Unexpected Sandwiched-Layer Structure of the Cocrystal Formed by Hexamethylbenzene with 1,3-Diiodotetrafluorobenzene: A Combined Theoretical and Crystallographic Study


Abstract
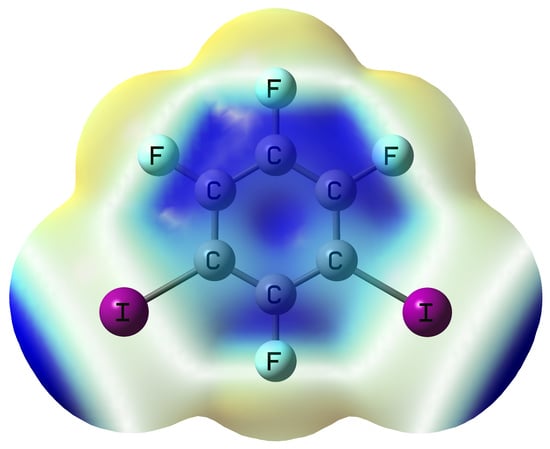
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Quantum Chemical Calculation
2.2. Crystal Preparation
2.3. Measurement
3. Results and Discussion
3.1. Noncovalent Interactions in the Gas Phase
3.2. Noncovalent Interactions in the Crystal Structure
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Desiraju, G.R. Crystal Engineering: The design of Organic Solids; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman & Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Hobza, P.; Müller-Dethlefs, K. Noncovalent Interactions. Theory and Experiment; RSC Theoretical and Computational Chemistry Series; Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Schreiner, P.R.; Chernish, L.V.; Gunchenko, P.A.; Tikhonchuk, E.Y.; Hausmann, H.; Serafin, M.; Schlecht, S.; Dahl, J.E.P.; Carlson, R.M.K.; Fokin, A.A. Overcoming Lability of Extremely Long Alkane Carbon-Carbon Bonds Through Dispersion Forces. Nature 2011, 477, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-Holes and Electrostatically-Driven Interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Thakuria, R.; Nath, N.K.; Saha, B.K. The Nature and Applications of π-π Interactions: A Perspective. Cryst. Growth Des. 2019, 19, 523–528. [Google Scholar] [CrossRef]
- Hunter, C.A.; Sanders, J.K.M. The Nature of π-π Interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Hernández-Trujillo, J.; Costas, M.; Vela, A. Quadrupole Interactions in Pure Non-dipolar Fluorinated or Methylated Benzenes and their Binary Mixtures. J. Chem. Soc. Faraday Trans. 1993, 89, 2441–2443. [Google Scholar] [CrossRef]
- Dahl, T. Crystal Structure of the Trigonal Form of the 1:1 Complex Between Hexamethylbenzene and Hexafluorobenzene. Acta Chem. Scand. 1972, 26, 1569–1575. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Pang, X.; Wang, H.; Wang, W.; Jin, W.J. Phosphorescent π-Hole···π Bonding Cocrystals of Pyrene with Haloperfluorobenzenes (F, Cl, Br, I). Cryst. Growth Des. 2015, 15, 4938–4945. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly Accurate Benchmark Calculations of the Interaction Energies in the Complexes C6H6···C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The Benzene···Naphthalene Complex: A more Challenging System than the Benzene Dimer for newly Developed Computational Methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef]
- Li, M.M.; Wang, Y.B.; Zhang, Y.; Wang, W. The Nature of the Noncovalent Interactions between Benzene and C60 Fullerene. J. Phys. Chem. A 2016, 120, 5766–5772. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Pitoňák, M.; Neogrády, P.; Řezáč, J.; Jurečka, P.; Urban, M.; Hobza, P. Benzene Dimer: High-Level Wave Function and Density Functional Theory Calculations. J. Chem. Theory Comput. 2008, 4, 1829–1834. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Wang, Y.B. The π···π Stacking Interactions between Homogeneous Dimers of C6FxI(6−x) (x = 0, 1, 2, 3, 4, and 5): A Comparative Study with the Halogen Bond. J. Phys. Chem. A 2012, 116, 12486–12491. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Uchimaru, T.; Wakisaka, A.; Ono, T. Magnitude and Directionality of Halogen Bond of Benzene with C6F5X, C6H5X, and CF3X (X = I, Br, Cl, and F). J. Phys. Chem. A 2016, 120, 7020–7029. [Google Scholar] [CrossRef]
- Nwachukwu, C.I.; Bowling, N.P.; Bosch, E. C–I···N and C–I···π Halogen Bonding in the Structures of 1-Benzyliodoimidazole Derivatives. Acta Cryst. 2017, C73, 2–8. [Google Scholar] [CrossRef]
- Tauer, T.P.; Sherrill, C.D. Beyond the Benzene Dimer: An Investigation of the Additivity of π-π Interactions. J. Phys. Chem. A 2005, 109, 10475–10478. [Google Scholar] [CrossRef]
- Saraswatula, V.G.; Sharada, D.; Saha, B.K. Stronger π···π Interaction Leads to a Smaller Thermal Expansion in Some Charge Transfer Complexes. Cryst. Growth Des. 2018, 18, 52–56. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, J.-G.; Wang, W. Unexpected Sandwiched-Layer Structure of the Cocrystal Formed by Hexamethylbenzene with 1,3-Diiodotetrafluorobenzene: A Combined Theoretical and Crystallographic Study. Crystals 2020, 10, 379. https://doi.org/10.3390/cryst10050379
Zhang Y, Wang J-G, Wang W. Unexpected Sandwiched-Layer Structure of the Cocrystal Formed by Hexamethylbenzene with 1,3-Diiodotetrafluorobenzene: A Combined Theoretical and Crystallographic Study. Crystals. 2020; 10(5):379. https://doi.org/10.3390/cryst10050379
Chicago/Turabian StyleZhang, Yu, Jian-Ge Wang, and Weizhou Wang. 2020. "Unexpected Sandwiched-Layer Structure of the Cocrystal Formed by Hexamethylbenzene with 1,3-Diiodotetrafluorobenzene: A Combined Theoretical and Crystallographic Study" Crystals 10, no. 5: 379. https://doi.org/10.3390/cryst10050379
APA StyleZhang, Y., Wang, J.-G., & Wang, W. (2020). Unexpected Sandwiched-Layer Structure of the Cocrystal Formed by Hexamethylbenzene with 1,3-Diiodotetrafluorobenzene: A Combined Theoretical and Crystallographic Study. Crystals, 10(5), 379. https://doi.org/10.3390/cryst10050379