Dahal, K.; Mao, J.; Huang, L.; Zhang, Q.; Ren, Z. Thermoelectric properties of n-type half-Heusler compounds (Hf0.25Zr0.75)1−xNbxNiSn. Acta Mater. 2016, 113, 41–47. [Google Scholar] [CrossRef]
- Fu, C.G.; Xie, H.H.; Liu, Y.T.; Zhu, T.J.; Xie, J.; Zhao, X.B. Thermoelectric properties of FeVSb half-Heusler compounds by levitation melting and spark plasma sintering. Intermetallics 2013, 32, 39. [Google Scholar] [CrossRef]
- Fu, C.G.; Xie, H.H.; Zhu, T.J.; Xie, J.; Zhao, X.B. Enhanced phonon scattering by mass and strain field fluctuations in Nb substituted FeVSb half-Heusler thermoelectric materials. J. Appl. Phys. 2012, 112, 124915. [Google Scholar] [CrossRef]
- Fu, C.G.; Zhu, T.J.; Pei, Y.Z.; Xie, H.H.; Wang, H.; Snyder, G.J.; Liu, Y.; Liu, Y.T.; Zhao, X.B. High band degeneracy contributing to high thermoelectric performance in p-type half-Heusler compounds. Adv. Energy Mater. 2014, 1400600. [Google Scholar] [CrossRef]
- Fu, C.G.; Liu, Y.T.; Xie, H.H.; Liu, X.; Zhao, X.; Jeffrey, S.G.; Xie, J.; Zhu, T. Electron and phonon transport in Co-doped FeV0.6Nb0.4Sb half-Heusler thermoelectric materials. J. Appl. Phys. 2013, 114, 134905. [Google Scholar] [CrossRef]
- Wang, G.; Wang, D. Electronic structure and thermoelectric properties of Pb-based half-Heusler compounds: ABPb (A = Hf, Zr; B = Ni, Pd). J. Alloys Compd. 2016, 682, 375–380. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Birch, F. Finite Elastic Strain of Cubic Crystals. Phys. Rev. 1947, 71, 809. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A. Elastic constants of beryllium: A first-principles investigation. J. Phys. Condens. Matter 2016, 28, 075401. [Google Scholar] [CrossRef] [PubMed]
- Voigt, W. Lehrbuck der Kristallphysik (B. B. Teubner, Leipzig, 1928). 2022, p. 739. Available online: https://www.worldcat.org/title/lehrbuch-der-kristallphysik-mit-ausschluss-der-kristalloptik/oclc/23322264 (accessed on 21 November 2022).
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. (Lond.) 1952, 65, 349. [Google Scholar] [CrossRef]
- Huntington, H.B. The Elastic Constants of Crystals. Solid State Phys. 1958, 7, 213–351. [Google Scholar]
- Galanakis, I.; Dederichs, P.H.; Papanikolaou, N. Slater-Pauling behavior and origin of the half-metallicity of the full-Heusler alloys. Phys. Rev. B 2002, 66, 174429. [Google Scholar] [CrossRef]
- Togo, A. Phonopy. Available online: http://phonopy.sourceforge.net/ (accessed on 22 November 2018).
- Matsumoto, A.; Koyama, Y.; Togo, A.; Choi, M.; Tanaka, I. Electronic structures of dynamically stable As2O3, Sb2O3, and Bi2O3 crystal polymorphs. Phys. Rev. B 2011, 83, 214110. [Google Scholar] [CrossRef]
- Wallace, D.C. Thermodynamics of Crystals; Dover Publications: Mineola, NY, USA, 1998. [Google Scholar]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford Clarendon Press: Oxford, UK, 1956; pp. 120–156. [Google Scholar]
- Pugh, S.F. Relations between the Elastic Moduli and the Plastic Properties of Polycrystalline Pure Metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823. [Google Scholar] [CrossRef]
- Zener, C. Elasticity and Anelasticity of Metals; University of Chicago Press: Chicago, IL, USA, 1948. [Google Scholar]
- Kaur, K.; Rai, D.P.; Thapa, R.K. Srivastava, S.; Structural, electronic, mechanical, and thermoelectric properties of a novel half Heusler compound HfPtPb. J. Appl Phys. 2017, 122, 045110. [Google Scholar] [CrossRef]
- Fang, T.; Zheng, S.; Zhou, T.; Yan, L.; Zhang, P. Computational prediction of high thermoelectric performance in p-type half-Heusler compounds with low band effective mass. Phys. Chem. Chem. Phys. 2017, 19, 4411. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Methods | |||
|---|---|---|---|---|
| VIrSi | Present Work | 5.69 | 218.3 | 4.22 |
| Experiment | - | - |
| C (GPa) | C (GPa) | C (GPa) | G (GPa) | B (GPa) | E (Gpa) | Pugh Ratio | Poisson Ratio | Zener Anisotropy | Debye Temp. (K) | |
|---|---|---|---|---|---|---|---|---|---|---|
| This work | 295.02 | 180.10 | 109.13 | 84.36 | 218.43 | 224.17 | 2.589 | 0.2768 | 1.899 | 391.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adebambo, P.O.; Adetunji, B.I.; Uto, O.T.; Kenmoe, S.; Adebayo, G.A. First-Principles Calculations of the Phonon, Mechanical and Thermoelectric Properties of Half-Heusler Alloy VIrSi Alloys. Crystals 2022, 12, 1838. https://doi.org/10.3390/cryst12121838
Adebambo PO, Adetunji BI, Uto OT, Kenmoe S, Adebayo GA. First-Principles Calculations of the Phonon, Mechanical and Thermoelectric Properties of Half-Heusler Alloy VIrSi Alloys. Crystals. 2022; 12(12):1838. https://doi.org/10.3390/cryst12121838
Chicago/Turabian StyleAdebambo, Paul O., Bamidele I. Adetunji, Oghenekevwe T. Uto, Stephane Kenmoe, and Gboyega A. Adebayo. 2022. "First-Principles Calculations of the Phonon, Mechanical and Thermoelectric Properties of Half-Heusler Alloy VIrSi Alloys" Crystals 12, no. 12: 1838. https://doi.org/10.3390/cryst12121838
APA StyleAdebambo, P. O., Adetunji, B. I., Uto, O. T., Kenmoe, S., & Adebayo, G. A. (2022). First-Principles Calculations of the Phonon, Mechanical and Thermoelectric Properties of Half-Heusler Alloy VIrSi Alloys. Crystals, 12(12), 1838. https://doi.org/10.3390/cryst12121838