
The Polymorphism of 2-Benzoyl-N,N-diethylbenzamide

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis, Solvent Screening and Recrystallization
2.2. Thermogravimetric Analysis (TGA)
2.3. Differential Scanning Calorimetry (DSC)
2.4. Single-Crystal X-ray Diffraction (SXD)
2.5. X-ray Powder Diffraction (XRPD)
2.6. Variable Temperature X-ray Powder Diffraction (VT-XRPD)
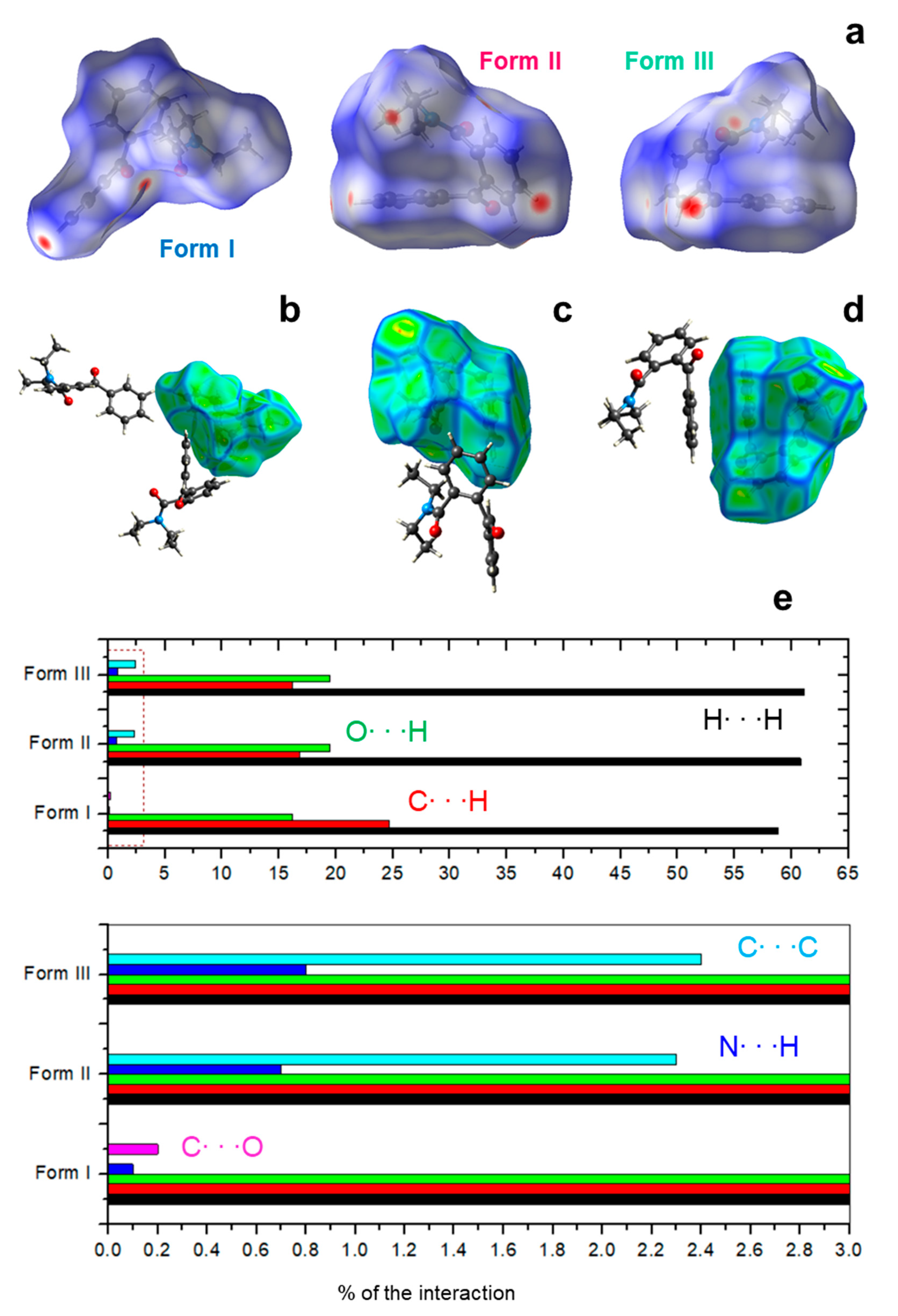
2.7. Hirshfeld Surfaces Calculations (HS)
3. Results and Discussion
3.1. Synthesis and Recrystallization
3.2. Phase Identification and Crystal Structures
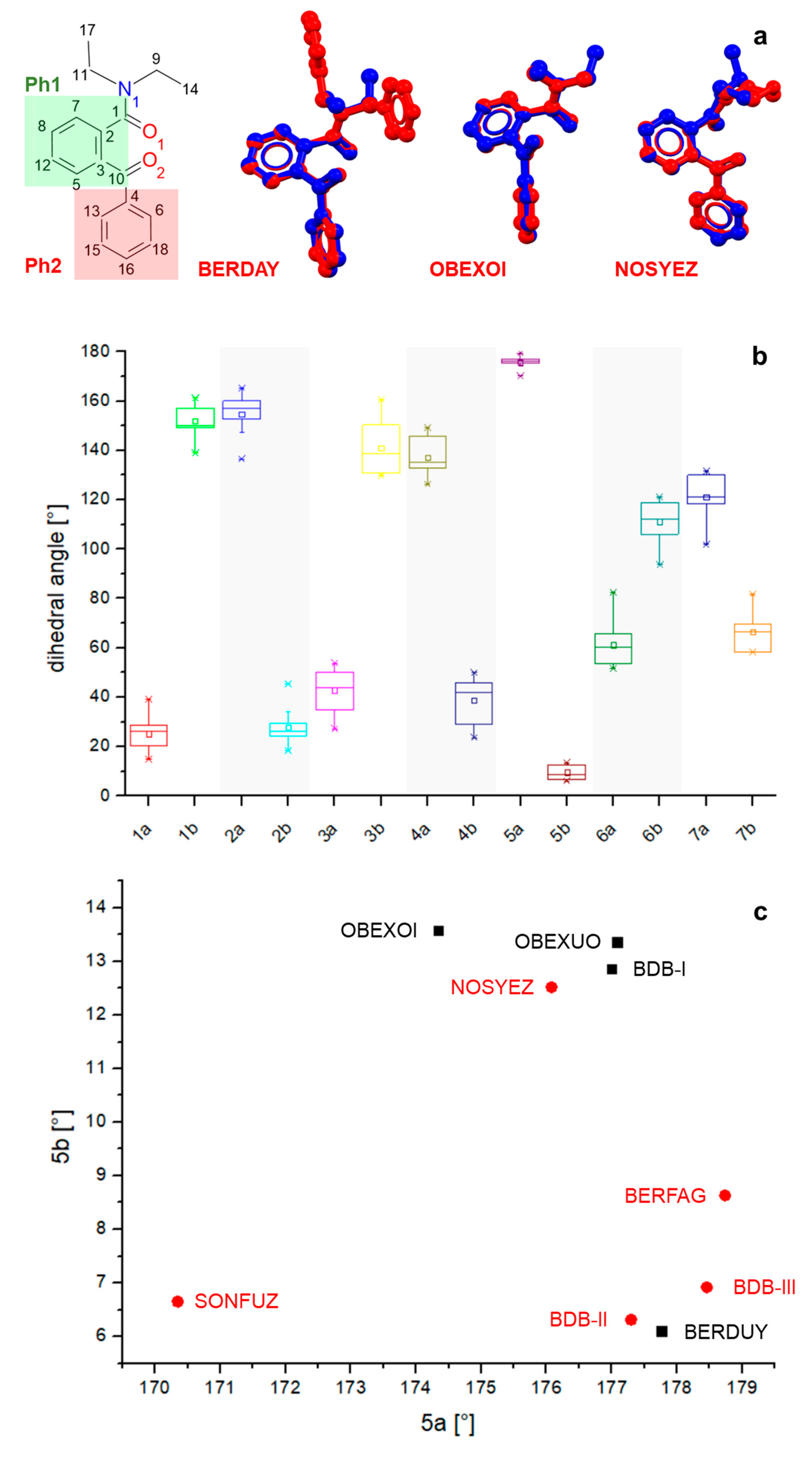
3.3. Database Survey
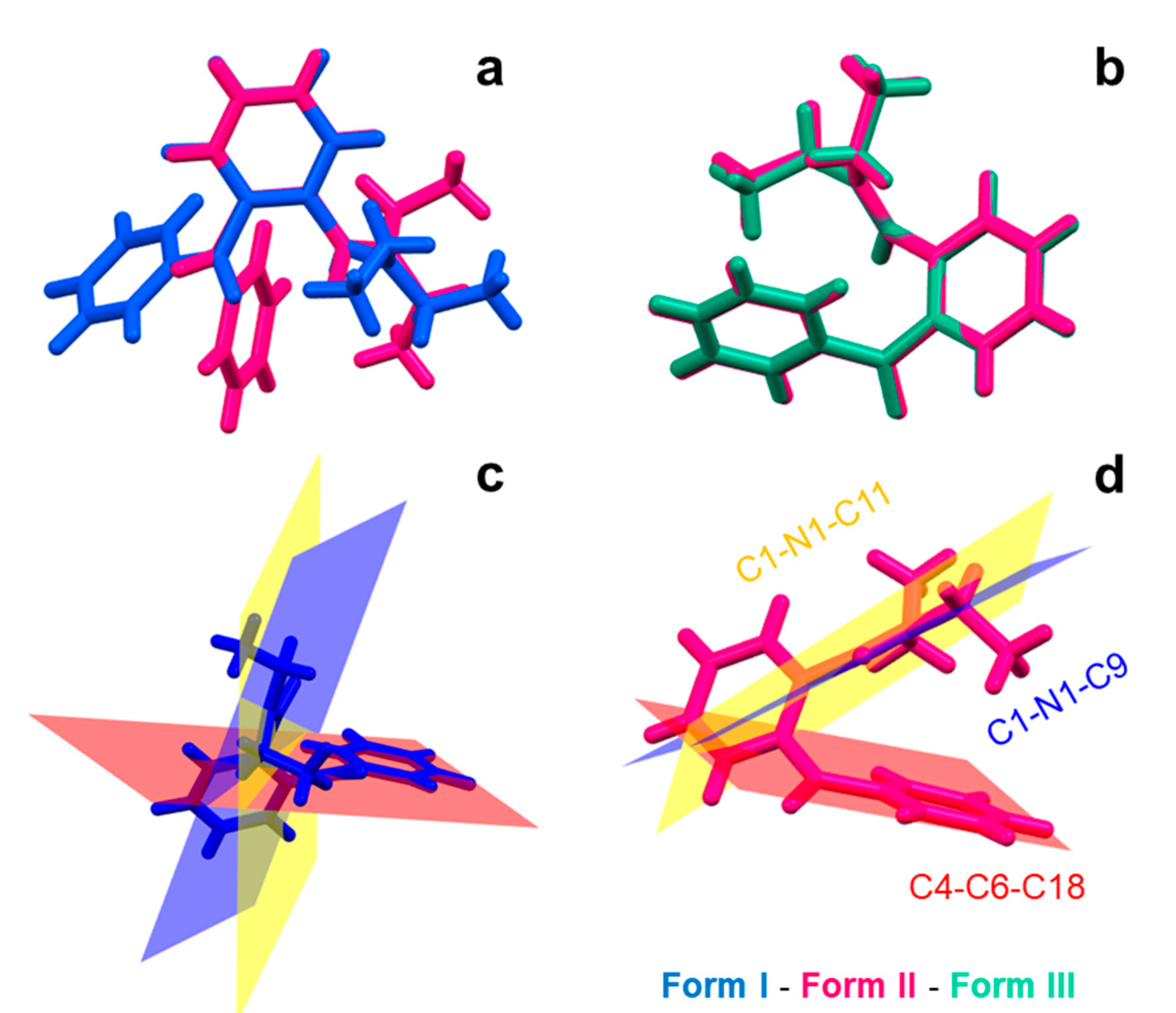
3.4. Crystal Structure Description
3.5. Thermal Characterization
3.6. Thermal Stability of Form II
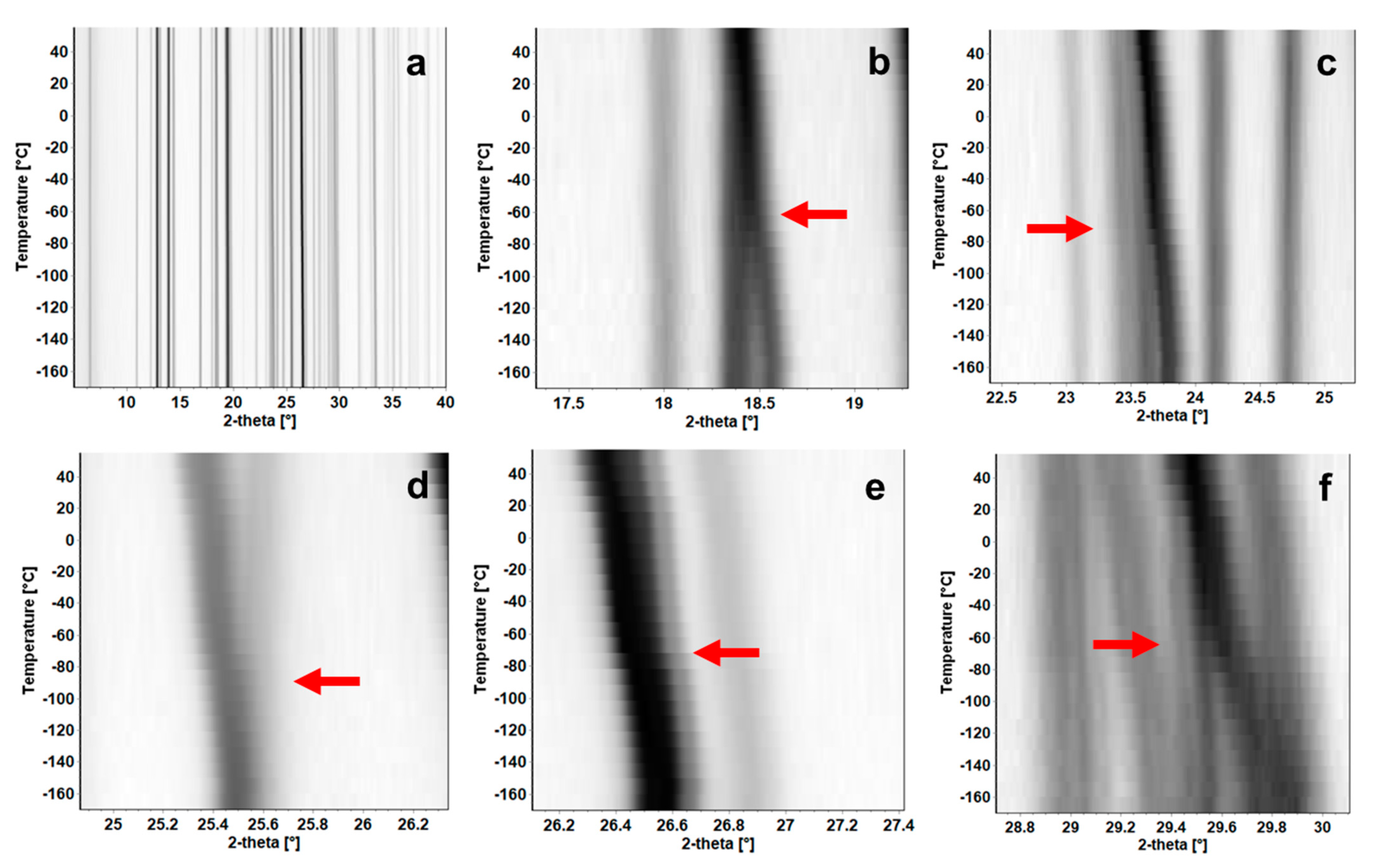
3.7. Temperature Induced Polymorphism of BDB
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olovsson, I.; Jönsson, P.-G. The Hydrogen Bond. Recent Developments in Theory and Experiments; North-Holland: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Caira, M.R. Crystalline Polymorphism of Organic Compounds. In Design of Organic Solids; Weber, E., Aoyama, Y., Caira, M.R., Desiraju, G.R., Glusker, J.P., Hamilton, A.D., Melendez, R.E., Nangia, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 163–208. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals; Oxford University Press: Oxford, UK, 2007; ISBN 9780199236565. [Google Scholar]
- Brog, J.-P.; Chanez, C.-L.; Crochet, A.; Fromm, K.M. Polymorphism, what it is and how to identify it: A systematic review. RSC Adv. 2013, 3, 16905–16931. [Google Scholar] [CrossRef] [Green Version]
- Sarma, B.; Chen, J.; Hsi, H.-Y.; Myerson, A.S. Solid forms of pharmaceuticals: Polymorphs, salts and cocrystals. Korean J. Chem. Eng. 2011, 28, 315–322. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Niazi, S.K. The scope of preformulation studies. In Handbook of Preformulation Chemical, Biological, and Botanical Drugs; Informa Healthcare: New York, NY, USA, 2007; pp. 57–86. [Google Scholar]
- Lee, R.; Yufit, D.S.; Probert, M.R.; Steed, J.W. High Pressure/Low Temperature Polymorphism in 2,6-Dimethylpyridine–Formic Acid Cocrystals. Cryst. Growth Des. 2017, 17, 1647–1653. [Google Scholar] [CrossRef]
- Cacela, C.; Baudot, A.; Duarte, M.; Matos-Beja, A.; Silva, M.R.; Paixão, J.A.; Fausto, R. Low temperature polymorphism in 3-amino-1-propanol. J. Mol. Struct. 2003, 649, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Maria, T.M.R.; Castro, R.A.E.; Silva, M.R.; Ramos, M.L.; Justino, L.; Burrows, H.D.; Canotilho, J.; Eusébio, M.E.S. Polymorphism and melt crystallisation of racemic betaxolol, a β-adrenergic antagonist drug. J. Therm. Anal. Calorim. 2013, 111, 2171–2178. [Google Scholar] [CrossRef]
- Delaney, S.P.; Smith, T.M.; Pan, D.; Yin, S.X.; Korter, T.M. Low-Temperature Phase Transition in Crystalline Aripiprazole Leads to an Eighth Polymorph. Cryst. Growth Des. 2014, 14, 5004–5010. [Google Scholar] [CrossRef]
- Foces-Foces, C.; Roux, M.V.; Notario, R.; Segura, M. Thermal behavior and polymorphism in medium–high temperature range of the sulfur containing amino acids l-cysteine and l-cystine. J. Therm. Anal. Calorim. 2011, 105, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Protiva, M.; Vejdělek, Z.J. Synthetic antispasmodics. I. Some new basic esters. Collect. Czechoslov. Chem. Commun. 1950, 15, 541–551. [Google Scholar] [CrossRef]
- Lynn, J.W.; English, J. Reaction of N,N-Dialkylbenzamides with Sodium. J. Org. Chem. 1951, 16, 1546–1555. [Google Scholar] [CrossRef]
- Sakamoto, M.; Kobaru, S.; Mino, T.; Fujita, T. Absolute asymmetric synthesis by nucleophilic carbonyl addition using chiral crystals of achiral amides. Chem. Commun. 2004, 4, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Yao, J.P.; Li, Z.Y.; Li, Q.L.; Lin, H.S.; Wang, G.W. Palladium-Catalyzed Decarboxylative ortho -Acylation of Benzamides with α-Oxocarboxylic Acids. J. Org. Chem. 2017, 82, 12715–12725. [Google Scholar] [CrossRef] [PubMed]
- Laha, J.K.; Patel, K.V.; Sharma, S. Palladium-Catalyzed Decarboxylative Ortho-Acylation of Tertiary Benzamides with Arylglyoxylic Acids. ACS Omega 2017, 2, 3806–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Park, E.; Kim, A.; Lee, Y.; Chi, K.W.; Kwak, J.H.; Jung, Y.H.; Kim, I.S. Rhodium-Catalyzed Oxidative ortho-Acylation of Benzamides with Aldehydes: Direct Functionalization of the sp 2 C–H Bond. Org. Lett. 2011, 13, 4390–4393. [Google Scholar] [CrossRef] [PubMed]
- Seganish, W.M.; DeShong, P. Application of Directed Orthometalation toward the Synthesis of Aryl Siloxanes. J. Org. Chem. 2004, 69, 6790–6795. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.-T.; Liu, Y.; Ye, L.-M.; Lei, R.-H.; Zhang, X.-J.; Yan, M. Rapid synthesis of isoquinolinones by intramolecular coupling of amides and ketones. Org. Biomol. Chem. 2015, 13, 817–824. [Google Scholar] [CrossRef]
- Hursthouse, M.B.; Coles, S.J. The UK National Crystallography Service; its origins, methods and science. Crystallogr. Rev. 2014, 20, 117–154. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPRO Software System; Version 1.171.40.82a; Rigaku Corporation: Oxford, UK, 2020. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. ORTEP -3 for Windows—A version of ORTEP -III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Cottrell, S.J.; Olsson, T.S.G.; Taylor, R.; Cole, J.C.; Liebeschuetz, J.W. Validating and Understanding Ring Conformations Using Small Molecule Crystallographic Data. J. Chem. Inf. Modeling 2012, 52, 956–962. [Google Scholar] [CrossRef]
- Bruno, I.; Cole, J.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, A.B.R.; Taylor, R.; Cooper, R.I.; And, S.E.H.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Albinati, A.; Willis, B.T.M. The Rietveld method in neutron and X-ray powder diffraction. J. Appl. Crystallogr. 1982, 15, 361–374. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Streek, J.V.D.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A.J. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. Appl. Cryst. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Visualisation and characterisation of voids in crystalline materials. CrystEngComm 2011, 13, 1804–1813. [Google Scholar] [CrossRef]
- Sakamoto, M.; Sekine, N.; Miyoshi, H.; Mino, T.; Fujita, T. Absolute Asymmetric Phthalide Synthesis via the Solid-State Photoreaction of N,N- Disubstituted 2-Benzoylbenzamides Involving a Radical Pair Intermediate. J. Am. Chem. Soc. 2000, 122, 10210–10211. [Google Scholar] [CrossRef]
- Valente, E.J.; Martin, S.B.; Sullivan, L.D. Pseudoacids. II. 2-Acylbenzoic Acid Derivatives. Acta Crystallogr. Sect. B Struct. Sci. 1998, 54, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Sun, L.; Liu, Y.; Ma, N.; Zhang, X.; Fan, X. Rh(III)-Catalyzed Oxidative Spirocyclization of Isoquinolones with α-Diazo-1,3-indandiones. Org. Lett. 2019, 21, 4082–4086. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Data | ||
|---|---|---|
| Label/REFCODE | Form II/BERDOS01 | Form III/BERDOS02 |
| Chemical Formula | C18H19NO2 | |
| MW (g/mol) | 281.34 | |
| a, b, c (Å) | 13.6350(6), 8.4424(3), 26.5721(9) | 13.629(3), 8.4793(6), 26.626(10) |
| β (°)/V (Å3) | 90.966(3), 3058.3 | 103.25(4), 2995.2 |
| Space Group | I 2/a | |
| Z, Z’ | 8/1 | |
| T (K) | 150 | 123 |
| Rint, Rw2, S | 0.0578, 0.1369, 1.026 | 0.0735, 0.1974, 1.075 |
| Recrystallization solvent | hexane: chloroform | dichloromethane |
| Pair | Atom Label | Slope | Intercept (°) | R2 | |
|---|---|---|---|---|---|
| 1 | a | C13-C4-C10-O2 | −0.95 (7) | 176 (2) | 0.9617 |
| b | C6-C4-C10-O2 | ||||
| 2 | a | C13-C4-C10-C3 | −0.92 (5) | 171 (8) | 0.9741 |
| b | C6-C4-C10-C3 | ||||
| 3 | a | C4-C10-C3-C5 | −1.18 (6) | 192 (3) | 0.9777 |
| b | C4-C10-C3-C2 | ||||
| 4 | a | O2-C10-C3-C5 | −1.16 (8) | 198 (12) | 0.9595 |
| b | O2-C10-C3-C2 | ||||
| 5 | a | C10-C3-C2-C7 | −0.04 (48) | 17 (85) | 0.0011 |
| b | C10-C3-C2-C1 | ||||
| 6 | a | C3-C2-C1-O1 | −0.89 (9) | 166 (6) | 0.9223 |
| b | O1-C1-C2-C7 | ||||
| 7 | a | C3-C2-C1-N1 | −0.82 (8) | 165 (10) | 0.9286 |
| b | N1-C1-C2-C7 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Moraes, L.S.; Liu, J.; Gopi, E.; Oketani, R.; Kennedy, A.R.; Geerts, Y.H. The Polymorphism of 2-Benzoyl-N,N-diethylbenzamide. Crystals 2021, 11, 1004. https://doi.org/10.3390/cryst11081004
de Moraes LS, Liu J, Gopi E, Oketani R, Kennedy AR, Geerts YH. The Polymorphism of 2-Benzoyl-N,N-diethylbenzamide. Crystals. 2021; 11(8):1004. https://doi.org/10.3390/cryst11081004
Chicago/Turabian Stylede Moraes, Lygia S., Jie Liu, Elumalai Gopi, Ryusei Oketani, Alan R. Kennedy, and Yves H. Geerts. 2021. "The Polymorphism of 2-Benzoyl-N,N-diethylbenzamide" Crystals 11, no. 8: 1004. https://doi.org/10.3390/cryst11081004
APA Stylede Moraes, L. S., Liu, J., Gopi, E., Oketani, R., Kennedy, A. R., & Geerts, Y. H. (2021). The Polymorphism of 2-Benzoyl-N,N-diethylbenzamide. Crystals, 11(8), 1004. https://doi.org/10.3390/cryst11081004