
Crystal Structure Prediction of the Novel Cr2SiN4 Compound via Global Optimization, Data Mining, and the PCAE Method

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Global and Local Optimization Using Empirical and Ab Initio Energy Functions
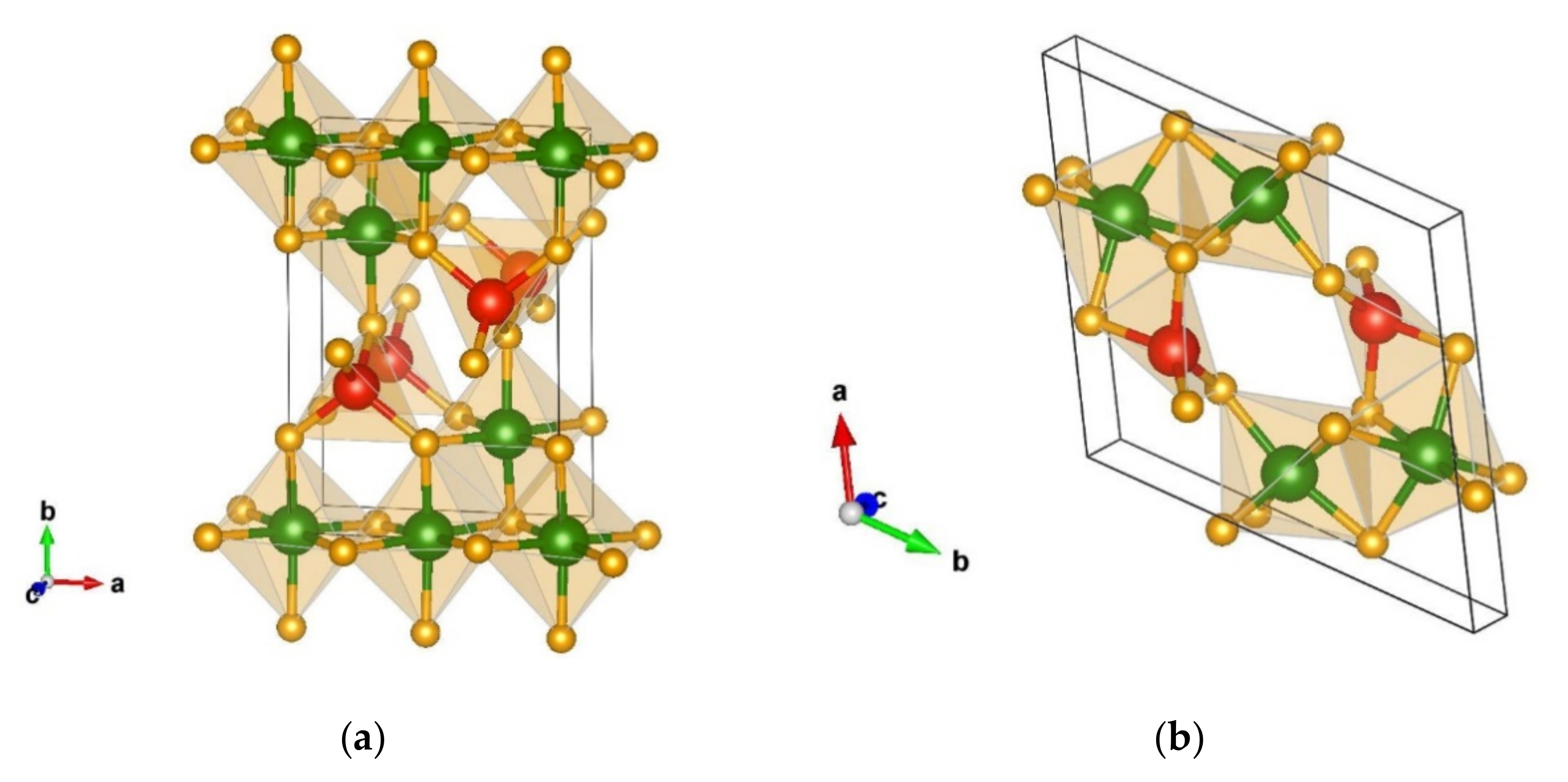
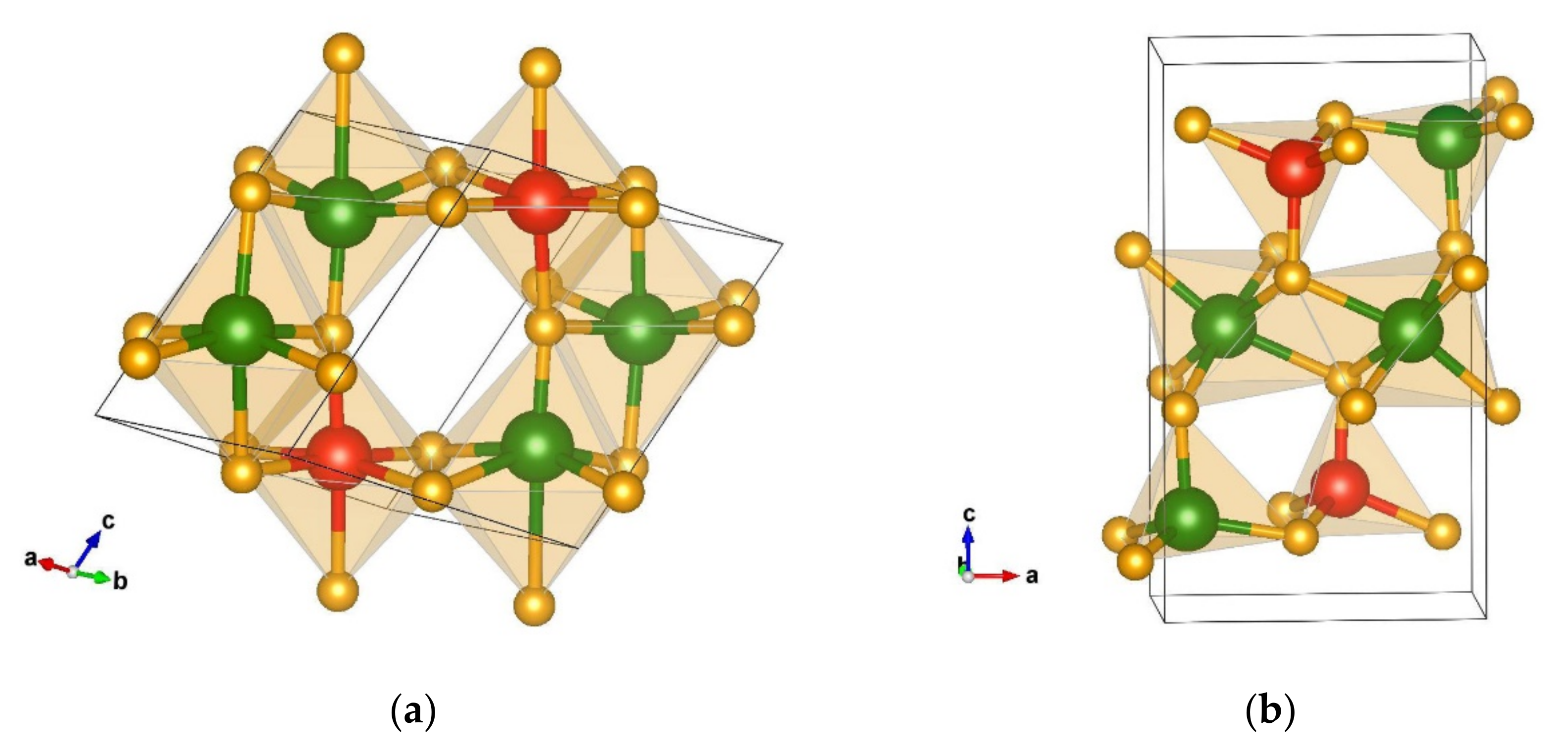
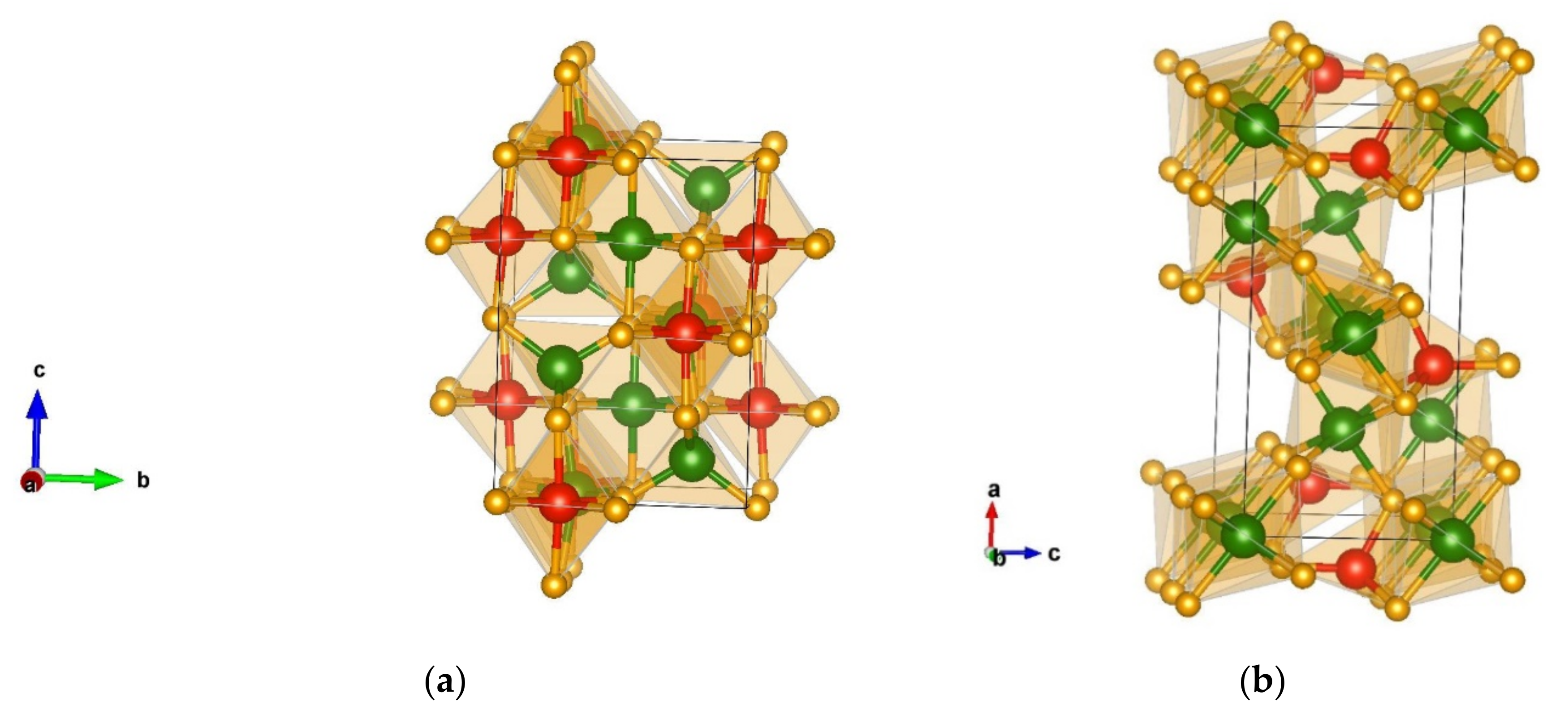
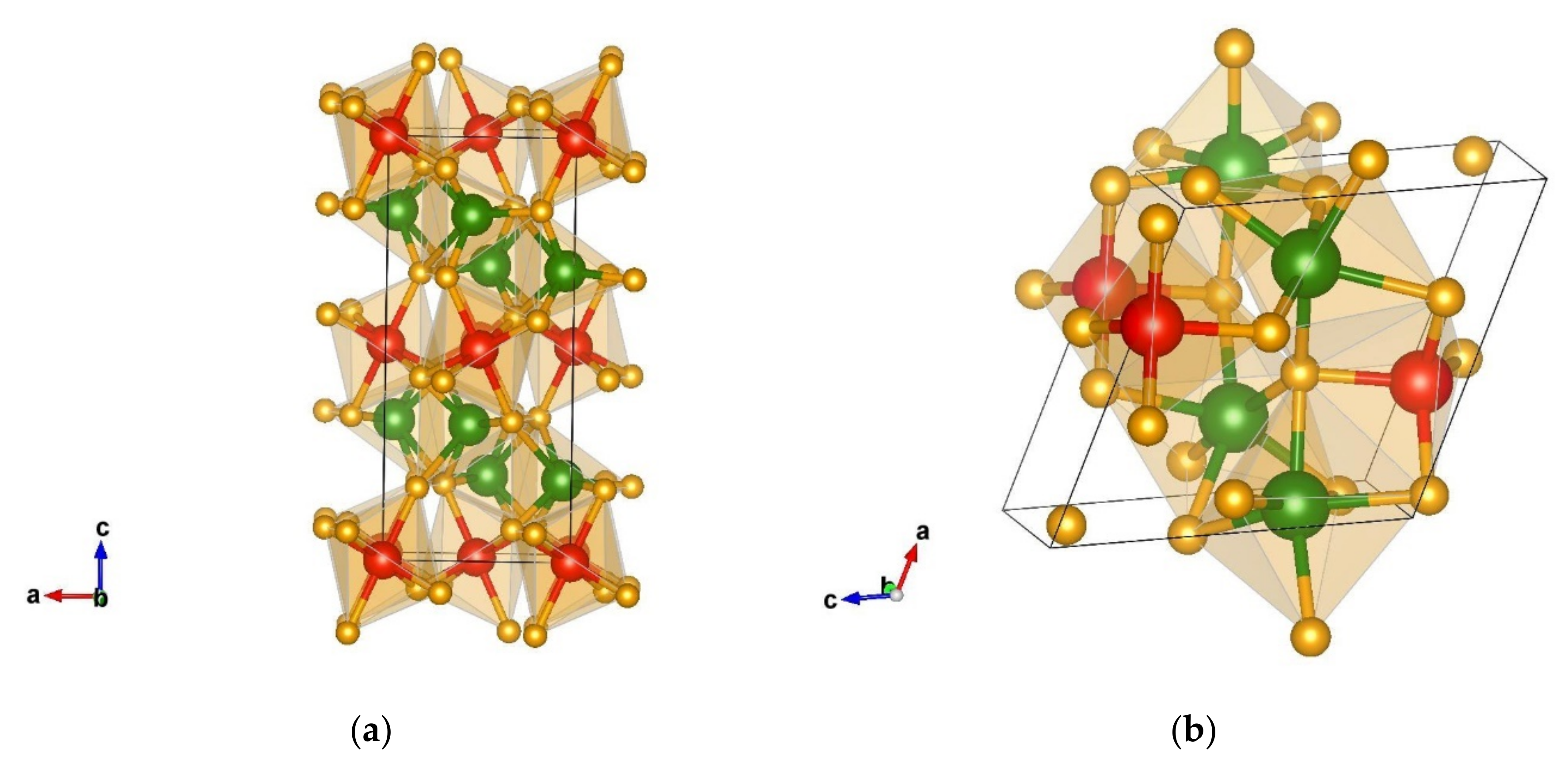
3.1.1. Structural Analysis of the Most Promising Modifications Found after Global Optimization (GO)
3.1.2. Structural Details of Non-Favorable Structures Found after a Global Search
3.2. Data Mining (DM) Based Searches Using the ICSD Database
3.2.1. Structural Analysis of Low-Energy Candidates from the DM-Based Searches
3.2.2. Structural Analysis of Non-Favorable Candidates Found after Data Mining
3.3. Structural Searches Using the PCAE Method
Structural Details of Candidates Found Using the PCAE Method
3.4. Energy Landscape of Cr2SiN4 on the Ab Initio Level
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modifications and Space Groups | Cell Parameters | Position of Atoms |
|---|---|---|
| Al2MgO4-spinel-type_LDA Fd-3m No 227 | a = 7.76 | Cr 0.000000 0.000000 0.000000 Si 0.625000 0.625000 0.625000 N 0.752556 0.752556 0.752556 |
| α-Cr2SiN4-type_LDA Pma2 No 28 | a = 5.45 b = 7.80 c = 2.76 | Cr 0.750000 0.241721 0.552936 Cr 0.500000 0.000000 0.898141 Si 0.750000 0.614777 0.000000 N 0.750000 0.006165 0.421951 N 0.750000 0.489793 0.499478 N 0.501351 0.244258 0.983987 |
| Na2MnCl4-type_LDA Pbam No 55 | a = 4.67 b = 8.58 c = 2.69 | Cr 0.432611 0.175929 0.500000 Si 0.000000 0.000000 0.000000 N 0.133271 0.203741 0.000000 N 0.257704 0.966687 0.500000 |
| γ-Cr2SiN4-type_LDA Cc No 9 | a = 5.56 b = 8.82 c = 5.25 β = 117.96 | Cr 0.499339 0.356416 0.521849 Cr 0.490863 0.639616 0.502192 Si 0.000000 0.578114 0.000000 N 0.817340 0.495466 0.665199 N 0.336682 0.499101 0.681972 N 0.677149 0.750383 0.851076 N 0.670732 0.238989 0.855745 |
| β-Cr2SiN4-type_LDA P-1 No 2 | a = 7.18 b = 7.69 c = 2.70 α = 94.01 β = 82.69 γ = 121.02 | Cr 0.350127 0.864148 0.734578 Cr 0.863665 0.518329 0.302902 Si 0.625158 0.737213 0.910010 N 0.050410 0.680530 0.762885 N 0.352905 0.668370 0.167669 N 0.662487 0.971411 0.739846 N 0.713401 0.667644 0.365727 |
| δ-Cr2SiN4-type_LDA P21/m No 11 | a = 6.14 b = 3.76 c = 5.41 β = 115.88 | Cr 0.091491 0.750000 0.659687 Cr 0.148173 0.750000 0.246670 Si 0.610570 0.750000 0.913336 N 0.638880 0.250000 0.419381 N 0.142755 0.250000 0.721438 N 0.367862 0.750000 0.044580 N 0.131910 0.250000 0.172992 |
| TiMn2O4-type_LDA P4322 No 95 | a = 5.56 c = 7.61 | Cr 0.500000 0.290089 0.000000 Cr 0.233800 0.233800 0.625000 Si 0.000000 0.260505 0.000000 N 0.238872 0.500821 0.998335 N 0.247363 0.028667 0.009638 |
| Mg2SiO4-type_LDA Pnma No 62 | a = 9.23 b = 5.35 c = 4.77 | Cr 0.500000 0.500000 0.500000 Cr 0.752388 0.750000 0.005094 Si 0.911386 0.750000 0.581141 N 0.915976 0.750000 0.228570 N 0.580994 0.750000 0.754642 N 0.831042 0.496221 0.748887 |
| ε-Cr2SiN4-type_LDA P21/m No 11 | a = 5.07 b = 2.86 c = 8.45 β = 90.40 | Cr 0.780604 0.250000 0.510721 Cr 0.923710 0.750000 0.845133 Si 0.427115 0.250000 0.804711 N 0.570806 0.750000 0.868258 N 0.885468 0.750000 0.109395 N 0.589494 0.750000 0.396942 N 0.052827 0.250000 0.362060 |
| λ-Cr2SiN4-type_LDA Pm No 6 | a = 4.96 b = 2.84 c = 8.88 β = 98.40 | Cr 0.914225 0.500000 0.418382 Cr 0.707639 0.500000 0.700020 Cr 0.303954 0.000000 0.721261 Cr 0.508118 0.500000 0.052804 Si 0.000000 0.000000 0.000000 Si 0.419874 0.000000 0.378878 N 0.545716 0.000000 0.572397 N 0.460020 0.500000 0.846268 N 0.704067 0.000000 0.081337 N 0.551407 0.500000 0.308625 N 0.057263 0.500000 0.603299 N 0.068995 0.000000 0.346274 N 0.943534 0.000000 0.807418 N 0.155450 0.500000 0.072091 |
| λ’-Cr2SiN4-type_LDA Pm No 6 | a = 4.99 b = 2.82 c = 8.84 β = 90.95 | Cr 0.175453 0.500000 0.329781 Cr 0.898546 0.500000 0.677506 Cr 0.311343 0.000000 0.668615 Cr 0.483688 0.500000 0.973214 Si 0.000000 0.000000 0.000000 Si 0.681904 0.000000 0.380834 N 0.683473 0.000000 0.573821 N 0.506564 0.500000 0.783113 N 0.663254 0.000000 0.037342 N 0.818343 0.500000 0.305474 N 0.181392 0.500000 0.535694 N 0.363881 0.000000 0.300294 N 0.023309 0.000000 0.806317 N 0.153474 0.500000 0.070996 |
| Modification and Space Group | Search Method | Total Energy (Eh) | Relative Energy (kcal/mol) |
|---|---|---|---|
| Al2MgO4-spinel-type_LDA | DM | −5180.729 | 0.0 |
| α-Cr2SiN4-type_LDA | GO | −5180.694 | 21.963 |
| Na2MnCl4-type_LDA | DM | −5180.672 | 35.768 |
| γ-Cr2SiN4-type_LDA | PCAE | −5180.667 | 38.906 |
| β-Cr2SiN4-type_LDA | GO | −5180.653 | 47.691 |
| δ-Cr2SiN4-type_LDA | GO | −5180.653 | 47.691 |
| TiMn2O4-type_LDA | DM | −5180.621 | 67.771 |
| Mg2SiO4-type_LDA | DM | −5180.620 | 68.399 |
| ε-Cr2SiN4-type_LDA | GO | −5180.615 | 71.536 |
| λ-Cr2SiN4-type_LDA | GO | −5180.606 | 77.184 |
| λ’-Cr2SiN4-type_LDA | GO | −5180.604 | 78.439 |
References
- Benlatreche, Y.; Nouveau, C.; Aknouche, H.; Imhoff, L.; Martin, N.; Gavoille, J.; Rousselot, C.; Rauch, J.-Y.; Pilloud, D. Physical and Mechanical Properties of CrAlN and CrSiN Ternary Systems for Wood Machining Applications. Plasma Process. Polym. 2009, 6, S113–S117. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ye, Y.; Wang, Y. Structure, corrosion, and tribological properties of CrSiN coatings with various Si contents in 3.5% NaCl solution. Surf. Interface Anal. 2018, 50, 471–479. [Google Scholar] [CrossRef]
- Yang, D.; Chen, H.; Ye, Y.; Wang, C.; Zhao, H.; Gong, D. Doping silicon to enhance the anti-corrosion and anti-wear abilities of chromium nitride coating in seawater. Surf. Topogr. Metrol. Prop. 2018, 6, 044001. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Mizuno, Y.; Nakayama, T.; Suzuki, T. Preparation of Cr–Si–N–O thin films epitaxially grown on MgO substrates by pulsed laser deposition. Vaccum 2020, 179, 109498. [Google Scholar] [CrossRef]
- Mercs, D.; Bonasso, N.; Naamane, S.; Bordes, J.-M.; Coddet, C. Mechanical and tribological properties of Cr–N and Cr–SI–N coatings reactively sputter deposited. Surf. Coat. Technol. 2005, 200, 403–407. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, K.H.; Lee, D.; Moore, J. Study on high-temperature oxidation behaviors of Cr–Si–N films. Surf. Coat. Technol. 2006, 200, 6702–6705. [Google Scholar] [CrossRef]
- Lee, J.-W.; Kuo, Y.-C.; Wang, C.-J.; Chang, L.-C.; Liu, K.-T. Effects of substrate bias frequencies on the characteristics of chromium nitride coatings deposited by pulsed DC reactive magnetron sputtering. Surf. Coat. Technol. 2008, 203, 721–725. [Google Scholar] [CrossRef]
- Dinu, M.; Mouele, E.S.M.; Parau, A.C.; Vladescu, A.; Petrik, L.F.; Braic, M. Enhancement of the Corrosion Resistance of 304 Stainless Steel by Cr–N and Cr(N,O) Coatings. Coatings 2018, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Yang, L.; Wu, Y.; Tan, J.; Dang, W.; He, X.; Ma, X. Study of the oxidation behavior of CrN coating on Zr alloy in air. J. Nucl. Mater. 2019, 515, 354–369. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, N.; Sproul, W.D.; Moore, J.J. A comparison of the oxidation behavior of CrN films deposited using continuous dc, pulsed dc and modulated pulsed power magnetron sputtering. Surf. Coat. Technol. 2012, 206, 3283–3290. [Google Scholar] [CrossRef]
- Shan, L.; Wang, Y.; Li, J.; Jiang, X.; Chen, J. Improving tribological performance of CrN coatings in seawater by structure design. Tribol. Int. 2015, 82, 78–88. [Google Scholar] [CrossRef]
- Shah, H.N.; Jayaganthan, R.; Kaur, D. Influence of reactive gas and temperature on structural properties of magnetron sputtered CrSiN coatings. Appl. Surf. Sci. 2011, 257, 5535–5543. [Google Scholar] [CrossRef]
- Wo, P.; Munroe, P.; Li, Z.; Jiang, Z.-T.; Xie, Z.; Zhou, Z.; Li, K.Y. Factors governing the mechanical behaviour of CrSiN coatings: Combined nanoindentation testing and transmission electron microscopy. Mater. Sci. Eng. A 2012, 534, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Bobzin, K.; Bagcivan, N.; Immich, P.; Bolz, S.; Cremer, R.; Leyendecker, T. Mechanical properties and oxidation behaviour of (Al,Cr)N and (Al,Cr,Si)N coatings for cutting tools deposited by HPPMS. Thin Solid Films 2008, 517, 1251–1256. [Google Scholar] [CrossRef]
- Vepřek, S. New development in superhard coatings: The superhard nanocrystalline-amorphous composites. Thin Solid Films 1998, 317, 449–454. [Google Scholar] [CrossRef]
- Morgiel, J.; Grzonka, J.; Mania, R.; Zimowski, S.; Lábár, J.L.; Fogarassy, Z. Relation between microstructure and hardness of nano-composite CrN/Si3N4 coatings obtained using CrSi single target magnetron system. Vaccum 2013, 90, 170–175. [Google Scholar] [CrossRef]
- Martinez, E.; Sanjinés, R.; Karimi, A.; Esteve, J.; Lévy, F. Mechanical properties of nanocomposite and multilayered Cr–Si–N sputtered thin films. Surf. Coat. Technol. 2004, 180–181, 570–574. [Google Scholar] [CrossRef]
- Park, J.H.; Chung, W.S.; Cho, Y.-R.; Kim, K.H. Synthesis and mechanical properties of Cr–Si–N coatings deposited by a hybrid system of arc ion plating and sputtering techniques. Surf. Coat. Technol. 2004, 188–189, 425–430. [Google Scholar] [CrossRef]
- Lin, H.-H.; Chou, C.-C.; Lee, J.-W. Tribological properties of Cr–Si–N nanocomposite film adherent silicon under various environments. Thin Solid Films 2010, 518, 7509–7514. [Google Scholar] [CrossRef]
- Azzi, M.; Benkahoul, M.; Szpunar, J.; Klemberg-Sapieha, J.; Martinu, L. Tribological properties of CrSiN-coated 301 stainless steel under wet and dry conditions. Wear 2009, 267, 882–889. [Google Scholar] [CrossRef]
- Lee, J.-W.; Chang, Y.-C. A study on the microstructures and mechanical properties of pulsed DC reactive magnetron sputtered Cr–Si–N nanocomposite coatings. Surf. Coat. Technol. 2007, 202, 831–836. [Google Scholar] [CrossRef]
- Lee, H.; Jung, W.; Han, J.; Seo, S.; Kim, J.; Bae, Y. The synthesis of CrSiN film deposited using magnetron sputtering system. Surf. Coat. Technol. 2005, 200, 1026–1030. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, L.; Wang, S.; Yan, P.; Xue, Q. Structure and mechanical properties of reactive sputtering CrSiN films. Appl. Surf. Sci. 2009, 255, 4425–4429. [Google Scholar] [CrossRef] [Green Version]
- Geng, Z.; Wang, H.; Wang, C.; Wang, L.; Zhang, G. Effect of Si content on the tribological properties of CrSiN films in air and water environments. Tribol. Int. 2014, 79, 140–150. [Google Scholar] [CrossRef]
- Shao, T.; Ge, F.; Pei, C.; Huang, F.; Sun, D.; Zhang, S. Effects of Si content on Tribo-corrosion behavior of Cr1−xSixN coatings prepared via magnetron sputtering. Surf. Coat. Technol. 2018, 356, 11–18. [Google Scholar] [CrossRef]
- Shan, L.; Zhang, Y.-R.; Wang, Y.-X.; Li, J.-L.; Jiang, X.; Chen, J.-M. Corrosion and wear behaviors of PVD CrN and CrSiN coatings in seawater. Trans. Nonferr. Met. Soc. China 2016, 26, 175–184. [Google Scholar] [CrossRef]
- Park, I.-W.; Kang, D.S.; Moore, J.J.; Kwon, S.C.; Rha, J.J.; Kim, K.H. Microstructures, mechanical properties, and tribological behaviors of Cr–Al–N, Cr–Si–N, and Cr–Al–Si–N coatings by a hybrid coating system. Surf. Coat. Technol. 2007, 201, 5223–5227. [Google Scholar] [CrossRef]
- Lu, S.; Wang, Y.; Gaoand, P.; Meng, D. Effect of Si Content on Structure and Friction and Wear Properties of CrSiN Coatings. IOP Conf. Ser. Earth Environ. Sci. 2020, 440, 022028. [Google Scholar] [CrossRef]
- Wang, Q.; Lin, Y.; Zhou, F.; Kong, J. The influence of Ni concentration on the structure, mechanical and tribological properties of Ni–CrSiN coatings in seawater. J. Alloy. Compd. 2020, 819, 152998. [Google Scholar] [CrossRef]
- Kim, G.; Kim, B.; Lee, S. High-speed wear behaviors of CrSiN coatings for the industrial applications of water hydraulics. Surf. Coat. Technol. 2005, 200, 1814–1818. [Google Scholar] [CrossRef]
- Chang, W.J.; Zhang, H.; Chen, Y.Y.; Li, J.; Zhang, X.; Jiang, P.Z.; Fan, X.W.; Duo, S.W. Tribological Performances of CrSiN Coatings Deposited by High Power Pulse Magnetron Sputtering. Solid State Phenom. 2018, 281, 540–545. [Google Scholar] [CrossRef]
- Lou, B.-S.; Chang, Y.-C.; Lee, J.-W. High Temperature Oxidation Behaviors of CrNx and Cr-Si-N Thin Films at 1000 °C. Coatings 2019, 9, 540. [Google Scholar] [CrossRef] [Green Version]
- Mikula, M.; Grančič, B.; Drienovsky, M.; Satrapinskyy, L.; Roch, T.; Hájovská, Z.; Gregor, M.; Plecenik, T.; Čička, R.; Kúš, P. Thermal stability and high-temperature oxidation behavior of Si–Cr–N coatings with high content of silicon. Surf. Coat. Technol. 2013, 232, 349–356. [Google Scholar] [CrossRef]
- Suzuki, K.; Suzuki, T.; Endo, T.; Nakayama, T.; Suematsu, H.; Niihara, K. Epitaxial growth of chromium nitride thin films with addition of silicon. Phys. Status Solidi 2015, 12, 545–548. [Google Scholar] [CrossRef]
- Xu, X.M.; Zhang, H.; Luo, F.F.; Zhou, Z.H.; Duo, S.W. The Properties of CrSiN Coatings of Different Si Content. Appl. Mech. Mater. 2014, 687–691, 4323–4326. [Google Scholar] [CrossRef]
- Wang, Q.M.; Kim, K.H. Microstructural control of Cr–Si–N films by a hybrid arc ion plating and magnetron sputtering process. Acta Mater. 2009, 57, 4974–4987. [Google Scholar] [CrossRef]
- Martínez, E.; Sanjinés, R.; Banakh, O.; Lévy, F. Electrical, optical and mechanical properties of sputtered CrNy and Cr1−xSixN1.02 thin films. Thin Solid Films 2004, 447–448, 332–336. [Google Scholar] [CrossRef]
- Ma, B.; Luo, B.; Wang, Z.; Meng, C.; He, X. Friction and Wear Properties of CrAl-Based Coatings for Nuclear Fuel Cladding. Front. Energy Res. 2021, 9, 24. [Google Scholar] [CrossRef]
- Zagorac, J.; Schön, J.C.; Matović, B.; Škundrić, T. Predicting Feasible Modifications of Ce2ON2 Using a Combination of Global Optimization and Data Mining. J. Phase Equilibria Diffus. 2020, 41, 538–549. [Google Scholar] [CrossRef]
- Zagorac, D.; Zagorac, J.; Schön, J.C.; Stojanović, N.; Matović, B. ZnO/ZnS (hetero)structures: Ab initio investigations of polytypic behavior of mixed ZnO and ZnS compounds. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2018, 74, 628–642. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Schön, J.C. Nanomaterials—What energy landscapes can tell us. Process. Appl. Ceram. 2015, 9, 157–168. [Google Scholar] [CrossRef]
- Schön, J.C.; Jansen, M. Determination of candidate structures for simple ionic compounds through cell optimisation. Comput. Mater. Sci. 1995, 4, 43–58. [Google Scholar] [CrossRef]
- Bergerhoff, G.; Brown, I.D. Crystallographic Databases; Allen, F.H., Bergerhoff, G., Sievers, R., Eds.; International Union of Crystallography: Chester, UK, 1987. [Google Scholar]
- Zagorac, D.; Müller, H.; Ruehl, S.; Rehme, S. Recent developments in the Inorganic Crystal Structure Database: Theoretical crystal structure data and related features. J. Appl. Crystallogr. 2019, 52, 918–925. [Google Scholar] [CrossRef] [Green Version]
- Zagorac, J.; Rosic, M.; Schön, J.C.; Matovic, B. Structure prediction of aluminum nitride combining data mining and quantum mechanics. CrystEngComm 2017, 19, 5259–5268. [Google Scholar] [CrossRef]
- Cvijović-Alagić, I.; Rakin, M.; Laketić, S.; Zagorac, D. Microstructural study of Ti45Nb alloy before and after HPT processing using experimental and ab initio data mining approach. Mater. Charact. 2020, 169, 110635. [Google Scholar] [CrossRef]
- Zagorac, J.; Jovanovic, D.; Volkov-Husovic, T.; Matovic, B.; Zagorac, D. Structure prediction, high pressure effect and properties investigation of superhard B6O. Model. Simul. Mater. Sci. Eng. 2020, 28. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Dovesi, R.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.; D’Arco, P.; Noël, Y.; Rérat, M.; Carbonnière, P.; et al. The CRYSTAL code, 1976–2020 and beyond, a long story. J. Chem. Phys. 2020, 152, 204111. [Google Scholar] [CrossRef] [PubMed]
- Doll, K.; Saunders, V.R.; Harrison, N.M. Analytical Hartree–Fock gradients for periodic systems. Int. J. Quantum Chem. 2001, 82, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Doll, K.; Dovesi, R.; Orlando, R. Analytical Hartree–Fock gradients with respect to the cell parameter for systems periodic in three dimensions. Theor. Chem. Acc. 2004, 112, 394–402. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Schön, J.C.; Čančarević, Ž.; Jansen, M. Structure prediction of high-pressure phases for alkali metal sulfides. J. Chem. Phys. 2004, 121, 2289–2304. [Google Scholar] [CrossRef] [PubMed]
- Zagorac, D.; Doll, K.; Schön, J.C.; Jansen, M. Ab initiostructure prediction for lead sulfide at standard and elevated pressures. Phys. Rev. B 2011, 84, 045206. [Google Scholar] [CrossRef]
- Zagorac, J.; Schön, J.C.; Jansen, M. Prediction of structure candidates for zinc oxide as a function of pressure and investigation of their electronic properties. Phys. Rev. B 2014, 89, 075201. [Google Scholar] [CrossRef]
- Catti, M.; Sandrone, G.; Valerio, G.; Dovesi, R. Electronic, magnetic and crystal structure of Cr2O3 by theoretical methods. J. Phys. Chem. Solids 1996, 57, 1735–1741. [Google Scholar] [CrossRef]
- Ruiz, E.; Llunell, M.; Alemany, P. Calculation of exchange coupling constants in solid state transition metal compounds using localized atomic orbital basis sets. J. Solid State Chem. 2003, 176, 400–411. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.; Orlando, R.; Roetti, C.; Ugliengo, P.; Dovesi, R. Vibration Frequencies of Mg3Al2Si3O12 Pyrope. An ab Initio Study with the CRYSTAL Code. J. Phys. Chem. B 2005, 109, 6146–6152. [Google Scholar] [CrossRef] [PubMed]
- Noel, Y.; Catti, M.; D’Arco, P.; Dovesi, R. The vibrational frequencies of forsterite Mg2SiO4: An all-electron ab initio study with the CRYSTAL code. Phys. Chem. Miner. 2006, 33, 383–393. [Google Scholar] [CrossRef]
- Dovesi, R.; Causa’, M.; Orlando, R.; Roetti, C.; Saunders, V.R. Ab initio approach to molecular crystals: A periodic Hartree–Fock study of crystalline urea. J. Chem. Phys. 1990, 92, 7402–7411. [Google Scholar] [CrossRef]
- Zagorac, D.; Djukic, M.; Jordanov, D.; Matović, B. Theoretical study of AlN mechanical behaviour under high pressure regime. Theor. Appl. Fract. Mech. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Hundt, R.; Schön, J.C.; Hannemann, A.; Jansen, M. Determination of symmetries and idealized cell parameters for simulated structures. J. Appl. Crystallogr. 1999, 32, 413–416. [Google Scholar] [CrossRef] [Green Version]
- Hannemann, A.; Hundt, R.; Schön, J.C.; Jansen, M. A New Algorithm for Space-Group Determination. J. Appl. Crystallogr. 1998, 31, 922–928. [Google Scholar] [CrossRef]
- Hundt, R.; Schön, J.C.; Jansen, M. CMPZ– an algorithm for the efficient comparison of periodic structures. J. Appl. Crystallogr. 2006, 39, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Hundt, R. KPLOT: A Program for Plotting and Analysing Crystal Structures; Technicum Scientific Publishing: Stuttgart, Germany, 2016. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Cui, L.; Hu, M.; Wang, Q.; Xu, B.; Yu, D.; Liu, Z.; He, J. Prediction of novel hard phases of Si3N4: First-principles calculations. J. Solid State Chem. 2015, 228, 20–26. [Google Scholar] [CrossRef]
- Yamanaka, T.; Takéuchi, Y. Order-disorder transition in MgAl2O4 spinel at high temperatures up to 1700 °C. Z. Krist. 1983, 165, 65–78. [Google Scholar] [CrossRef]
- Kroll, P. Pathways to metastable nitride structures. J. Solid State Chem. 2003, 176, 530–537. [Google Scholar] [CrossRef]
- Goodyear, J.; Ali, S.A.D.; Steigmann, G.A. The crystal structure of Na2MnCl4. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1971, 27, 1672–1674. [Google Scholar] [CrossRef]
- Bertaut, E.; Vincent, H. Etude par diffraction neutronique de la forme ordonnee de l’orthotitanate de manganese-structure cristalline et structure magnetique. Solid State Commun. 1968, 6, 269–275. [Google Scholar] [CrossRef]
- Smyth, J.; Hazen, R. The crystal structures of forsterite and hortonolite at several temperatures up to 900 °C. Am. Mineral. 1973, 58, 588–593. [Google Scholar]
- Friedt, O.; Braden, M.; André, G.; Adelmann, P.; Nakatsuji, S.; Maeno, Y. Structural and magnetic aspects of the metal-insulator transition inCa2−xSrxRuO4. Phys. Rev. B 2001, 63, 174432. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.N.; Norby, P.; Hanson, J.C. A crystal structure determination of HgC2O4 from synchrotron X-ray and neutron powder diffraction data. Z. Krist. Cryst. Mater. 1994, 209, 874–877. [Google Scholar] [CrossRef]
- Matar, S.F.; Etourneau, J. (CaO)nIrO2 (n = 1, 2, 4) family: Chemical scissors effects of CaO on structural characteristics correlated to physical properties. Ab initio study. J. Solid State Chem. 2017, 255, 82–88. [Google Scholar] [CrossRef]
- Shashkin, D.P.; Simonov, M.A.; Belov, N.V. Crystalline structure of calciborite—CaB2O4=Ca2[BO3BO]2. Dokl. Akad. Nauk SSSR 1970, 195, 345–348. [Google Scholar]
- Partik, M.; Stingl, T.; Lutz, H.D.; Sabrowsky, H.; Vogt, P. Strukturverfeinerung und magnetische Messungen an Mn2SnS4. Z. Anorg. Allg. Chem. 1995, 621, 1600–1604. [Google Scholar] [CrossRef]
- Hahn, H.; Klingler, W. Zur Struktur des βCu2HgJ4 und des β-Ag2HgJ4. Z. Anorg. Allg. Chem. 1955, 279, 271–280. [Google Scholar] [CrossRef]
- Schneider, J.; Johnson, F.N.; Laschke, K. Structure refinements of β-Si3N4 at temperatures up to 1360 °C by X-ray powder investigation. Z. Krist. Cryst. Mater. 1994, 209, 328–333. [Google Scholar] [CrossRef]
- Yang, P.; Fun, H.K.; Rahman, I.A.; Saleh, M. Two phase refinements of the structures of α-Si3N4 and β-Si3N4 made from rice husk by Rietveld analysis. Ceram. Int. 1995, 21, 137–142. [Google Scholar] [CrossRef]
- Toraya, H. Crystal structure refinement of α-Si3N4 using synchrotron radiation powder diffraction data: Unbiased refinement strategy. J. Appl. Crystallogr. 2000, 33, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Kroll, P.; Milko, M. Theoretical Investigation of the Solid State Reaction of Silicon Nitride and Silicon Dioxide forming Silicon Oxynitride (Si2N2O) under Pressure. Z. Anorg. Allg. Chem. 2003, 629, 1737–1750. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Kaneta, Y.; Iwata, S. First-principles investigation of the structural, electronic and optical properties of olivine-Si3N4 and olivine-Ge3N4. J. Phys. Condens. Matter 2006, 18, 10663–10676. [Google Scholar] [CrossRef]
- Wang, R.; Sun, Z.; Pal, U.B.; Gopalan, S.; Basu, S.N. Mitigation of chromium poisoning of cathodes in solid oxide fuel cells employing CuMn1.8O4 spinel coating on metallic interconnect. J. Power Sources 2018, 376, 100–110. [Google Scholar] [CrossRef]
- Hassan, M.A.; Mamat, O.B. Mitigation of Chromium Poisoning of Ferritic Interconnect from Annealed Spinel of CuFe2O4. Processes 2020, 8, 1113. [Google Scholar] [CrossRef]
- Rose, L. On the Degradation of Porous Stainless Steel in Low and Intermediate Temperature Solid Oxide Fuel Cell Support Materials; University of British Columbia: Vancouver, BC, Canada, 2011. [Google Scholar]




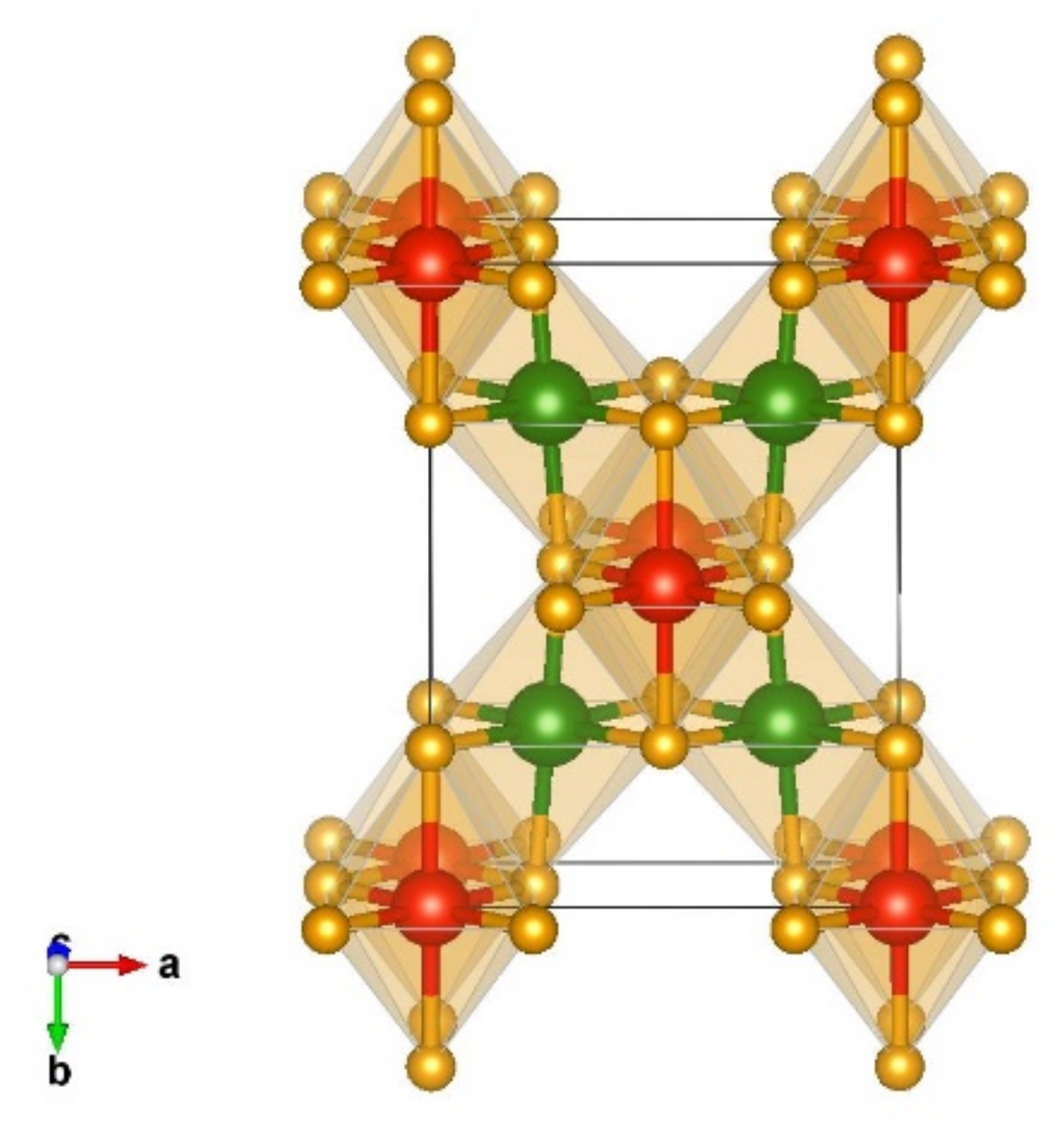



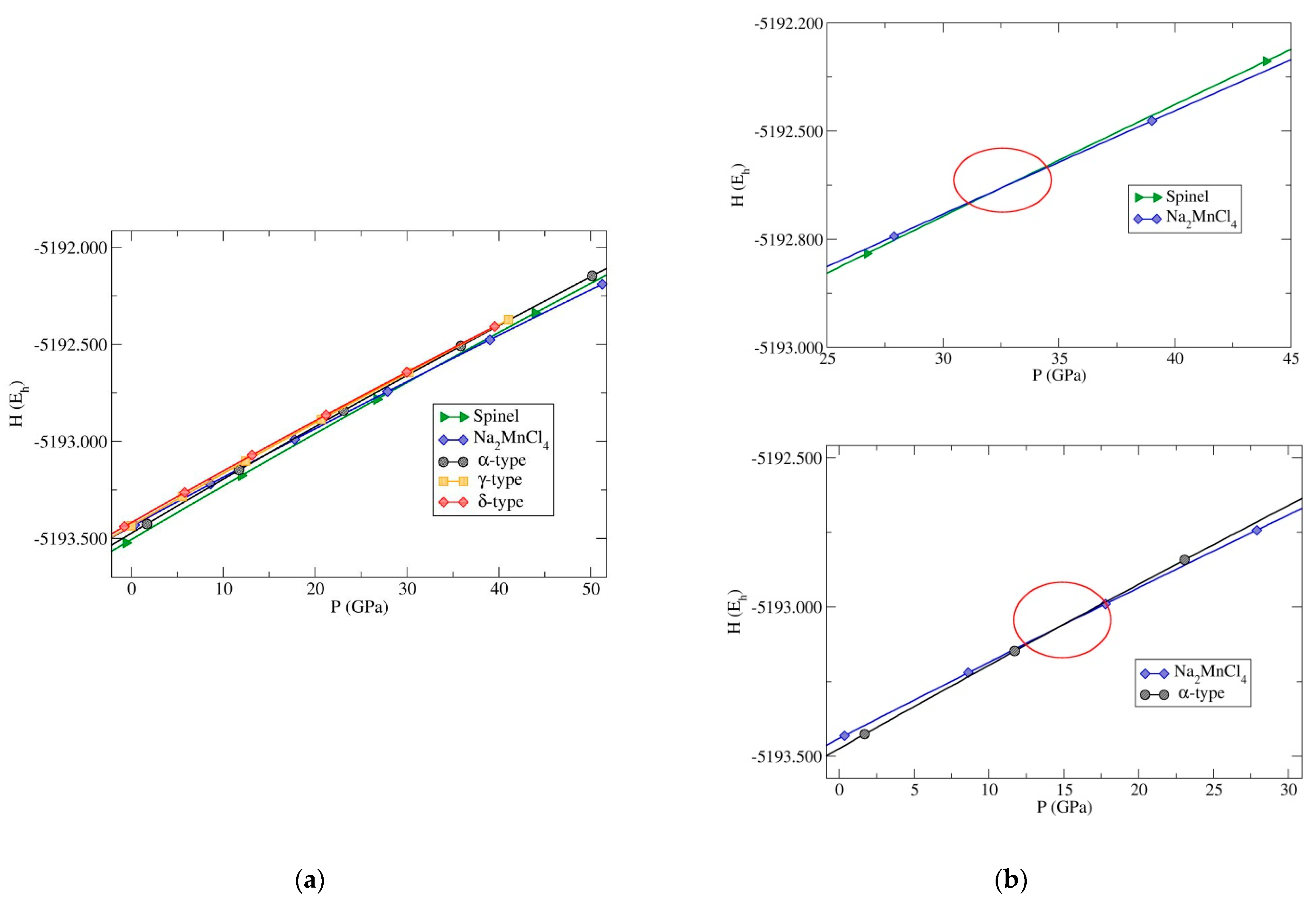
| Modification | Total Energy (Eh) | Relative Energy (kcal/mol) |
|---|---|---|
| α-Cr2SiN4-type | −5193.474 | 20.708 |
| β-Cr2SiN4-type | −5193.438 | 43.298 |
| δ-Cr2SiN4-type | −5193.419 | 55.221 |
| ε-Cr2SiN4-type | −5193.413 | 58.986 |
| λ-Cr2SiN4-type | −5193.407 | 62.750 |
| λʹ-Cr2SiN4-type | −5193.404 | 64.634 |
| nf1-Cr2SiN4-type | −5193.398 | 68.399 |
| nf2-Cr2SiN4-type | −5193.388 | 74.674 |
| nf3-Cr2SiN4-type | −5193.385 | 76.556 |
| nf4-Cr2SiN4-type | −5193.364 | 89.734 |
| nf5-Cr2SiN4-type | −5193.364 | 89.734 |
| Modifications and Space Group | Cell Parameters | Position of Atoms |
|---|---|---|
| α-Cr2SiN4-type Pma2 No 28 | a = 5.54 b = 7.91 c = 2.81 | Cr 0.750000 0.245688 0.560883 Cr 0.500000 0.000000 0.891696 Si 0.750000 0.616760 0.000000 N 0.750000 0.011076 0.423540 N 0.750000 0.493745 0.499960 N 0.501383 0.244129 0.983362 |
| β-Cr2SiN4-type P-1 No 2 | a = 7.28 b = 7.79 c = 2.74 α = 93.66 β = 82.48 γ = 120.64 | Cr 0.347419 0.861114 0.730714 Cr 0.862711 0.520348 0.305878 Si 0.627078 0.740684 0.913120 N 0.050229 0.677562 0.759435 N 0.353428 0.666353 0.160724 N 0.660109 0.973354 0.749444 N 0.717565 0.676085 0.369516 |
| δ-Cr2SiN4-type P21/m No 11 | a = 6.21 b = 3.82 c = 5.54 β = 116.24 | Cr 0.088537 0.750000 0.660564 Cr 0.155145 0.750000 0.253988 Si 0.613292 0.750000 0.917590 N 0.639615 0.250000 0.413635 N 0.137168 0.250000 0.721296 N 0.368220 0.750000 0.042876 N 0.136099 0.250000 0.176958 |
| ε-Cr2SiN4-type P21/m No 11 | a = 5.09 b = 2.89 c = 8.90 β = 90.20 | Cr 0.778189 0.250000 0.506779 Cr 0.918790 0.750000 0.827219 Si 0.419972 0.250000 0.798720 N 0.567333 0.750000 0.861544 N 0.897872 0.750000 0.121156 N 0.589340 0.750000 0.395794 N 0.060536 0.250000 0.368680 |
| λ-Cr2SiN4-type Pm No 6 | a = 5.07 b = 2.88 c = 9.27 β = 99.77 | Cr 0.951900 0.500000 0.415607 Cr 0.698619 0.500000 0.708230 Cr 0.291904 0.000000 0.722310 Cr 0.507805 0.500000 0.040389 Si 0.000000 0.000000 0.000000 Si 0.455229 0.000000 0.394275 N 0.535616 0.000000 0.585846 N 0.455096 0.500000 0.844293 N 0.704956 0.000000 0.076886 N 0.586474 0.500000 0.329108 N 0.050793 0.500000 0.599924 N 0.108035 0.000000 0.351718 N 0.939400 0.000000 0.811337 N 0.163111 0.500000 0.068461 |
| λ’-Cr2SiN4-type Pm No 6 | a = 5.06 b = 2.87 c = 9.18 β = 90.97 | Cr 0.166652 0.500000 0.355602 Cr 0.889145 0.500000 0.685518 Cr 0.307908 0.000000 0.681019 Cr 0.485319 0.500000 0.980324 Si 0.000000 0.000000 0.000000 Si 0.670339 0.000000 0.395017 N 0.676765 0.000000 0.583978 N 0.498298 0.500000 0.794695 N 0.665262 0.000000 0.039248 N 0.812605 0.500000 0.324037 N 0.176521 0.500000 0.552065 N 0.351051 0.000000 0.317489 N 0.015000 0.000000 0.810727 N 0.151227 0.500000 0.066773 |
| Modifications and Space Group | Cell Parameters | Position of Atoms |
|---|---|---|
| nf1-Cr2SiN4-type P21/m No 11 | a = 5.03 b = 2.89 c = 9.25 β = 100.34 | Cr 0.780740 0.750000 0.501443 Cr 0.025858 0.250000 0.824580 Si 0.517842 0.750000 0.797882 N 0.959527 0.250000 0.630184 N 0.552291 0.250000 0.393501 N 0.609701 0.750000 0.135709 N 0.136923 0.250000 0.122932 |
| nf2-Cr2SiN4-type Cc No 9 | a = 5.06 b = 14.14 c = 4.77 β = 121.05 | Cr 0.622331 0.095134 0.360180 Cr 0.380879 0.098904 0.725602 Si 0.000000 0.191719 0.000000 N 0.487148 0.004091 0.555289 N 0.511786 0.374204 0.196387 N 0.720274 0.147401 0.064041 N 0.358846 0.192090 0.368624 |
| nf3-Cr2SiN4-type Pm No 6 | a = 6.79 b = 3.09 c = 6.88 β = 109.29 | Cr 0.319643 0.500000 0.397294 Cr 0.308694 0.500000 0.793094 Cr 0.034585 0.000000 0.406174 Cr 0.733922 0.500000 0.593240 Si 0.000000 0.000000 0.000000 Si 0.588865 0.000000 0.169164 N 0.515186 0.500000 0.666342 N 0.819399 0.000000 0.531146 N 0.551036 0.500000 0.283640 N 0.215615 0.000000 0.242977 N 0.384133 0.000000 0.929487 N 0.000403 0.500000 0.870324 N 0.122011 0.500000 0.540245 N 0.829057 0.000000 0.140470 |
| nf4-Cr2SiN4-type Pm No 6 | a = 7.37 b = 3.05 c = 7.56 β = 115.96 | Cr 0.779088 0.000000 0.218259 Cr 0.459516 0.500000 0.327876 Cr 0.253478 0.000000 0.544973 Cr 0.396471 0.500000 0.935322 Si 0.000000 0.500000 0.000000 Si 0.852505 0.000000 0.603621 N 0.330961 0.500000 0.474928 N 0.889506 0.500000 0.738026 N 0.613308 0.000000 0.401256 N 0.355218 0.000000 0.802744 N 0.932400 0.000000 0.070113 N 0.262965 0.500000 0.082041 N 0.643042 0.500000 0.129399 N 0.988794 0.000000 0.464379 |
| nf5-Cr2SiN4-type P-1 No 2 | a = 7.17 b = 3.06 c = 7.41 α = 89.69 β = 66.68 γ = 88.06 | Cr 0.654478 0.292004 0.523373 Cr 0.288987 0.742844 0.838266 Si 0.865354 0.748679 0.767528 N 0.255163 0.244316 0.972502 N 0.804235 0.255276 0.687399 N 0.477326 0.210250 0.368818 N 0.127792 0.732554 0.709190 |
| Modification | Total Energy (Eh) | Relative Energy (kcal/mol) |
|---|---|---|
| Al2MgO4-spinel-type | −5193.507 | 0.0 |
| Na2MnCl4-type | −5193.436 | 44.553 |
| TiMn2O4-type | −5193.414 | 58.358 |
| Mg2SiO4-type | −5193.403 | 65.261 |
| Ca2RuO4-type | −5193.402 | 65.889 |
| HgC2O4-like | −5193.400 | 67.144 |
| Ca2IrO4-type | −5193.349 | 99.147 |
| CaB2O4-like | −5193.347 | 100.402 |
| Mn2SnS4-type | −5193.342 | 103.539 |
| Modifications and Space Group | Cell Parameters | Position of Atoms |
|---|---|---|
| Al2MgO4-spinel-type Fd-3m No 227 | a = 7.88 | Cr 0.000000 0.000000 0.000000 Si 0.625000 0.625000 0.625000 N 0.752483 0.752483 0.752483 |
| Na2MnCl4-type Pbam No 55 | a = 4.74 b = 8.70 c = 2.73 | Cr 0.433387 0.175989 0.500000 Si 0.000000 0.000000 0.000000 N 0.133667 0.203146 0.000000 N 0.256351 0.966597 0.500000 |
| TiMn2O4-type P4322 No 95 | a = 5.64 c = 7.74 | Cr 0.500000 0.288490 0.000000 Cr 0.234522 0.234522 0.625000 Si 0.000000 0.259306 0.000000 N 0.239859 0.499775 0.998379 N 0.246452 0.027106 0.010564 |
| Mg2SiO4-type Pnma No 62 | a = 9.42 b = 5.45 c = 4.82 | Cr 0.500000 0.500000 0.500000 Cr 0.751225 0.750000 0.005741 Si 0.911733 0.750000 0.580414 N 0.915086 0.750000 0.227101 N 0.579248 0.750000 0.754925 N 0.831733 0.498905 0.747339 |
| Ca2RuO4-type Pbca No 61 | a = 4.55 b = 4.88 c = 10.32 | Cr 0.951271 0.088328 0.314395 Si 0.000000 0.000000 0.000000 N 0.189814 0.299961 0.071415 N 0.821277 0.910789 0.170556 |
| HgC2O4-like P21 No 4 | a = 5.34 b = 5.09 c = 5.36 β = 115.62 | Cr 0.009785 0.463302 0.266681 Cr 0.242643 0.853311 0.472408 Si 0.651871 0.000000 0.959796 N 0.021251 0.353294 0.916553 N 0.626651 0.296871 0.103741 N 0.597719 0.030584 0.610720 N 0.079595 0.179181 0.498154 |
| Ca2IrO4-type P-62m No 189 | a = 8.33 c = 2.70 | Cr 0.000000 0.000000 0.000000 Cr 0.333333 0.666667 0.500000 Cr 0.699230 0.000000 0.500000 Si 0.337748 0.000000 0.000000 N 0.173727 0.000000 0.500000 N 0.473996 0.000000 0.500000 N 0.448912 0.247641 0.000000 |
| CaB2O4-like Pccn No 56 | a = 7.98 b = 14.42 c = 4.85 | Cr 0.931376 0.565538 0.393557 Cr 0.170647 0.535703 0.989804 Si 0.857355 0.679567 0.867484 N 0.864237 0.698217 0.499220 N 0.303715 0.441717 0.754883 N 0.009972 0.618966 0.076401 N 0.377123 0.568169 0.113939 |
| Mn2SnS4-type Cmmm No 65 | a = 5.58 b = 7.82 c = 2.76 | Cr 0.750000 0.750000 0.500000 Si 0.000000 0.000000 0.000000 N 0.000000 0.247324 0.000000 N 0.220861 0.000000 0.500000 |
| Modification | Total Energy (Eh) | Relative Energy (kcal/mol) |
|---|---|---|
| γ-Cr2SiN4-type | −5193.435 | 45.181 |
| Cr2SiN4-PCAE-1-type | −5193.385 | 76.556 |
| Cr2SiN4-PCAE-2-type | −5193.374 | 83.459 |
| Modification and Space Group | Cell Parameters | Position of Atoms |
|---|---|---|
| γ-Cr2SiN4-type Cc (no. 9) | a = 5.62 b = 8.96 c = 5.36 Å, β = 117.93 | Cr 0.505134 0.354319 0.539894 Cr 0.490988 0.640587 0.506788 Si 0.000000 0.574728 0.000000 N 0.820626 0.494637 0.668512 N 0.339534 0.498028 0.687817 N 0.683793 0.745757 0.858424 N 0.673275 0.237591 0.860703 |
| Cr2SiN4-PCAE-1-type Pm (no. 6) | a = 7.04 b = 3.04 c = 7.03 β = 110.72 | Cr 0.338406 0.500000 0.381358 Cr 0.068281 0.000000 0.404484 Cr 0.778353 0.500000 0.589017 Cr 0.381419 0.500000 0.771064 Si 0.000000 0.000000 0.000000 Si 0.613136 0.000000 0.165992 N 0.569205 0.500000 0.274738 N 0.859037 0.000000 0.522005 N 0.985060 0.500000 0.869495 N 0.164949 0.500000 0.544984 N 0.227376 0.000000 0.231306 N 0.437810 0.000000 0.912940 N 0.551129 0.500000 0.636169 N 0.853139 0.000000 0.153320 |
| Cr2SiN4-PCAE-2-type P1 (no. 1) | a = 7.88 b = 7.96 c = 5.79 α = 89.97 β = 89.86 γ = 120.26 | Cr 0.179061 0.774468 0.214657 Cr 0.576899 0.834251 0.216822 Cr 0.514893 0.179271 0.216823 Cr 0.596707 0.344636 0.708045 Cr 0.010451 0.684706 0.713360 Cr 0.678545 0.756495 0.700070 Cr 0.343219 0.429431 0.991834 Cr 0.923504 0.343769 0.996260 Si 0.000000 0.000000 0.000000 Si 0.263900 0.513623 0.496139 Si 0.839296 0.185951 0.496185 Si 0.163373 0.091041 0.497914 N 0.743253 0.861393 0.985305 N 0.495442 0.322138 0.989374 N 0.052018 0.604045 0.985425 N 0.711195 0.932465 0.477493 N 0.419187 0.211059 0.484630 N 0.145337 0.648381 0.483835 N 0.406296 0.578413 0.243870 N 0.773280 0.267856 0.250745 N 0.088745 0.948405 0.247062 N 0.418188 0.578819 0.734852 N 0.770928 0.262416 0.742269 N 0.078587 0.945416 0.743265 N 0.427572 0.932217 0.132893 N 0.761872 0.590844 0.634583 N 0.087863 0.251396 0.996965 N 0.090039 0.265519 0.498530 |
| Modification | Search Method | Total Energy (Eh) | Relative Energy (kcal/mol) |
|---|---|---|---|
| Al2MgO4-spinel-type | DM | −5193.507 | 0.0 |
| α-Cr2SiN4-type | GO | −5193.474 | 20.708 |
| β-Cr2SiN4-type | GO | −5193.438 | 43.298 |
| Na2MnCl4-type | DM | −5193.436 | 44.553 |
| γ-Cr2SiN4-type | PCAE | −5193.435 | 45.181 |
| δ-Cr2SiN4-type | GO | −5193.419 | 55.221 |
| TiMn2O4-type | DM | −5193.414 | 58.358 |
| ε-Cr2SiN4-type | GO | −5193.413 | 58.986 |
| λ-Cr2SiN4-type | GO | −5193.407 | 62.750 |
| λʹ-Cr2SiN4-type | GO | −5193.404 | 64.634 |
| Mg2SiO4-type | DM | −5193.403 | 65.261 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Škundrić, T.; Zagorac, D.; Schön, J.C.; Pejić, M.; Matović, B. Crystal Structure Prediction of the Novel Cr2SiN4 Compound via Global Optimization, Data Mining, and the PCAE Method. Crystals 2021, 11, 891. https://doi.org/10.3390/cryst11080891
Škundrić T, Zagorac D, Schön JC, Pejić M, Matović B. Crystal Structure Prediction of the Novel Cr2SiN4 Compound via Global Optimization, Data Mining, and the PCAE Method. Crystals. 2021; 11(8):891. https://doi.org/10.3390/cryst11080891
Chicago/Turabian StyleŠkundrić, Tamara, Dejan Zagorac, Johann Christian Schön, Milan Pejić, and Branko Matović. 2021. "Crystal Structure Prediction of the Novel Cr2SiN4 Compound via Global Optimization, Data Mining, and the PCAE Method" Crystals 11, no. 8: 891. https://doi.org/10.3390/cryst11080891
APA StyleŠkundrić, T., Zagorac, D., Schön, J. C., Pejić, M., & Matović, B. (2021). Crystal Structure Prediction of the Novel Cr2SiN4 Compound via Global Optimization, Data Mining, and the PCAE Method. Crystals, 11(8), 891. https://doi.org/10.3390/cryst11080891