
First Principle Study on Mg2X (X = Si, Ge, Sn) Intermetallics by Bi Micro-Alloying


Abstract
1. Introduction
2. Model and Calculation Method
3. Results and Discussion
3.1. Lattice Parameters
3.2. Elastic Properties
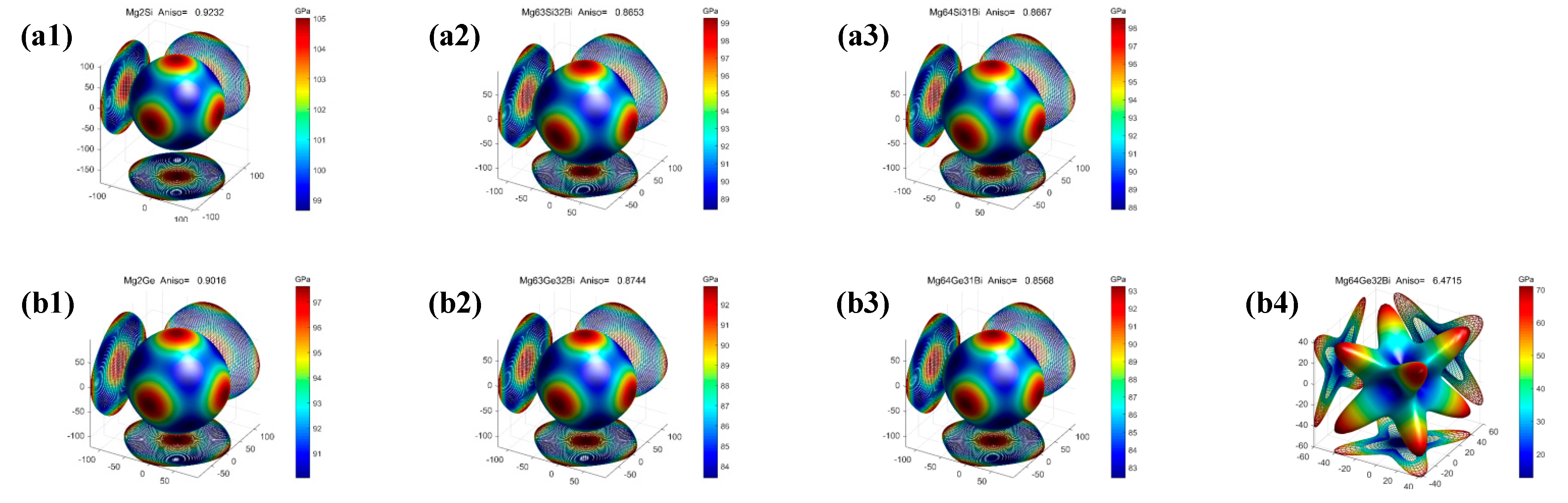
3.3. Electronic Properties
4. Conclusions
- (1)
- The lattice parameters of Mg2X are smaller than those of Bi-doped Mg2X, because the radius of doping element Bi is larger than that of alloying element X and Mg. The ΔHf of Mg64X31Bi is smaller than that of others, which indicates that the element Bi preferentially occupies the position of the X (X = Si, Ge, Sn) atom than other positions.
- (2)
- Mg2X (X = Si, Ge, Sn), Mg63X32Bi, Mg64X31Bi, Mg64Ge32Bi, and Mg64Sn32Bi are mechanically stable, while Mg64Si32Bi indicates that it cannot exist stably. The ability of Mg2Si to resist deformation after doping is enhanced, and Mg63Si32Bi has stronger deformation resistance. The doping of alloy element Bi makes the Mg2X (X = Si, Ge, Sn) alloy convert from brittle material to ductile material, and results in plasticity enhancement and stiffness reduction.
- (3)
- The pure and Bi-doped Mg2X (X = Si, Ge, Sn) exhibit elastic anisotropic properties. The anisotropy of Bi-doped the Mg2X (X = Si, Ge) phase is larger than that of Mg2X, whereas the anisotropy of Bi-doped Mg2Sn is smaller than that of Mg2Sn. Mg64Ge32Bi shows strong anisotropy among these phases.
- (4)
- In an energy range from −10 to 0 eV, there is no significant difference in the shape of TDOS between the pure and doped Mg2X phases. The contribution of Bi orbitals of Mg63X32Bi, Mg64X31Bi, and Mg63X32Bi are different, resulting in different hybridization effects in three types of Bi-doped Mg2X.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Potzies, C.; Kainer, K. Fatigue of Magnesium Alloys. Adv. Eng. Mater. 2004, 6, 281–289. [Google Scholar] [CrossRef]
- Schumann, S.; Friedrich, H.E. Current and Future Use of Magnesium in the Automobile Industry. Mater. Sci. Forum 2003, 419, 51–56. [Google Scholar] [CrossRef]
- Kulekci, M.K. Magnesium and its alloys applications in automotive industry. Int. J. Adv. Manuf. Technol. 2008, 39, 851–865. [Google Scholar] [CrossRef]
- Brungs, D. Light weight design with light metal castings. Mater. Des. 1997, 18, 285–291. [Google Scholar] [CrossRef]
- Gao, X.; Nie, J. Characterization of strengthening precipitate phases in a Mg-Zn alloy. Scr. Mater. 2007, 56, 645–648. [Google Scholar] [CrossRef]
- Friedrich, H.; Schumann, S. Research for a “new age of magnesium” in the automotive industry. J. Mater. Process. Technol. 2001, 117, 276–281. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Y.; Hou, H.; Zhang, T.; Liang, J.; Li, M.; Li, J. Development of AZ91D magnesium alloy-graphene nanoplatelets composites using thixomolding process. J. Alloy. Compd. 2019, 778, 359–374. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.; Chen, L.; Liang, J.; Li, M.; Hou, H. Graphene Nanoplatelets Reinforced Magnesium Matrix Composites Fabricated by Thixomolding. Acta Metall. Sin. 2019, 55, 638–646. [Google Scholar] [CrossRef]
- Cheng, P.; Zhao, Y.; Lu, R.; Hou, H. Effect of the morphology of long-period stacking ordered phase on mechanical properties and corrosion behavior of cast Mg-Zn-Y-Ti alloy. J. Alloy. Compd. 2018, 764, 226–238. [Google Scholar] [CrossRef]
- Li, G.H.; Gill, H.S.; Varin, R.A. Magnesium silicide intermetallic alloys. Met. Mater. Trans. A 1993, 24, 2383–2391. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, J.; Xu, S.; Peng, P. Thermal stability and elastic properties of Mg2X (X = Si, Ge, Sn, Pb) phases from first-principle calculations. Comput. Mater. Sci. 2012, 51, 409–414. [Google Scholar] [CrossRef]
- Martin, J. Thermal conductivity of Mg2Si, Mg2Ge and Mg2Sn. J. Phys. Chem. Solids 1972, 33, 1139–1148. [Google Scholar] [CrossRef]
- Jund, P.; Viennois, R.; Colinet, C.; Hug, G.; Fèvre, M.; Tédenac, J.-C. Lattice stability and formation energies of intrinsic defects in Mg2Si and Mg2Ge via first principles simulations. J. Physics Condens. Matter 2012, 25, 035403. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.; Polymeris, G.; Hatzikraniotis, E.; Paraskevopoulos, K.; Kyratsi, T. Effect of Bi-doping and Mg-excess on the thermoelectric properties of Mg2Si materials. J. Phys. Chem. Solids 2014, 75, 984–991. [Google Scholar] [CrossRef]
- Li, W.X. Magnesium and Magnesium Alloys; Central South University Press: Changsha, China, 2005; p. 102. [Google Scholar]
- Zhao, Y.H.; Zhao, X.M.; Yang, H.M.; Sui, H.; Hou, P.D. First-Principles Study on Elastic Properties and Electronic Structure of Ca, Sr and Ba Doped Mg2Si. Rare Metal. Mat. Eng. 2015, 44, 638–643. [Google Scholar]
- Wang, C.; Fu, H.; Jiang, L.; Xue, D.; Xie, J. A property-oriented design strategy for high performance copper alloys via machine learning. npj Comput. Mater. 2019, 5, 87. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, J.; Bai, G.; Zhang, L.; Hou, H. First Principles Study on the Thermodynamic and Elastic Mechanical Stability of Mg2X (X = Si,Ge) Intermetallics with (anti) Vacancy Point Defects. Crystals 2020, 10, 234. [Google Scholar] [CrossRef]
- Lisitsyn, V.; Ben-Hamu, G.; Eliezer, D.; Shin, K. The role of Ca microalloying on the microstructure and corrosion behavior of Mg-6Zn-Mn-(0.5-2)Si alloys. Corros. Sci. 2009, 51, 776–784. [Google Scholar] [CrossRef]
- Akrami, A.; Emamy, M.; Mousavian, H. The effect of Bi addition on the microstructure and tensile properties of cast Al-15%Mg2Si composite. Mater. Werkst. 2013, 44, 431–435. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, B.; Hou, H.; Chen, W.; Wang, M. Phase-field simulation for the evolution of solid/liquid interface front in directional solidification process. J. Mater. Sci. Technol. 2019, 35, 1044–1052. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, Y.; Guo, H.; Tian, X.; Hou, H. Early Stages of Precipitation in γ’ Phase of a Ni-Al-Ti Model Alloy: Phase-Field and First-Principles Study. Sci. Adv. Mater. 2020, 12, 746–754. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Hou, H.; Han, P. First-principles investigation of the structural, mechanical and thermodynamic properties of Al2Cu phase under various pressure and temperature conditions. Solid State Commun. 2017, 268, 44–50. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Deng, S.; Yang, W.; Lian, D.; Hou, H. First-principle studies on the mechanical, thermodynamic and electronic properties of β″-Mg3Gd and β′-Mg7Gd alloys under pressure. J. Phys. Chem. Solids 2019, 125, 115–122. [Google Scholar] [CrossRef]
- Fan, W.; Chen, R.; Wang, L.; Han, P.; Meng, Q. First-Principles and Experimental Studies of Y-Doped Mg2Si Prepared Using Field-Activated Pressure-Assisted Synthesis. J. Electron. Mater. 2011, 40, 1209–1214. [Google Scholar] [CrossRef]
- Meng, Q.; Fan, W.; Chen, R.; Munir, Z. Thermoelectric properties of Sc- and Y-doped Mg2Si prepared by field-activated and pressure-assisted reactive sintering. J. Alloy. Compd. 2011, 509, 7922–7926. [Google Scholar] [CrossRef]
- Stewart, J.C.; Matthew, D.S.; Chris, J.P.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First Principles Methods Using Castep. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, S.; Liu, H.; Zhang, J.; Guo, Z.; Hou, H. First-principle investigation of pressure and temperature influence on structural, mechanical and thermodynamic properties of Ti3AC2 (A = Al and Si). Comput. Mater. Sci. 2018, 154, 365–370. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Hendrik, J.M.; James, D.P. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Fischer, T.H.; Almlof, J. General methods for geometry and wave function optimization. J. Phys. Chem. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Murtaza, G.; Sajid, A.; Rizwan, M.; Takagiwa, Y.; Khachai, H.; Jibran, M.; Khenata, R.; Bin-Omran, S. First principles study of Mg2X (X = Si, Ge, Sn, Pb): Elastic, optoelectronic and thermoelectric properties. Mater. Sci. Semicond. Process. 2015, 40, 429–435. [Google Scholar] [CrossRef]
- Tani, J.-I.; Kido, H. Lattice dynamics of Mg2Si and Mg2Ge compounds from first-principles calculations. Comput. Mater. Sci. 2008, 42, 531–536. [Google Scholar] [CrossRef]
- Grosch, G.; Range, K. Studies on AB2-type Intermetallic Compounds, I. Mg2Ge and Mg2Sn: Single-crystal Structure Refinement and Abinitio Calculations. J. Alloy. Compd. 1996, 235, 250–255. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, W.-C.; Li, D.; Zeng, X.-Q.; Xu, C.-S. Predictions of the structural, electronic and thermodynamic properties of the anti-fluorite-type Mg2Sn under pressure from first principles. Phys. Scr. 2013, 88, 045–302. [Google Scholar] [CrossRef]
- Benhelal, O.; Chahed, A.; Laksari, S. First-principles calculations of the structural, electronic and optical properties of IIA-IV antifluorite compounds. Phys. Status Solidi B 2005, 242, 2022–2032. [Google Scholar] [CrossRef]
- Duan, L.J.; Liu, Y.C. Relationships Between Elastic Constants and EAM/FS Potential Functions for Cubic Crystals. Acta Metall. Sin. 2020, 56, 112–118. [Google Scholar] [CrossRef]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford University Press: New York, NY, USA, 1985. [Google Scholar]
- Zhang, X.-D.; Jiang, W. Lattice stabilities, mechanical and thermodynamic properties of Al3Tm and Al3Lu intermetallics under high pressure from first-principles calculations. Chin. Phys. B 2016, 25, 338–347. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Wang, B.; Hou, H.; Zhang, Y. The structural, mechanical and thermodynamic properties of Ti-B compounds under the influence of temperature and pressure: First-principles study. Mater. Chem. Phys. 2018, 209, 200–207. [Google Scholar] [CrossRef]
- Aydin, S.; Şimşek, M. First-principles calculations of elemental crystalline boron phases under high pressure: Orthorhombic B28 and tetragonal B48. J. Alloy. Compd. 2011, 509, 5219–5229. [Google Scholar] [CrossRef]
- Madelung, O.; Landbolt, B. Numerical Data and Functional Relationships in Science and Technology; Springer: Berlin, Germany, 1983. [Google Scholar]
- Na-Na, L.; Ren-Bo, S.; Han-Ying, S.; Da-Wei, D. The electronic structure and thermodynamic properties of Mg2Sn from first-principles calculations. Acta Phys. Sin. 2008, 57, 7145–7150. [Google Scholar] [CrossRef]
- Tani, J.-I.; Takahashi, M.; Kido, H. First-principles calculation of impurity doping into Mg2Ge. J. Alloy. Compd. 2009, 485, 764–768. [Google Scholar] [CrossRef]
- Davis, L.; Whitten, W.; Danielson, G. Elastic constants and calculated lattice vibration frequencies of Mg2Sn. J. Phys. Chem. Solids 1967, 28, 439–447. [Google Scholar] [CrossRef]
- Boulet, P.; Verstraete, M.; Crocombette, J.-P.; Briki, M.; Record, M.-C. Electronic properties of the Mg2Si thermoelectric material investigated by linear-response density-functional theory. Comput. Mater. Sci. 2011, 50, 847–851. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, S.; Zhang, B.; Yuan, Y.; Guo, Q.; Hou, H. The anisotropy of three-component medium entropy alloys in AlCoCrFeNi system: First-principle studies. J. Solid State Chem. 2019, 276, 232–237. [Google Scholar] [CrossRef]
- Liu, Q.-J.; Liu, Z.-T.; Feng, L.-P.; Tian, H.; Liu, L.; Liu, W.-T. Mechanical and thermodynamic properties of seven phases of SrHfO3: First-principles calculations. Comput. Mater. Sci. 2010, 48, 677–679. [Google Scholar] [CrossRef]
- Tian, J.; Zhao, Y.; Wen, Z.; Hou, H.; Han, P. Physical properties and Debye temperature of Al7Cu2Fe alloy under various pressures analyzed by first-principles. Solid State Commun. 2017, 257, 6–10. [Google Scholar] [CrossRef]
- Li, C.; Hoe, J.L.; Wu, P. Empirical correlation between melting temperature and cohesive energy of binary Laves phases. J. Phys. Chem. Solids 2003, 64, 201–212. [Google Scholar] [CrossRef]
- Fu, C.; Wang, X.; Ye, Y.; Ho, K. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation. Intermetallics 1999, 7, 179–184. [Google Scholar] [CrossRef]
- Otero-De-La-Roza, A.; Abbasi-Pérez, D.; Luaña, V. Gibbs2: A new version of the quasiharmonic model code. II. Models for solid-state thermodynamics, features and implementation. Comput. Phys. Commun. 2011, 182, 2232–2248. [Google Scholar] [CrossRef]
- Wen, Z.; Zhao, Y.; Hou, H.; Wang, B.; Han, P. The mechanical and thermodynamic properties of Heusler compounds Ni2XAl (X = Sc, Ti, V) under pressure and temperature: A first-principles study. Mater. Des. 2017, 114, 398–403. [Google Scholar] [CrossRef]
- Huang, Z.; Zhao, Y.; Hou, H.; Han, P. Electronic structural, elastic properties and thermodynamics of Mg17Al12, Mg2Si and Al2Y phases from first-principles calculations. Phys. B Condens. Matter 2012, 407, 1075–1081. [Google Scholar] [CrossRef]
- Ji, D.; Chong, X.; Ge, Z.-H.; Feng, J. First-principles study of pressure-induced phase transformations in thermoelectric Mg2Si. J. Alloy. Compd. 2019, 773, 988–996. [Google Scholar] [CrossRef]
- Wang, W.; Ren, Y.; Li, Y. First Principles Study of Structural Stability, Elastic Properties, and Electronic Structures of Y-Doped Mg2Si. J. Electron. Mater. 2018, 48, 1582–1589. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Lattice Constants a/Å | ΔHf (eV/atom) | ||
|---|---|---|---|---|
| This Work | Cal | Exp | ||
| pure Mg2Si | 6.371 | 6.30 [10] | 6.35 [34] | –0.170 |
| Mg64Si32 | 12.741 | –0.170 | ||
| Mg63Si32Bi | 12.791 | - | - | −0.191 |
| Mg64Si31Bi | 12.804 | - | - | −0.204 |
| Mg64Si32Bi | 12.823 | - | - | −0.175 |
| pure Mg2Ge | 6.355 | 6.318 [35] | 6.3849 [36] | −0.259 |
| Mg64Ge32 | 12.710 | −0.259 | ||
| Mg63Ge32Bi | 12.906 | - | - | −0.279 |
| Mg64Ge31Bi | 12.909 | - | - | −0.290 |
| Mg64Ge32Bi | 12.940 | - | - | −0.264 |
| pure Mg2Sn | 6.843 | 6.829 [37] | 6.759 [38] | −0.196 |
| Mg64Sn32 | 13.685 | −0.196 | ||
| Mg63Sn32Bi | 13.688 | - | - | −0.228 |
| Mg64Sn31Bi | 13.670 | - | - | −0.237 |
| Mg64Sn32Bi | 13.711 | - | - | −0.220 |
| Phase | C11 | C12 | C44 | B | G | E | G/B | ν | Az |
|---|---|---|---|---|---|---|---|---|---|
| Mg64Si32 | 111.97 | 21.55 | 41.74 | 51.69 | 43.10 | 101.17 | 0.834 | 0.174 | 0.923z |
| Exp. [44] | 126.00 | 26.00 | 48.50 | 59.00 | - | - | - | - | - |
| Cal. [11] | 121.20 | 23.70 | 49.50 | 56.20 | 49.20 | 113.50 | - | - | - |
| Cal. [45] | 118.80 | 22.27 | 44.96 | - | - | - | - | - | - |
| Mg63Si32Bi | 108.53 | 24.84 | 36.21 | 52.74 | 38.37 | 92.64 | 0.728 | 0.207 | 0.865 |
| Mg64Si31Bi | 107.64 | 24.50 | 36.03 | 52.21 | 38.15 | 92.03 | 0.731 | 0.206 | 0.867 |
| Mg64Si32Bi | 37.44 | 57.65 | 32.58 | - | - | - | - | - | - |
| Mg64Ge32 | 103.88 | 19.65 | 37.97 | 49.73 | 39.57 | 93.01 | 0.829 | 0.175 | 0.902 |
| Exp. [44] | 117.90 | 23.00 | 46.50 | 54.06 | - | - | - | - | - |
| Cal. [11] | 118.10 | 23.60 | 48.00 | 55.10 | 47.70 | 111.10 | - | 0.164 | - |
| Cal. [46] | 116.10 | 20.60 | 44.00 | 52.50 | 45.40 | 105.9 | - | 0.164 | - |
| Mg63Ge32Bi | 101.58 | 23.26 | 34.24 | 49.37 | 36.13 | 87.14 | 0.732 | 0.206 | 0.874 |
| Mg64Ge31Bi | 101.25 | 22.21 | 33.86 | 48.56 | 36.02 | 86.64 | 0.742 | 0.203 | 0.857 |
| Mg64Ge32Bi | 52.54 | 43.72 | 28.52 | 46.66 | 13.91 | 37.96 | 0.298 | 0.364 | 6.472 |
| Mg64Sn32 | 68.36 | 29.39 | 34.20 | 40.38 | 28.11 | 68.46 | 0.696 | 0.217 | 1.630 |
| Exp. [47] | 82.40 | 20.80 | 36.60 | - | - | - | - | - | - |
| Cal. [11] | 83.71 | 39.79 | 21.69 | 42.36 | 21.79 | 74.78 | 0.51 | 0.206 | - |
| Cal. [44] | 81.10 | 20.16 | 34.85 | 43.73 | 31.70 | - | - | - | - |
| Mg63Sn32Bi | 66.42 | 27.11 | 27.42 | 40.21 | 24.00 | 60.04 | 0.597 | 0.251 | 1.395 |
| Mg64Sn31Bi | 67.51 | 25.96 | 28.63 | 39.81 | 25.18 | 62.38 | 0.632 | 0.239 | 1.378 |
| Mg64Sn32Bi | 58.06 | 29.27 | 23.25 | 38.97 | 19.18 | 49.41 | 0.493 | 0.288 | 1.615 |
| Non-Spin Polarized | Spin Polarized | |
|---|---|---|
| Total Energy (eV) | −64958.92186 | −64958.92179 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, G.; Tian, J.; Guo, Q.; Li, Z.; Zhao, Y. First Principle Study on Mg2X (X = Si, Ge, Sn) Intermetallics by Bi Micro-Alloying. Crystals 2021, 11, 142. https://doi.org/10.3390/cryst11020142
Bai G, Tian J, Guo Q, Li Z, Zhao Y. First Principle Study on Mg2X (X = Si, Ge, Sn) Intermetallics by Bi Micro-Alloying. Crystals. 2021; 11(2):142. https://doi.org/10.3390/cryst11020142
Chicago/Turabian StyleBai, Guoning, Jinzhong Tian, Qingwei Guo, Zhiqiang Li, and Yuhong Zhao. 2021. "First Principle Study on Mg2X (X = Si, Ge, Sn) Intermetallics by Bi Micro-Alloying" Crystals 11, no. 2: 142. https://doi.org/10.3390/cryst11020142
APA StyleBai, G., Tian, J., Guo, Q., Li, Z., & Zhao, Y. (2021). First Principle Study on Mg2X (X = Si, Ge, Sn) Intermetallics by Bi Micro-Alloying. Crystals, 11(2), 142. https://doi.org/10.3390/cryst11020142