
Nuclear Resonance Vibrational Spectroscopy: A Modern Tool to Pinpoint Site-Specific Cooperative Processes


Abstract
1. Introduction
1.1. NRVS Transitions and Selection Rules
1.2. Historic Evolution of NRVS
1.3. NRVS Advantages
2. Experimental Aspects (I): Instruments
2.1. Detecting Inelastic Scattering
2.2. Light Sources for NRVS
2.3. High-Resolution Monochromators
2.4. X-Ray Interaction with NRVS Sample
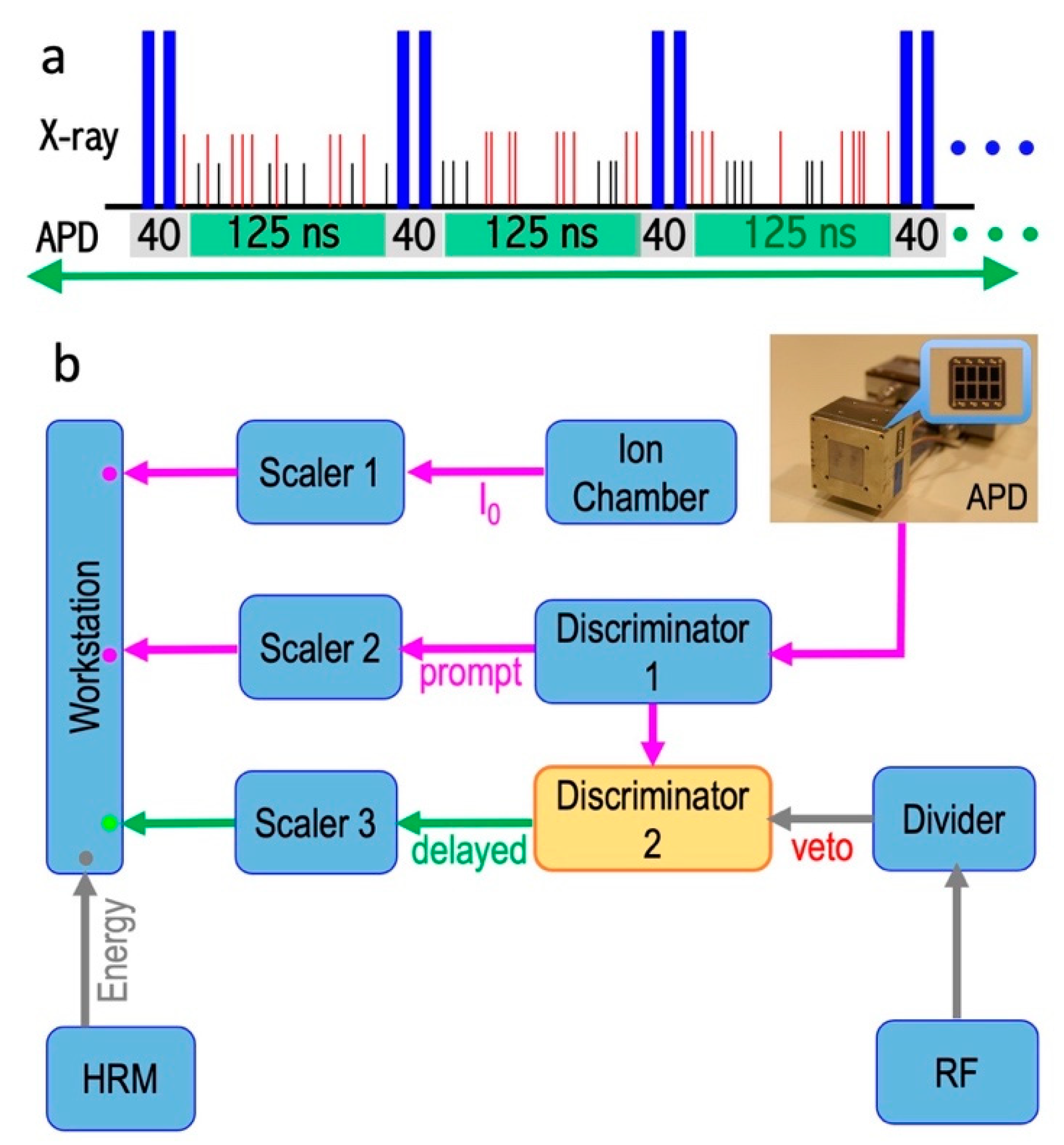
2.5. Detectors
2.6. Data-Acquisition Electronics
3. Experimental Aspects (II): Measurements
3.1. Checking for Dark Count Rate
3.2. Energy Calibration
3.3. Signal-to-Noise Ratio and Detection Limit
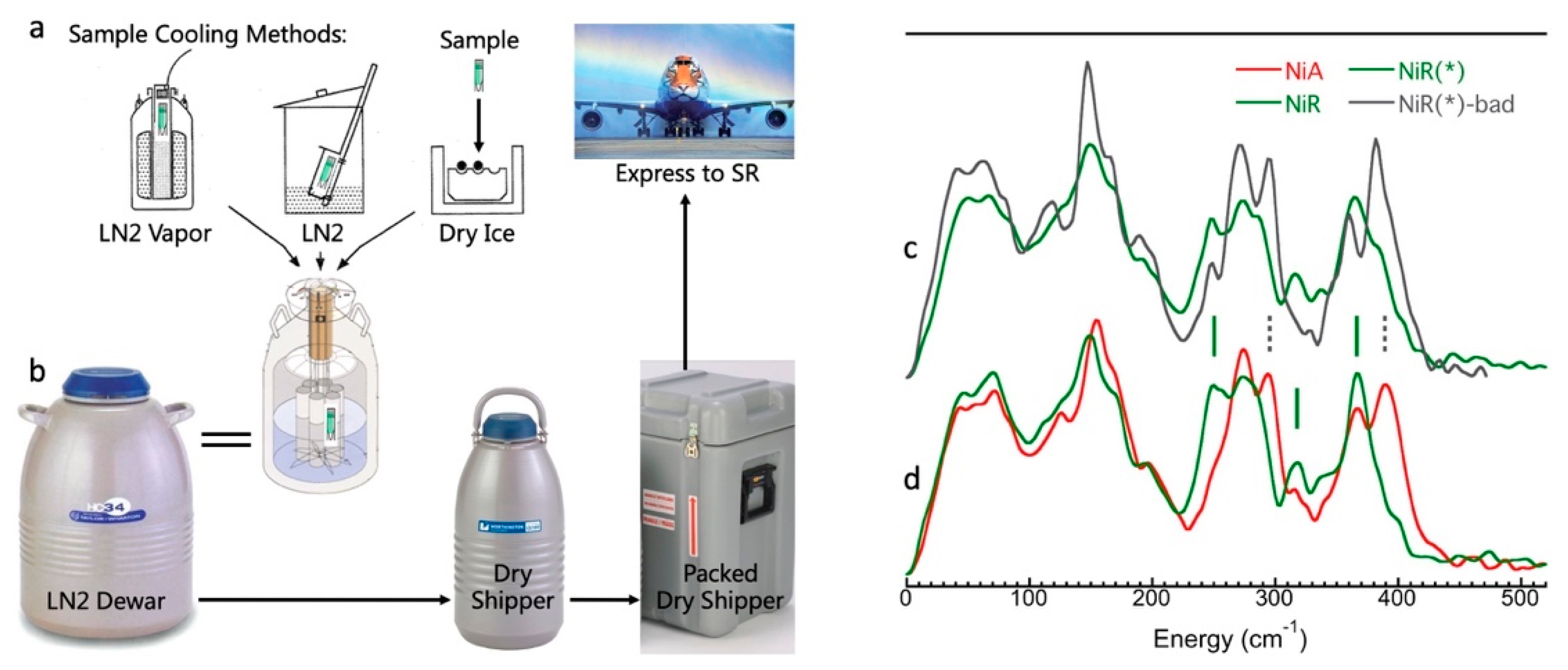
3.4. Making and Monitoring Samples
3.5. Real Sample Temperatures
3.6. Photolysis and NRVS
4. Experimental Aspects (III): Data Process
4.1. Converting to PVDOS
- sum rule analysis for LM factor, etc.,
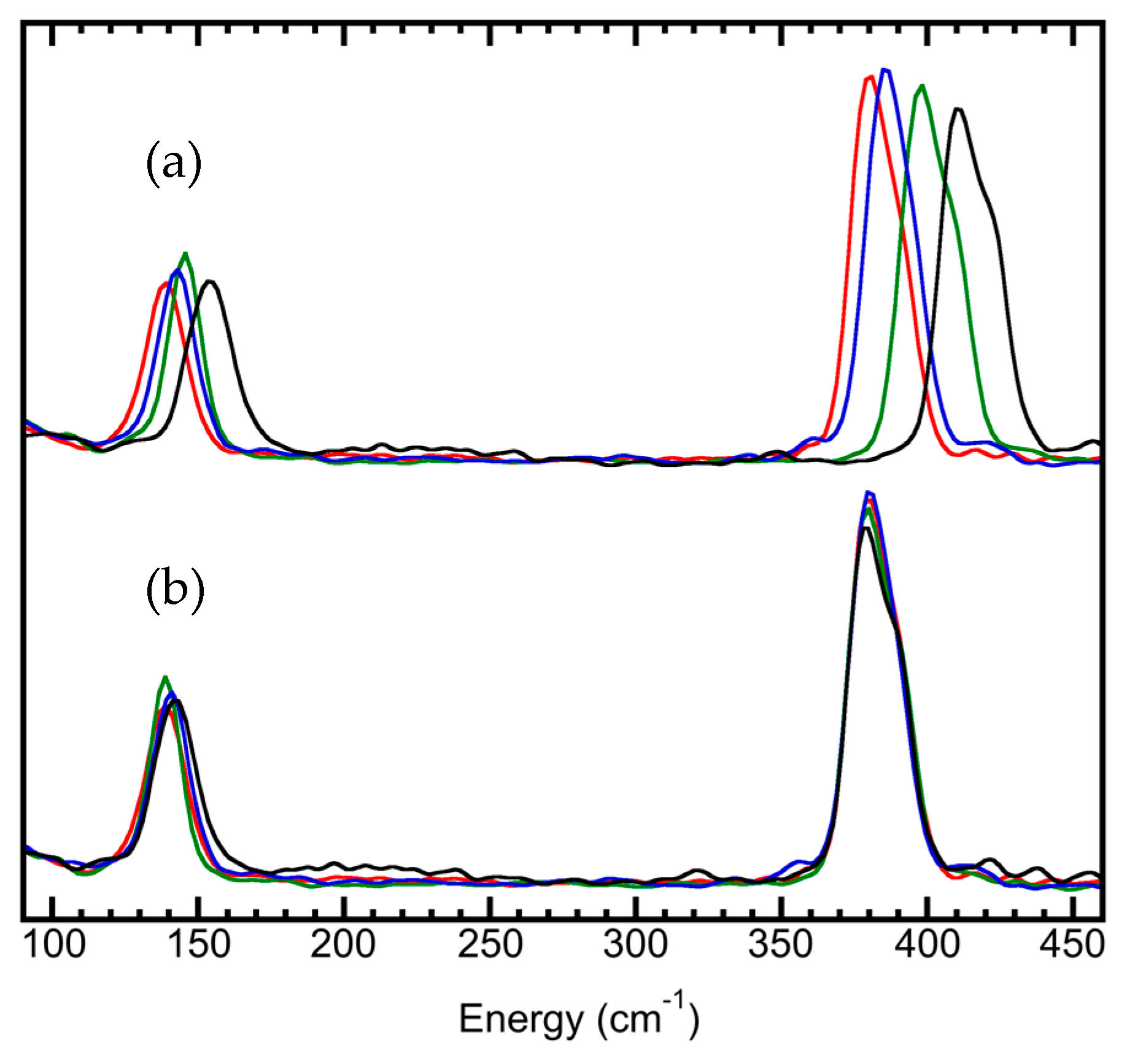
- chemical shifts and isotope shifts,
- interpretation via empirical NMA simulation,
- interpretation via ab initio DFT calculations.
4.2. Understanding Vibrational Modes
4.3. Normal Mode Analysis (NMA)
4.4. Density Function Theory (DFT)
5. NRVS Applications
5.1. Examples in Materials Sciences
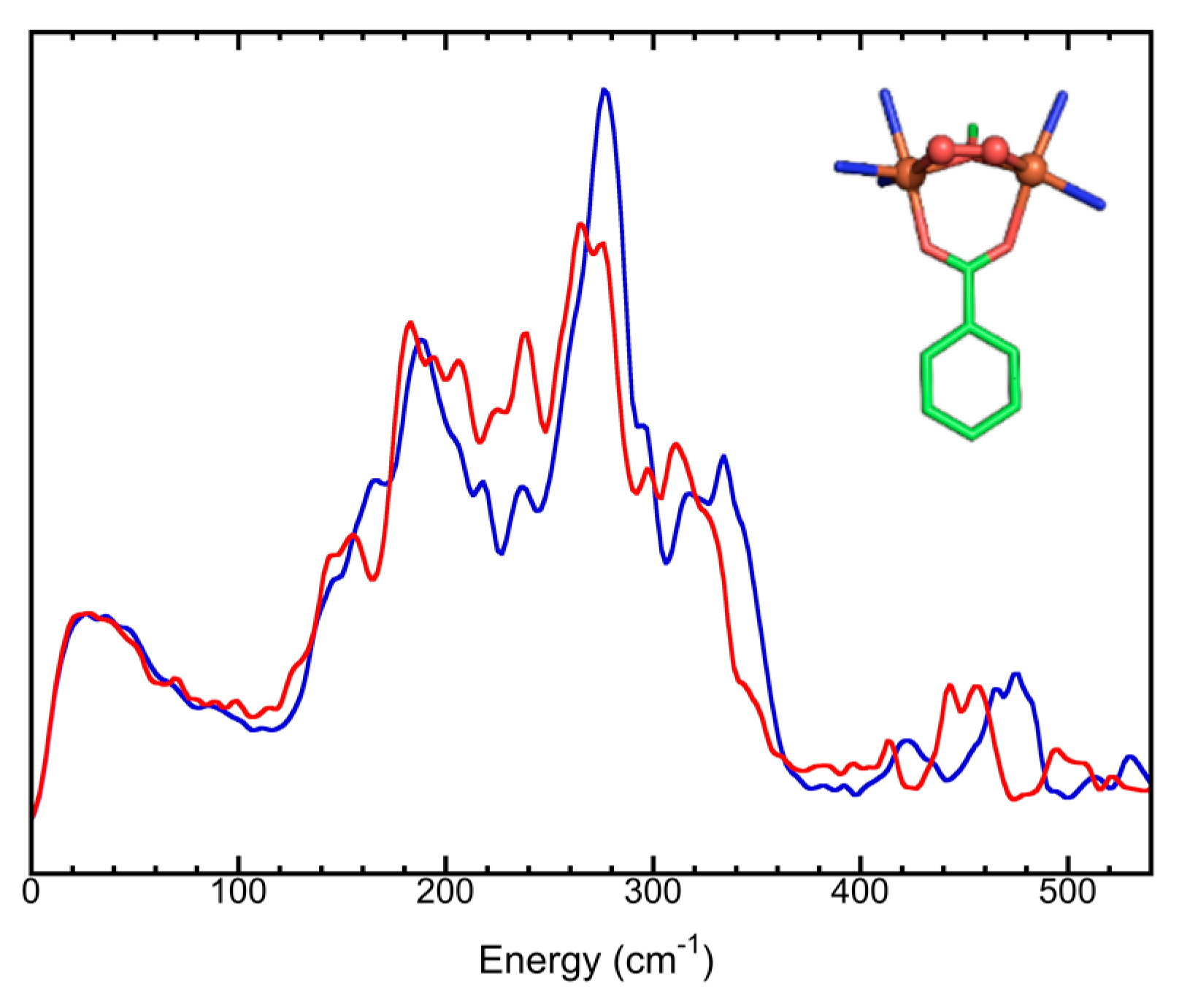
5.2. Simple Chemical Complexes
5.3. A Simple Iron Protein: Solution and Crystal
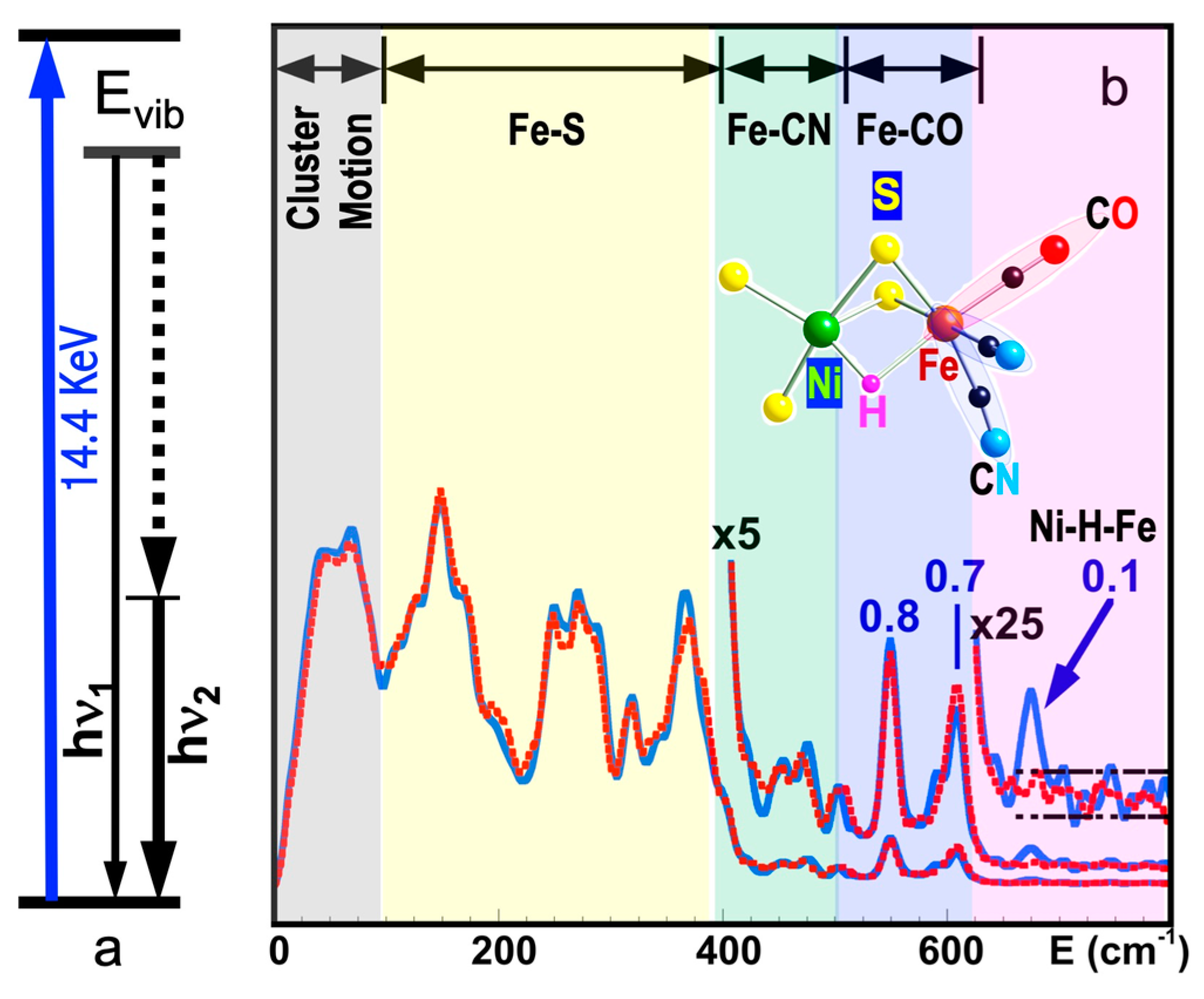
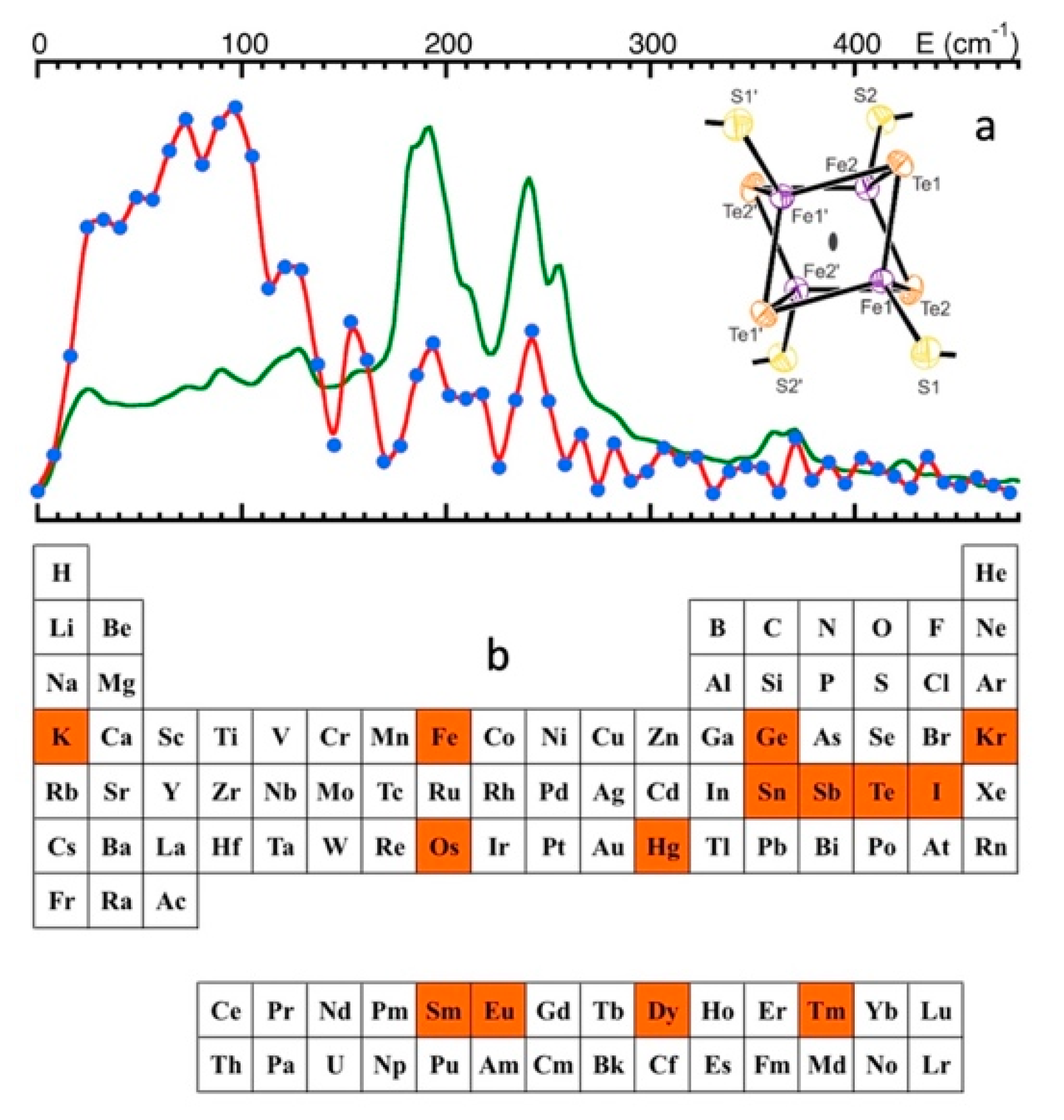
5.4. FeS Cluster Compositions
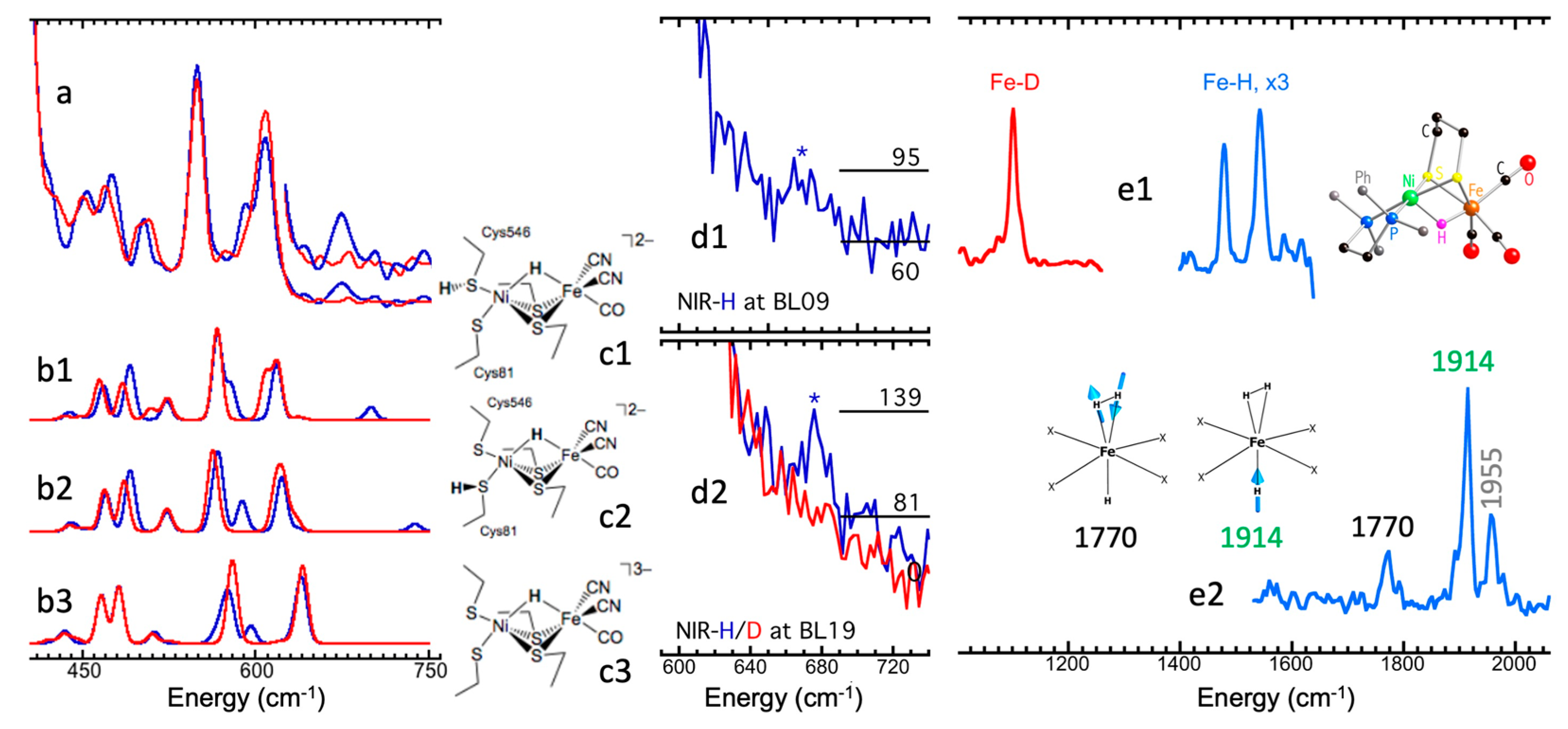
5.5. Fe–H Vibrations and Structures
5.6. Site-Selectivity Labeling
6. Future Perspectives
6.1. Non-57Fe NRVS
6.2. NRVS with Spatial Resolution
6.3. NRVS at Future SR Sources
6.4. More Prospective Applications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seto, M.; Yoda, Y.; Kikuta, S.; Zhang, X.W.; Ando, M. Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett. 1995, 74, 3828–3831. [Google Scholar] [CrossRef] [PubMed]
- Sturhahn, W.; Toellner, T.S.; Alp, E.E.; Zhang, X.; Ando, M.; Yoda, Y.; Kikuta, S.; Seto, M.; Kimball, C.W.; Dabrowski, B. Phonon Density of States Measured by Inelastic Nuclear Resonant Scattering. Phys. Rev. Lett. 1995, 74, 3832–3835. [Google Scholar] [CrossRef] [PubMed]
- Sage, J.T.; Durbin, S.M.; Sturhahn, W.; Wharton, D.C.; Champion, P.M.; Hession, P.; Sutter, J.; Alp, E.E. Long-Range Reactive Dynamics in Myoglobin. Phys. Rev. Lett. 2001, 86, 4966–4969. [Google Scholar] [CrossRef] [PubMed]
- Chumakov, A.I.; Rüffer, R.; Grünsteudel, H.F.; Grübel, G.; Metge, J.; Leupold, O.; Goodwin, H.A. Energy Dependence of Nuclear Recoil Measured with Incoherent Nuclear Scattering of Synchrotron Radiation. EPL (Europhys. Lett.) 1995, 30, 427–432. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, H.; George, S.; Smith, M.C.; Adams, M.W.W.; Jenney, F.E.; Sturhahn, W.; Alp, E.E.; Zhao, J.; Yoda, Y.; et al. Normal Mode Analysis of Pyrococcus furiosus Rubredoxin via Nuclear Resonance Vibrational Spectroscopy (NRVS) and Resonance Raman Spectroscopy. J. Am. Chem. Soc. 2005, 127, 14596–14606. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Smith, M.C.; Newton, W.; Case, D.A.; George, S.; Wang, H.; Sturhahn, W.; Alp, E.; Zhao, J.; Yoda, Y.; et al. How Nitrogenase Shakes—Initial Information about P-Cluster and FeMo-cofactor Normal Modes from Nuclear Resonance Vibrational Spectroscopy (NRVS). J. Am. Chem. Soc. 2006, 128, 7608–7612. [Google Scholar] [CrossRef]
- Ogata, H.; Krämer, T.; Wang, H.; Schilter, D.; Pelmentschikov, V.; Van Gastel, M.; Neese, F.; Rauchfuss, T.B.; Gee, L.B.; Scott, A.D.; et al. Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy. Nat. Commun. 2015, 6, 7890. [Google Scholar] [CrossRef]
- Wang, H.; Yoda, Y.; Ogata, H.; Tanaka, Y.; Lubitz, W. A strenuous experimental journey searching for spectroscopic evidence of a bridging nickel-iron-hydride in [NiFe] hydrogenase. J. Synchrotron Radiat. 2015, 22, 1334–1344. [Google Scholar] [CrossRef]
- Kamali, S.; Wang, H.; Mitra, D.; Ogata, H.; Lubitz, W.; Manor, B.C.; Rauchfuss, T.B.; Byrne, D.; Bonnefoy, V.; Jenney, F.E., Jr.; et al. Observation of the Fe-CN and Fe-CO Vibrations in the Active Site of [NiFe] Hydrogenase by Nuclear Resonance Vibrational Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 724–728. [Google Scholar] [CrossRef]
- Alp, E.E.; Sturhahn, W.; Toellner, T.S.; Zhao, J.; Hu, M.; Brown, D.E. Vibrational Dynamics Studies by Nuclear Resonant Inelastic X-Ray Scattering. Hyperfine Interact. 2002, 144, 3–20. [Google Scholar] [CrossRef]
- Bi, W.; Zhao, J.; Lin, J.-F.; Jia, Q.; Hu, M.Y.; Jin, C.; Ferry, R.; Yang, W.; Struzhkin, V.; Alp, E.E. Nuclear resonant inelastic X-ray scattering at high pressure and low temperature. J. Synchrotron Radiat. 2015, 22, 760–765. [Google Scholar] [CrossRef]
- Yoda, Y. X-ray beam properties available at the nuclear resonant scattering beamline at Spring-8. Hyperfine Interact. 2019, 240, 72. [Google Scholar] [CrossRef]
- Lin, J.-F.; Sturhahn, W.; Zhao, J.; Shen, G.; Mao, H.-k.; Hemley, R.J. Chapter 19—Nuclear Resonant Inelastic X-Ray Scattering and Synchrotron Mössbauer Spectroscopy with Laser-Heated Diamond Anvil Cells. In Advances in High-Pressure Technology for Geo-Physical Applications; Chen, J., Wang, Y., Duffy, T.S., Shen, G., Dobrzhinetskaya, L.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 397–411. [Google Scholar]
- Gee, L.B.; Wang, H.X.; Cramer, S.P. 9. Nuclear Resonance Vibrational Spectroscopy. In Bioorganometallic Chemistry; Weigand, W., Apfel, U.-P., Eds.; De Gruyter: Berlin, Germany, 2020; pp. 353–394. [Google Scholar]
- Aldon, L.; Perea, A.; Womes, M.; Ionica-Bousquet, C.M.; Jumas, J.C. Determination of the Lamb-Mössbauer factors of LiFePO4 and FePO4 for electrochemical in situ and operando measurements in Li-ion batteries. J. Solid State Chem. 2010, 183, 218–222. [Google Scholar] [CrossRef]
- Chumakov, A.I.; Rüffer, R.; Leupold, O.; Sergueev, I. Insight to Dynamics of Molecules with Nuclear Inelastic Scattering. Struct. Chem. 2003, 14, 109–119. [Google Scholar] [CrossRef]
- Sturhahn, W. CONUSS and PHOENIX: Evaluation of nuclear resonant scattering data. Hyperfine Interact. 2000, 125, 149–172. [Google Scholar] [CrossRef]
- Mössbauer, R.L. Kernresonanzfluoreszenz von Gammastrahlung in Ir191. Eur. Phys. J. A 1958, 151, 124–143. [Google Scholar] [CrossRef]
- Mössbauer, R.L. Kernresonanzabsorption von γ-Strahlung in Ir191. Z. Nat. A 1959, 14, 211–216. [Google Scholar] [CrossRef]
- Singwi, K.S.; Sjölander, A. Resonance Absorption of Nuclear Gamma Rays and the Dynamics of Atomic Motions. Phys. Rev. 1960, 120, 1093–1102. [Google Scholar] [CrossRef]
- Visscher, W.M. Study of lattice vibrations by resonance absorption of nuclear gamma rays. Ann. Phys. 1960, 9, 194–210. [Google Scholar] [CrossRef]
- Cramer, S.P. X-ray Spectroscopy with Synchrotron Radiation: Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Willmott, P. An Introduction to Synchrotron Radiation: Techniques and Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Ruby, S.L. Mössbauer Experiments Without Conventional Sources. J. Phys. Colloq. 1974, 35, C6–C209. [Google Scholar] [CrossRef]
- Gerdau, E.; Rüffer, R.; Winkler, H.; Tolksdorf, W.; Klages, P.; Hannon, J.P. Nuclear Bragg diffraction of synchrotron radiation in yttrium iron garnet. Phys. Rev. Lett. 1985, 54, 835–838. [Google Scholar] [CrossRef]
- Achterhold, K.; Keppler, C.; Van Bürck, U.; Potzel, W.; Schindelmann, P.; Knapp, E.-W.; Melchers, B.; Chumakov, A.I.; Baron, A.Q.R.; Rüffer, R.; et al. Temperature dependent inelastic X-ray scattering of synchrotron radiation on myoglobin analyzed by the Mössbauer effect. Eur. Biophys. J. 1996, 25, 43–46. [Google Scholar] [CrossRef]
- Keppler, C.; Achterhold, K.; Ostermann, A.; Van Bürck, U.; Potzel, W.; Chumakov, A.I.; Baron, A.Q.R.; Rüffer, R.; Parak, F. Determination of the phonon spectrum of iron in myoglobin using inelastic X-ray scattering of synchrotron radiation. Eur. Biophys. J. 1997, 25, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, W.R.; Li, J.; Sage, J.T. What Can Be Learned from Nuclear Resonance Vibrational Spectroscopy: Vibrational Dynamics and Hemes. Chem. Rev. 2017, 117, 12532–12563. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, L.; Wang, H.; Horch, M.; Gee, L.; Yoda, Y.; Tanaka, Y.; Zebger, I.; Lenz, O.; Cramer, S.P. Nuclear resonance vibrational spectroscopy reveals the FeS cluster composition and active site vibrational properties of an O2-tolerant NAD+-reducing [NiFe] hydrogenase. Chem. Sci. 2015, 6, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Kuchenreuther, J.M.; Guo, Y.; Wang, H.; Myers, W.K.; George, S.; Boyke, C.A.; Yoda, Y.; Alp, E.E.; Zhao, J.; Britt, R.D.; et al. Nuclear Resonance Vibrational Spectroscopy and Electron Paramagnetic Resonance Spectroscopy of 57Fe-Enriched [FeFe] Hydrogenase Indicate Stepwise Assembly of the H-Cluster. Biochemistry 2013, 52, 818–826. [Google Scholar] [CrossRef][Green Version]
- Gilbert-Wilson, R.; Siebel, J.F.; Adamska-Venkatesh, A.; Pham, C.C.; Reijerse, E.; Wang, H.; Cramer, S.P.; Lubitz, W.; Rauchfuss, T.B. Spectroscopic Investigations of [FeFe] Hydrogenase Maturated with [57Fe2(adt)(CN)2(CO)4]2−. J. Am. Chem. Soc. 2015, 137, 8998–9005. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, H.; Xiao, Y.; Vogt, S.; Thauer, R.K.; Shima, S.; Volkers, P.I.; Rauchfuss, T.B.; Pelmentschikov, V.; Case, D.A.; et al. Characterization of the Fe Site in Iron−Sulfur Cluster-Free Hydrogenase (Hmd) and of a Model Compound via Nuclear Resonance Vibrational Spectroscopy (NRVS). Inorg. Chem. 2008, 47, 3969–3977. [Google Scholar] [CrossRef]
- Scott, A.D.; Pelmentschikov, V.; Guo, Y.; Yan, L.; Wang, H.; George, S.; Dapper, C.H.; Newton, W.E.; Yoda, Y.; Tanaka, Y.; et al. Structural Characterization of CO-Inhibited Mo-Nitrogenase by Combined Application of Nuclear Resonance Vibrational Spectroscopy, Extended X-ray Absorption Fine Structure, and Density Functional Theory: New Insights into the Effects of CO Binding and the Role of the Interstitial Atom. J. Am. Chem. Soc. 2014, 136, 15942–15954. [Google Scholar] [CrossRef]
- Wong, S.D.; Srnec, M.; Matthews, M.L.; Liu, L.V.; Kwak, Y.; Park, K.; Iii, C.B.B.; Alp, E.E.; Zhao, J.; Yoda, Y.; et al. Elucidation of the Fe(iv)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nat. Cell Biol. 2013, 499, 320–323. [Google Scholar] [CrossRef]
- Serrano, P.N.; Wang, H.; Crack, J.C.; Prior, C.; Hutchings, M.; Thomson, A.J.; Kamali, S.; Yoda, Y.; Zhao, J.; Hu, M.Y.; et al. Nitrosylation of Nitric-Oxide-Sensing Regulatory Proteins Containing [4Fe-4S] Clusters Gives Rise to Multiple Iron–Nitrosyl Complexes. Angew. Chem. Int. Ed. 2016, 55, 14575–14579. [Google Scholar] [CrossRef]
- Mitra, D.; Pelmentschikov, V.; Guo, Y.; Case, D.A.; Wang, H.; Dong, W.; Tan, M.-L.; Ichiye, T.; Jenney, F.E.; Adams, M.W.W.; et al. Dynamics of the [4Fe-4S] Cluster inPyrococcus furiosusD14C Ferredoxin via Nuclear Resonance Vibrational and Resonance Raman Spectroscopies, Force Field Simulations, and Density Functional Theory Calculations. Biochemistry 2011, 50, 5220–5235. [Google Scholar] [CrossRef]
- Xiao, Y.; Tan, M.-L.; Ichiye, T.; Wang, H.; Guo, Y.; Smith, M.C.; Meyer, J.; Sturhahn, W.; Alp, E.E.; Zhao, J.; et al. Dynamics ofRhodobacter capsulatus [2Fe-2S] Ferredoxin VI andAquifex aeolicusFerredoxin 5 via Nuclear Resonance Vibrational Spectroscopy (NRVS) and Resonance Raman Spectroscopy. Biochemistry 2008, 47, 6612–6627. [Google Scholar] [CrossRef] [PubMed]
- Lübbers, R.; Pleines, M.; Hesse, H.-J.; Wortmann, J.; Grünsteudel, H.F.; Rüffer, R.; Leupold, O.; Zukrowski, J. Magnetism under high pressure studied by 57Fe and 151Eu nuclear scattering of syn-chrotron radiation. Hyperfine Interact. 1999, 120, 49–58. [Google Scholar] [CrossRef]
- Wittkamp, F.; Mishra, N.; Wang, H.; Wille, H.-C.; Steinbrügge, R.; Kaupp, M.; Cramer, S.P.; Apfel, U.-P.; Pelmenschikov, V. Insights from 125Te and 57Fe nuclear resonance vibrational spectroscopy: A [4Fe-4Te] cluster from two points of view. Chem. Sci. 2019, 10, 7535–7541. [Google Scholar] [CrossRef] [PubMed]
- Alp, E.E.; Mooney, T.M.; Toellner, T.; Sturhahn, W.; Witthoff, E.; Röhlsberger, R.; Gerdau, E.; Homma, H.; Kentjana, M. Time resolved nuclear resonant scattering fromSn119nuclei using synchrotron radiation. Phys. Rev. Lett. 1993, 70, 3351–3354. [Google Scholar] [CrossRef] [PubMed]
- Vértes, A.; Klenesár, Z.; Vanko, G.; Marek, T.; Süvegh, K.; Homonnay, Z.; Kuzmann, E. Nuclear Techniques in the Elucidation of Chemical Structure. J. Radioanal. Nucl. Chem. 2000, 243, 241–253. [Google Scholar] [CrossRef]
- Tinberg, C.E.; Tonzetich, Z.J.; Wang, H.; Do, L.H.; Yoda, Y.; Cramer, S.P.; Lippard, S.J. Characterization of Iron Dinitrosyl Species Formed in the Reaction of Nitric Oxide with a Biological Rieske Center. J. Am. Chem. Soc. 2010, 132, 18168–18176. [Google Scholar] [CrossRef]
- Fontecilla-Camps, J.C.; Volbeda, A.; Cavazza, C.; Nicolet, Y. Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. (Chem. Rev. 2007, 107, 4273−4303. Published on the Web September 13, 2007.). Chem. Rev. 2007, 107, 5411. [Google Scholar] [CrossRef]
- Pelmentschikov, V.; Birrell, J.A.; Pham, C.; Mishra, N.; Wang, H.; Sommer, C.; Reijerse, E.; Richers, C.P.; Tamasaku, K.; Yoda, Y.; et al. Reaction Coordinate Leading to H2Production in [FeFe]-Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory. J. Am. Chem. Soc. 2017, 139, 16894–16902. [Google Scholar] [CrossRef]
- Pham, C.C.; Mulder, D.W.; Pelmenschikov, V.; King, P.W.; Ratzloff, M.W.; Wang, H.; Mishra, N.; Alp, E.E.; Zhao, J.; Michael, Y.H.; et al. Terminal Hydride Species in [FeFe]-Hydrogenases are Vibrationally Coupled to the Active Site Environment. Ang. Chem. 2018, 130, 10765–10769. [Google Scholar] [CrossRef]
- Wang, H.; Alp, E.E.; Yoda, Y.; Cramer, S.P. A Practical Guide for Nuclear Resonance Vibrational Spectroscopy (NRVS) of Biochemical Samples and Model Compounds. Methods Mol. Biol. 2014, 1122, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Reijerse, E.J.; Pham, C.; Pelmentschikov, V.; Gilbert-Wilson, R.; Adamska-Venkatesh, A.; Siebel, J.F.; Gee, L.B.; Yoda, Y.; Tamasaku, K.; Lubitz, W.; et al. Direct Observation of an Iron-Bound Terminal Hydride in [FeFe]-Hydrogenase by Nuclear Resonance Vibrational Spectroscopy. J. Am. Chem. Soc. 2017, 139, 4306–4309. [Google Scholar] [CrossRef]
- Smith, M.C.; Xiao, Y.; Wang, H.; George, S.; Coucouvanis, D.; Koutmos, M.; Sturhahn, W.; Alp, E.E.; Zhao, J.; Cramer, S.P. Normal-Mode Analysis of FeCl4-and Fe2S2Cl42-via Vibrational Mössbauer, Resonance Raman, and FT-IR Spectroscopies. Inorg. Chem. 2005, 44, 5562–5570. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Wang, H.; Olmstead, M.M.; Fettinger, J.C.; Nix, J.; Uchiyama, H.; Tsutsui, S.; Baron, A.Q.R.; Dowty, E.; Cramer, S.P. Inelastic X-ray Scattering of a Transition-Metal Complex (FeCl4–): Vibrational Spectroscopy for All Normal Modes. Inorg. Chem. 2013, 52, 6767–6769. [Google Scholar] [CrossRef][Green Version]
- Baron, A.Q.R. High-Resolution Inelastic X-Ray Scattering I: Context, Spectrometers, Samples, and Superconductors. In Syn-chrotron Light Sources and Free-Electron Lasers: Accelerator Physics, Instrumentation and Science Applications; Jaeschke, E.J., Khan, S., Schneider, J.R., Hastings, J.B., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1643–1719. [Google Scholar]
- Wang, H.; Chen, X.; Weiner, B.R. Laser photodissociation dynamics of thionyl chloride: Concerted and stepwise cleavage of S-Cl bonds. J. Phys. Chem. 1993, 97, 12260–12268. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Weiner, B.R. SO(X3Σ−) production from the 193 nm laser photolysis of thionyl fluoride. Chem. Phys. Lett. 1993, 216, 537–543. [Google Scholar] [CrossRef]
- He, M.; Wang, H.; Weiner, B.R. Production and laser-induced fluorescence spectrum of aluminum sulfide. Chem. Phys. Lett. 1993, 204, 563–566. [Google Scholar] [CrossRef]
- Wang, H.; Salzberg, A.P.; Weiner, B.R. Laser ablation of aluminum at 193, 248, and 351 nm. Appl. Phys. Lett. 1991, 59, 935–937. [Google Scholar] [CrossRef]
- Hara, T.; Yabashi, M.; Tanaka, T.; Bizen, T.; Goto, S.; MaréchalX, M.; Seike, T.; Tamasaku, K.; Ishikawa, T.; Kitamura, H. The brightest x-ray source: A very long undulator at SPring-8. Rev. Sci. Instrum. 2002, 73, 1125–1128. [Google Scholar] [CrossRef]
- Gu, W.; Jacquamet, L.; Patil, D.; Wang, H.-X.; Evans, D.; Smith, M.; Millar, M.; Koch, S.; Eichhorn, D.; Latimer, M.; et al. Refinement of the nickel site structure in Desulfovibrio gigas hydrogenase using range-extended EXAFS spectroscopy. J. Inorg. Biochem. 2003, 93, 41–51. [Google Scholar] [CrossRef]
- Toellner, T. Monochromatization of synchrotron radiation for nuclear resonant scattering experiments. Hyperfine Interact. 2000, 125, 3–28. [Google Scholar] [CrossRef]
- Wang, H.; Yoda, Y.; Dong, W.; Huang, S.D. Energy calibration issues in nuclear resonant vibrational spectroscopy: Observing small spectral shifts and making fast calibrations. J. Synchrotron Radiat. 2013, 20, 683–690. [Google Scholar] [CrossRef]
- Toellner, T.S.; Collins, J.L.; Goetze, K.; Hu, M.Y.; Preissner, C.; Trakhtenberg, E.; Yan, L. Ultra-stable sub-meV monochromator for hard X-rays. J. Synchrotron Radiat. 2015, 22, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y.; Rai, S.B. Blue and Red Luminescence from Europium-Doped Barium TetraBorate Crystals. J. Am. Ceram. Soc. 2010, 93, 727–731. [Google Scholar] [CrossRef]
- Vicentini, G.; Zinner, L.; Zukerman-Schpector, J.; Zinner, K. Luminescence and structure of europium compounds. Coord. Chem. Rev. 2000, 196, 353–382. [Google Scholar] [CrossRef]
- Yoda, Y.; Imai, Y.; Kobayashi, H.; Goto, S.; Takeshita, K.; Seto, M. Upgrade of the nuclear resonant scattering beamline, BL09XU in SPring-8. ICAME 2011 2013, 206, 379–382. [Google Scholar] [CrossRef]
- Kujala, N.; Marathe, S.; Shu, D.; Shi, B.; Qian, J.; Maxey, E.; Finney, L.; Macrander, A.; Assoufid, L. Kirkpatrick–Baez mirrors to focus hard X-rays in two dimensions as fabricated, tested and installed at the Advanced Photon Source. J. Synchrotron Radiat. 2014, 21, 662–668. [Google Scholar] [CrossRef]
- Birrell, J.A.; Pelmenschikov, V.; Mishra, N.; Wang, H.; Yoda, Y.; Tamasaku, K.; Rauchfuss, T.B.; Cramer, S.P.; Lubitz, W.; DeBeer, S. Spectroscopic and Computational Evidence that [FeFe] Hydrogenases Operate Exclusively with CO-Bridged Intermediates. J. Am. Chem. Soc. 2020, 142, 222–232. [Google Scholar] [CrossRef]
- Gee, L.B.; Pelmenschikov, V.; Wang, H.; Mishra, N.; Liu, Y.-C.; Yoda, Y.; Tamasaku, K.; Chiang, M.-H.; Cramer, S.P. Vibrational characterization of a diiron bridging hydride complex—A model for hydrogen catalysis. Chem. Sci. 2020, 11, 5487–5493. [Google Scholar] [CrossRef]
- Caserta, G.; Pelmenschikov, V.; Lorent, C.; Waffo, A.F.T.; Katz, S.; Lauterbach, L.; Schoknecht, J.; Wang, H.; Yoda, Y.; Tamasaku, K.; et al. Hydroxy-bridged resting states of a [NiFe]-hydrogenase unraveled by cryogenic vibrational spectroscopy and DFT computations. Chem. Sci. 2021, 12, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.-H.; Pelmenschikov, V.; Gee, L.B.; Liu, Y.-C.; Hsieh, C.-C.; Wang, H.; Yoda, Y.; Matsuura, H.; Li, L.; Cramer, S.P. High-Frequency Fe–H and Fe–H2 Modes in a trans-Fe(η2-H2)(H) Complex: A Speed Record for Nuclear Resonance Vibrational Spectroscopy. Inorg. Chem. 2021, 60, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Pelmenschikov, V.; Birrell, J.A.; Gee, L.B.; Richers, C.P.; Reijerse, E.J.; Wang, H.; Arragain, S.; Mishra, N.; Yoda, Y.; Matsuura, H.; et al. Vibrational Perturbation of the [FeFe] Hydrogenase H-Cluster Revealed by 13C2H-ADT Labeling. J. Am.Chem. Soc. 2021. [Google Scholar] [CrossRef]
- Baron, A. Report on the X-ray efficiency and time response of a 1 cm2 reach through avalanche diode. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 1995, 352, 665–667. [Google Scholar] [CrossRef]
- Kishimoto, S. High time resolution x-ray measurements with an avalanche photodiode detector. Rev. Sci. Instrum. 1992, 63, 824–827. [Google Scholar] [CrossRef]
- Wang, H.; Ralston, C.Y.; Patil, D.S.; Jones, R.M.; Gu, W.; Verhagen, M.; Adams, M.; Ge, P.; Riordan, C.; Marganian, C.A.; et al. Nickel L-Edge Soft X-ray Spectroscopy of Nickel−Iron Hydrogenases and Model CompoundsEvidence for High-Spin Nickel(II) in the Active Enzyme. J. Am. Chem. Soc. 2000, 122, 10544–10552. [Google Scholar] [CrossRef]
- Funk, T.; Deb, A.; George, S.J.; Wang, H.; Cramer, S.P. X-ray magnetic circular dichroism—a high energy probe of magnetic properties. Coord. Chem. Rev. 2005, 249, 3–30. [Google Scholar] [CrossRef]
- Cody, G.D.; Alexander, C.M.O.D.; Wirick, S. Approaches to establishing the chemical structure of extraterrestrial organic solids. In Proceedings of the Workshop on Cometary Dust in Astrophysics, Crystal Mountain, Washington, DC, USA, 10–15 August 2003; pp. 60571–60572. [Google Scholar]
- Gee, L.; Lin, C.-Y.; Jenney, F.E.; Adams, M.W.; Yoda, Y.; Masuda, R.; Saito, M.; Kobayashi, Y.; Tamasaku, K.; Lerche, M.; et al. Synchrotron-based Nickel Mössbauer Spectroscopy. Inorg. Chem. 2016, 55, 6866–6872. [Google Scholar] [CrossRef]
- Wang, H.; Yoda, Y.; Kamali, S.; Zhou, Z.-H.; Cramer, S.P. Real sample temperature: A critical issue in the experiments of nuclear resonant vibrational spectroscopy on biological samples. J. Synchrotron Radiat. 2012, 19, 257–263. [Google Scholar] [CrossRef]
- Owen, R.; Rudino-Pinera, E.; Garman, E. Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc. Natl. Acad. Sci. USA 2006, 103, 4912–4917. [Google Scholar] [CrossRef]
- Garman, E.F.; Nave, C.J. Radiation damage in protein crystals examined under various conditions by different methods. Synchrotron Rad. 2009, 16, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Kohn, V.; Chumakov, A. DOS: Evaluation of phonon density of states from nuclear resonant inelastic absorption. Hyperfine Interact. 2000, 125, 205–221. [Google Scholar] [CrossRef]
- Gee, L.B. Spectra Tools. Available online: http://spectra.tools (accessed on 30 June 2021).
- Dowty, E. Vibrational interactions of tetrahedra in silicate glasses and crystals. Phys. Chem. Miner. 1987, 14, 122–138. [Google Scholar] [CrossRef]
- Tan, M.-L.; Bizzarri, A.R.; Xiao, Y.; Cannistraro, S.; Ichiye, T.; Manzoni, C.; Cerullo, G.; Adams, M.W.; Jenney, F.E.; Cramer, S.P. Observation of terahertz vibrations in Pyrococcus furiosus rubredoxin via impulsive coherent vibrational spectroscopy and nuclear resonance vibrational spectroscopy—Interpretation by molecular mechanics. J. Inorg. Biochem. 2007, 101, 375–384. [Google Scholar] [CrossRef]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Phys. 1927, 46, 455–472. [Google Scholar] [CrossRef]
- Dirac, P.A.M. Quantum mechanics of many-electron systems. In Proceedings of the Royal Society of London. A. Mathematical and Physical Sciences; The Royal Society: London, UK, 1929; Volume 123, pp. 714–733. [Google Scholar]
- Sawatzky, G.A.; Van Der Woude, F.; Morrish, A.H. Recoilless-Fraction Ratios for Fe57 in Octahedral and Tetrahedral Sites of a Spinel and a Garnet. Phys. Rev. 1969, 183, 383–386. [Google Scholar] [CrossRef]
- Rykov, A.I.; Nomura, K.; Sawada, T.; Mitsui, T.; Seto, M.; Tamegai, T.; Tokunaga, M. Phonon density of states inSr2FeCoO6−δandBaSrFeCoO6−δ: Effects induced by magnetic order and transport coherence. Phys. Rev. B 2003, 68, 224401. [Google Scholar] [CrossRef]
- Nomura, K.; Tokumistu, K.; Hayakawa, T.; Homonnay, Z. The Influence of Mechanical Treatment on the Absorption of CO2 by Perovskite Oxides. J. Radioanal. Nucl. Chem. 2000, 246, 69–77. [Google Scholar] [CrossRef]
- Zhu, B.; Fan, L.; Deng, H.; He, Y.; Afzal, M.; Dong, W.; Yaqub, A.; Janjua, N.K. LiNiFe-based layered structure oxide and composite for advanced single layer fuel cells. J. Power Sources 2016, 316, 37–43. [Google Scholar] [CrossRef]
- Błachowski, A.; Jasek, A.K.; Komędera, K.; Pierzga, A.; Ruebenbauer, K.; Żukrowski, J.; Rang, T.; Northwood, D.; Brebbia, C.A. Mössbauer Studies of Iron-Based Superconductors. In Materials and Contact Characterisation VIII; Witpress Ltd.: Billerica, MA, USA, 2017; Volume 1, pp. 151–160. [Google Scholar]
- Waldron, R.D. Infrared Spectra of Ferrites. Phys. Rev. 1955, 99, 1727–1735. [Google Scholar] [CrossRef]
- Eissa, N.A.; Bahgat, A.A. Mössbauer effect studies of the thermal properties in Ni-ferrite. Phys. Status Solidi A 1974, 21, 317–321. [Google Scholar] [CrossRef]
- Hu, M.Y.; Sturhahn, W.; Toellner, T.S.; Mannheim, P.D.; Brown, D.E.; Zhao, J.; Alp, E.E. Measuring velocity of sound with nuclear resonant inelastic x-ray scattering. Phys. Rev. B 2003, 67, 67. [Google Scholar] [CrossRef]
- Lin, J.-F.; Tse, J.S.; Alp, E.E.; Zhao, J.; Lerche, M.; Sturhahn, W.; Xiao, Y.; Chow, P. Phonon density of states of Fe2O3across high-pressure structural and electronic transitions. Phys. Rev. B 2011, 84, 064424. [Google Scholar] [CrossRef]
- Do, L.H.; Wang, H.; Tinberg, C.E.; Dowty, E.; Yoda, Y.; Cramer, S.P.; Lippard, S.J. Characterization of a synthetic peroxodiiron(iii) protein model complex by nuclear resonance vibrational spectroscopy. Chem. Commun. 2011, 47, 10945–10947. [Google Scholar] [CrossRef][Green Version]
- Dong, Y.; Menage, S.; Brennan, B.A.; Elgren, T.E.; Jang, H.G.; Pearce, L.; Que, L. Dioxygen binding to diferrous centers. Models for diiron-oxo proteins. J. Am. Chem. Soc. 1993, 115, 1851–1859. [Google Scholar] [CrossRef]
- Park, K.; Tsugawa, T.; Furutachi, H.; Kwak, Y.; Liu, L.V.; Wong, S.D.; Yoda, Y.; Kobayashi, Y.; Saito, M.; Kurokuzu, M.; et al. Nuclear Resonance Vibrational Spectroscopy and DFT study of Peroxo-Bridged Biferric Complexes: Structural Insight into Peroxo Intermediates of Binuclear Non-heme Iron Enzymes. Angew. Chem. Int. Ed. 2013, 52, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Li, N.; Kwak, Y.; Srnec, M.; Bell, C.B.; Liu, L.V.; Wong, S.D.; Yoda, Y.; Kitao, S.; Seto, M.; et al. Peroxide Activation for Electrophilic Reactivity by the Binuclear Non-heme Iron Enzyme AurF. J. Am. Chem. Soc. 2017, 139, 7062–7070. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Brecht, E.; Aznavour, K.; Nix, J.C.; Xiao, Y.; Wang, H.; George, S.; Bau, R.; Keable, S.; Peters, J.; et al. Nuclear resonance vibrational spectroscopy (NRVS) of rubredoxin and MoFe protein crystals. Hyperfine Interact. 2013, 222, 77–90. [Google Scholar] [CrossRef]
- Lehnert, N.; Sage, J.T.; Silvernail, N.; Scheidt, W.R.; Alp, E.E.; Sturhahn, W.; Zhao, J. Oriented Single-Crystal Nuclear Resonance Vibrational Spectroscopy of [Fe(TPP)(MI)(NO)]: Quantitative Assessment of the trans Effect of NO. Inorg. Chem. 2010, 49, 7197–7215. [Google Scholar] [CrossRef] [PubMed]
- Horch, M.; Lauterbach, L.; Lenz, O.; Hildebrandt, P.; Zebger, I. NAD(H)-coupled hydrogen cycling—Structure-function relationships of bidirectional [NiFe] hydrogenases. FEBS Lett. 2012, 586, 545–556. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Guo, Y.; Wang, H.; Cramer, S.P.; Case, D.A. Fe-H/D stretching and bending modes in nuclear resonant vibrational, Raman and infrared spectroscopies: Comparisons of density functional theory and experiment. Faraday Discuss. 2011, 148, 409–420. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Gee, L.B.; Wang, H.; MacLeod, K.C.; McWilliams, S.F.; Skubi, K.L.; Cramer, S.P.; Holland, P.L. High-Frequency Fe-H Vibrations in a Bridging Hydride Complex Characterized by NRVS and DFT. Angew. Chem. Int. Ed. 2018, 57, 9367–9371. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, P.; Jacquamet, L.; Bergmann, U.; de Groot, F.M.F.; Cramer, S.P. Site-Selective EXAFS in Mixed-Valence Compounds Using High-Resolution Fluorescence Detection: A Study of Iron in Prussian Blue. Inorg. Chem. 2002, 41, 3121–3127. [Google Scholar] [CrossRef] [PubMed]
- Ba, L.A.; Döring, M.; Jamier, V.; Jacob, C. Tellurium: An element with great biological potency and potential. Org. Biomol. Chem. 2010, 8, 4203–4216. [Google Scholar] [CrossRef]
- Braun, A.; Chen, Q. Experimental neutron scattering evidence for proton polaron in hydrated metal oxide proton conductors. Nat. Commun. 2017, 8, 15830. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Chen, Q.; Yelon, A. Hole and Protonic Polarons in Perovskites. Chim. Int. J. Chem. 2019, 73, 936–942. [Google Scholar] [CrossRef]
- Colomban, P.; Slodczyk, A. The structural and dynamics neutron study of proton conductors: Difficulties and improvement procedures in protonated perovskite. Eur. Phys. J. Spéc. Top. 2012, 213, 171–193. [Google Scholar] [CrossRef]
- Spahr, E.J.; Wen, L.; Stavola, M.; Boatner, L.; Feldman, L.C.; Tolk, N.H.; Lüpke, G. Giant Enhancement of Hydrogen Transport in RutileTiO2at Low Temperatures. Phys. Rev. Lett. 2010, 104, 205901. [Google Scholar] [CrossRef]
- Spahr, E.J.; Wen, L.; Stavola, M.; Boatner, L.A.; Feldman, L.C.; Tolk, N.H.; Lüpke, G. Proton Tunneling: A Decay Channel of the O-H Stretch Mode in KTaO3. Phys. Rev. Lett. 2009, 102, 075506. [Google Scholar] [CrossRef]
- Yoshida, Y.; Suzuki, K.; Hayakawa, K.; Yukihira, K.; Soejima, H. Mössbauer spectroscopic microscope. Hyperfine Interact. 2008, 188, 121–126. [Google Scholar] [CrossRef]
- Yan, L.; Zhao, J.; Toellner, T.S.; Divan, R.; Xu, S.; Cai, Z.; Boesenberg, J.S.; Friedrich, J.M.; Cramer, S.P.; Alp, E.E. Exploration of synchrotron Mössbauer microscopy with micrometer resolution: Forward and a new backscattering modality on natural samples. J. Synchrotron Radiat. 2012, 19, 814–820. [Google Scholar] [CrossRef] [PubMed]
- McCammon, C. Mössbauer Spectroscopy with High Spatial Resolution: Spotlight on Geoscience. In Topics in Applied Physics; Springer Science and Business Media LLC: Berlin, Germany, 2021; pp. 221–266. [Google Scholar]
- Fleischer, I.; Klingelhöfer, G.; Schröder, C.; Morris, R.V.; Hahn, M.; Rodionov, D.; Gellert, R.; de Souza, P. Depth selective Mössbauer spectroscopy: Analysis and simulation of 6.4 keV and 14.4 keV spectra obtained from rocks at Gusev Crater, Mars, and layered laboratory samples. J. Geophys. Res. Space Phys. 2008, 113. [Google Scholar] [CrossRef]
- Braun, A.; Wang, H.; Bergmann, U.; Tucker, M.; Gu, W.; Cramer, S.; Cairns, E. Origin of chemical shift of manganese in lithium battery electrode materials—a comparison of hard and soft X-ray techniques. J. Power Sources 2002, 112, 231–235. [Google Scholar] [CrossRef]
- Döring, F.; Robisch, A.; Eberl, C.; Osterhoff, M.; Ruhlandt, A.; Liese, T.; Schlenkrich, F.; Hoffmann, S.; Bartels, M.; Salditt, T.; et al. Sub-5 nm hard x-ray point focusing by a combined Kirkpatrick-Baez mirror and multilayer zone plate. Opt. Express 2013, 21, 19311–19323. [Google Scholar] [CrossRef]
- Ade, H.; Zhang, X.; Cameron, S.; Costello, C.; Kirz, J.; Williams, S. Chemical contrast in X-ray microscopy and spatially resolved XANES spectroscopy of organic specimens. Science 1992, 258, 972–975. [Google Scholar] [CrossRef]
- Eriksson, T.A.; Doeff, M.M. A study of layered lithium manganese oxide cathode materials. J. Power Sources 2003, 119-121, 145–149. [Google Scholar] [CrossRef][Green Version]
- Roth, T.; Leupold, O.; Wille, H.-C.; Rüffer, R.; Quast, K.W.; Röhlsberger, R.; Burkel, E. Coherent nuclear resonant scattering byNi61using the nuclear lighthouse effect. Phys. Rev. B 2005, 71, 140401. [Google Scholar] [CrossRef]
- Shao, W.; Wang, F.; Feng, X.-L.; Oh, C.H. Ionic vibration induced transparency and Autler–Townes splitting. Laser Phys. Lett. 2017, 14, 045203. [Google Scholar] [CrossRef]
- Qin, H.; Ding, M.; Yin, Y. Induced Transparency with Optical Cavities. Adv. Photon. Res. 2020, 1. [Google Scholar] [CrossRef]
- Radeonychev, Y.V.; Khairulin, I.; Vagizov, F.G.; Scully, M.; Kocharovskaya, O. Observation of Acoustically Induced Transparency for γ-Ray Photons. Phys. Rev. Lett. 2020, 124, 163602. [Google Scholar] [CrossRef] [PubMed]
- Khairulin, I.R.; Radeonychev, Y.V.; Antonov, V.A.; Kocharovskaya, O. Acoustically induced transparency for synchrotron hard X-ray photons. Sci. Rep. 2021, 11, 7930. [Google Scholar] [CrossRef] [PubMed]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SR/Beamlines | E (GeV) | Emittance (nm∙rad) | Studied Isotopes |
|---|---|---|---|
| ESRF/18ID | 6 | 4 | 57Fe, 151Eu, 119Sn, 149Sm, 161Dy, 121Sb, 125Te, 129Xe |
| APS/16ID | 7 | 3.1 | 57Fe |
| APS/03ID | 7 | 3.1 | 57Fe, 151Eu, 119Sn, 83Kr, 161Dy |
| SPring-8 /BL09XU | 8 | 3.4 | 181Ta, 57Fe, 151Eu, 149Sm, 119Sn, 40K, 125Te, 121Sb, 61Ni |
| SPring-8 /BL11XU | 8 | 3.4 | 57Fe, 121Sb, 149Sm, 40K, 158Gd |
| SPring-8 /BL19LXU | 8 | 3.4 | 57Fe |
| Petra-III/P01 | 6 | 1 | 57Fe, 125Te, 119Sn, 121Sb, 193Ir |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Braun, A.; Cramer, S.P.; Gee, L.B.; Yoda, Y. Nuclear Resonance Vibrational Spectroscopy: A Modern Tool to Pinpoint Site-Specific Cooperative Processes. Crystals 2021, 11, 909. https://doi.org/10.3390/cryst11080909
Wang H, Braun A, Cramer SP, Gee LB, Yoda Y. Nuclear Resonance Vibrational Spectroscopy: A Modern Tool to Pinpoint Site-Specific Cooperative Processes. Crystals. 2021; 11(8):909. https://doi.org/10.3390/cryst11080909
Chicago/Turabian StyleWang, Hongxin, Artur Braun, Stephen P. Cramer, Leland B. Gee, and Yoshitaka Yoda. 2021. "Nuclear Resonance Vibrational Spectroscopy: A Modern Tool to Pinpoint Site-Specific Cooperative Processes" Crystals 11, no. 8: 909. https://doi.org/10.3390/cryst11080909
APA StyleWang, H., Braun, A., Cramer, S. P., Gee, L. B., & Yoda, Y. (2021). Nuclear Resonance Vibrational Spectroscopy: A Modern Tool to Pinpoint Site-Specific Cooperative Processes. Crystals, 11(8), 909. https://doi.org/10.3390/cryst11080909