Effect of Choice of Solvent on Crystallization Pathway of Paracetamol: An Experimental and Theoretical Case Study




Abstract
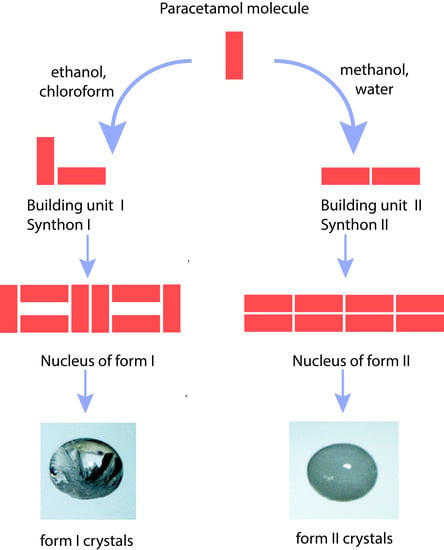
1. Introduction
2. Materials and Methods
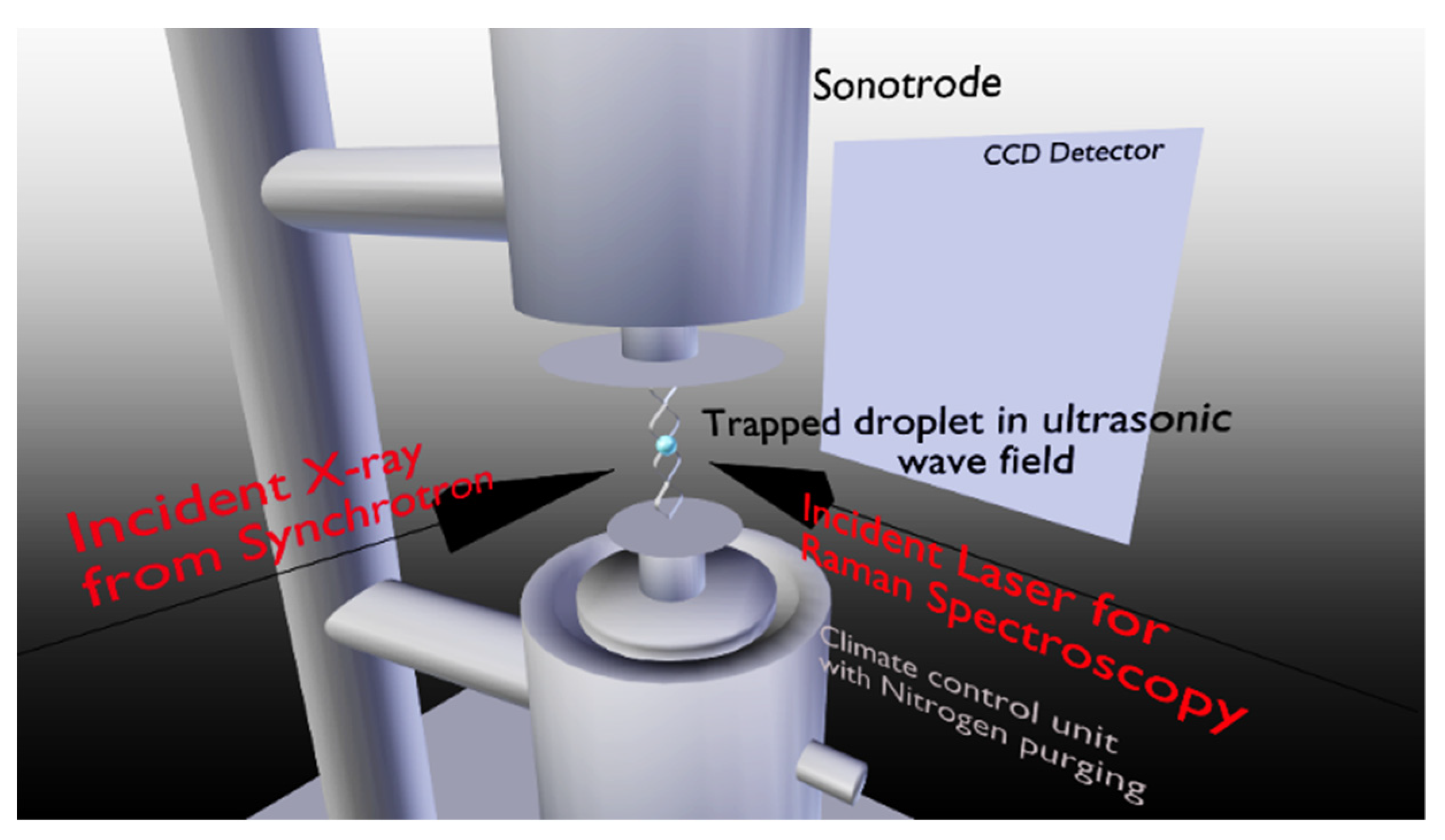
2.1. Crystallization Experiments
2.2. Molecular Dynamics Simulations
2.2.1. Conformer Preparation
2.2.2. Simulation Protocol
2.3. Analysis of the Trajectory
3. Results
4. Discussion
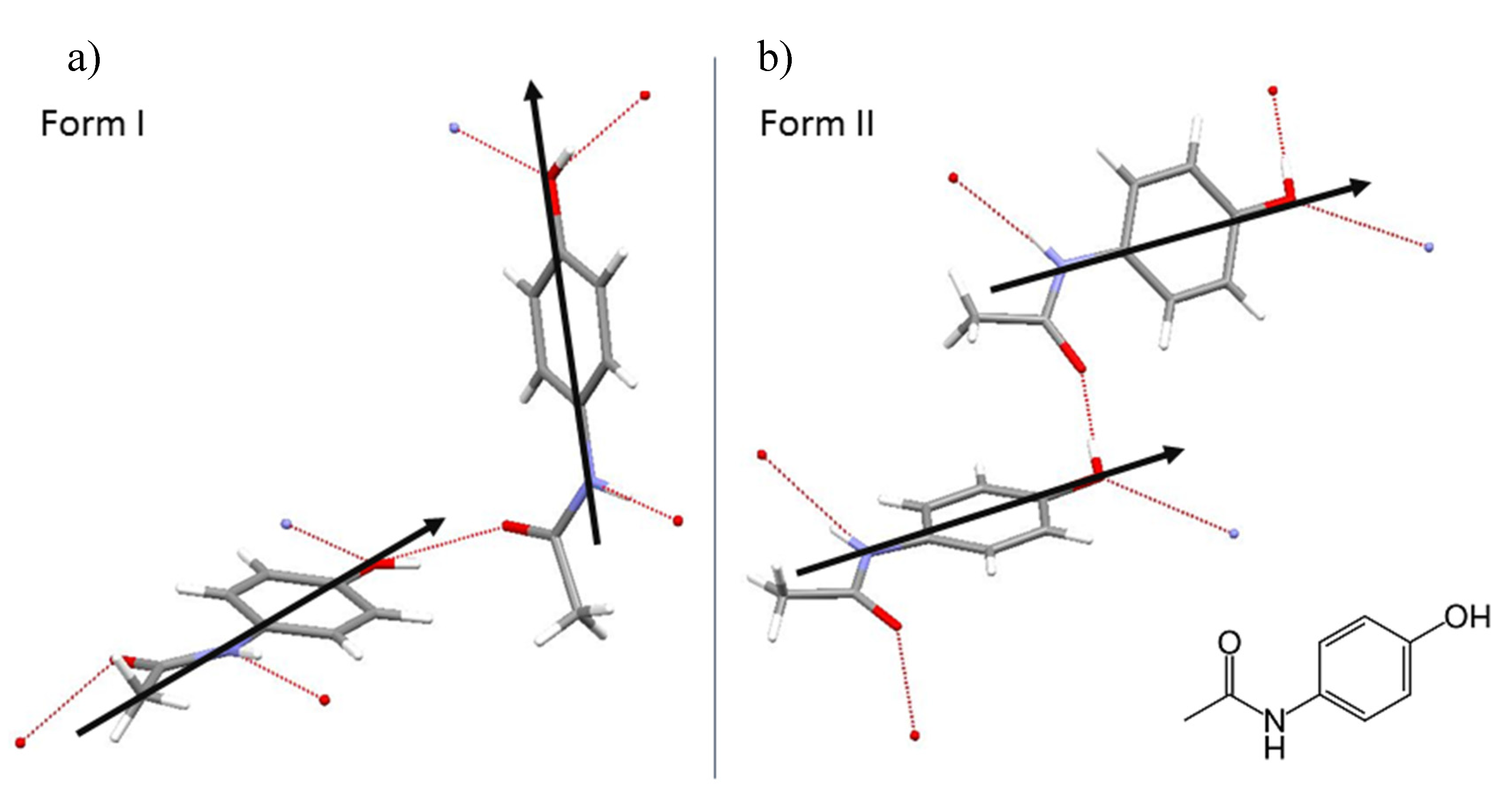
4.1. MD Simulation
4.2. Wet Lab Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wille, R.L.; Lutton, E.S. Polymorphism of Cocoa Butter. J. Am. Oil Chem. Soc. 1966, 43, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Roth, K. Von vollmilch bis bitter, edelste polymorphie. Chem. Unserer Zeit 2005, 39, 416–428. [Google Scholar] [CrossRef]
- Bernstein, J. Polymorphism—A Perspective. Cryst. Growth Des. 2011, 11, 632–650. [Google Scholar] [CrossRef]
- Bučar, D.K.; Lancaster, R.W.; Bernstein, J. Disappearing Polymorphs Revisited. Angew. Chem. Int. Ed. Engl. 2015, 54, 6972–6993. [Google Scholar] [CrossRef] [PubMed]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Hilfiker, R. Polymorphism: In the Pharmaceutical Industry; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Ochsenbein, P.; Schenk, K.J. Crystallography for Polymorphs. In Polymorphism; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 139–166. [Google Scholar] [CrossRef]
- Davey, R.J.; Schroeder, S.L.M.; Ter Horst, J.H. Nucleation of Organic Crystals—A Molecular Perspective. Angew. Chem. Int. Ed. 2013, 52, 2167–2179. [Google Scholar] [CrossRef]
- Pan, W.; Kolomeisky, A.B.; Vekilov, P.G.; Pan, W. Nucleation of Ordered Solid Phases of Proteins via a Disordered High-Density State: Phenomenological Approach. J. Chem. Phys. 2005, 122, 174905. [Google Scholar] [CrossRef]
- Karthika, S.; Radhakrishnan, T.K.; Kalaichelvi, P. A Review of Classical and Nonclassical Nucleation Theories. Cryst. Growth Des. 2016, 16, 6663–6681. [Google Scholar] [CrossRef]
- Volmer, M. Kinetic Der Phasenbildung; Verlag Von Theodor Steinkopff: Dresden, Germany, 1939; Chapter 4. [Google Scholar]
- Becker, R.; Döring, W. Kinetische Behandlung Der Keimbildung in Übersättigten Dämpfen. Ann. Phys. 1935, 416, 719–752. [Google Scholar] [CrossRef]
- Frenkel, J. A General Theory of Heterophase Fluctuations and Pretransition Phenomena. J. Chem. Phys. 1939, 7, 538–547. [Google Scholar] [CrossRef]
- ten Wolde, P.R.; Frenkel, D. Enhancement of Protein Crystal Nucleation by Critical Censity Fluctuations. Science 1997, 277, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Gliko, O.; Neumaier, N.; Pan, W.; Haase, I.; Fischer, M.; Bacher, A.; Weinkauf, S.; Vekilov, P.G. A Metastable Prerequisite for the Growth of Lumazine Synthase Crystals. J. Am. Chem. Soc. 2005, 127, 3433–3438. [Google Scholar] [CrossRef] [PubMed]
- Galkin, O.; Vekilov, P.G. Control of Protein Crystal Nucleation around the Metastable Liquid–Liquid Phase Boundary. Proc. Natl. Acad. Sci. USA 2000, 97, 6277–6281. [Google Scholar] [CrossRef]
- Maes, D.; Vorontsova, M.A.; Potenza, M.A.C.; Sanvito, T.; Sleutel, M.; Giglio, M.; Vekilov, P.G. Do Protein Crystals Nucleate within Dense Liquid Clusters? Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.H.; Aloni, S.; De Yoreo, J.J. In Situ TEM Imaging of CaCO3 Nucleation Reveals Coexistence of Direct and Indirect Pathways. Science 2014, 345, 1158–1162. [Google Scholar] [CrossRef]
- Leiterer, J.; Leitenberger, W.; Emmerling, F.; Thünemann, A.F.; Panne, U. The Use of an Acoustic Levitator to Follow Crystallization in Small Droplets by Energy-Dispersive X-Ray Diffraction. J. Appl. Crystallogr. 2006, 39, 771–773. [Google Scholar] [CrossRef]
- Leiterer, J.; Delissen, F.; Emmerling, F.; Thunemann, A.F.; Panne, U. Structure Analysis Using Acoustically Levitated Droplets. Anal. Bioanal. Chem. 2008, 391, 1221–1228. [Google Scholar] [CrossRef]
- Leiterer, J.; Emmerling, F.; Panne, U.; Christen, W.; Rademann, K. Tracing Coffee Tabletop Traces. Langmuir 2008, 24, 7970–7978. [Google Scholar] [CrossRef]
- Matsumoto, M.; Saito, S.; Ohmine, I. Molecular Dynamics Simulation of the Ice Nucleation and Growth Process Leading to Water Freezing. Nature 2002, 416, 409–413. [Google Scholar] [CrossRef]
- Kimura, T.; Maruyama, S. Molecular Dynamics Simulation of Heterogeneous Nucleation of a Liquid Droplet on a Solid Surface. Microscale Thermophys. Eng. 2002, 6, 3–13. [Google Scholar] [CrossRef]
- Sanz, E.; Vega, C.; Espinosa, J.R.; Caballero-Bernal, R.; Abascal, J.L.F.; Valeriani, C. Homogeneous Ice Nucleation at Moderate Supercooling from Molecular Simulation. J. Am. Chem. Soc. 2013, 135, 15008–15017. [Google Scholar] [CrossRef]
- Chakraborty, D.; Patey, G.N. How Crystals Nucleate and Grow in Aqueous NaCl Solution. J. Phys. Chem. Lett. 2013. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Thi, Y.; Rademann, K.; Emmerling, F. Direct Evidence of Polyamorphism in Paracetamol. CrystEngComm 2015, 17, 9029–9036. [Google Scholar] [CrossRef]
- Nguyen, T.Y.; Roessler, E.A.; Rademann, K.; Emmerling, F. Control of Organic Polymorph Formation: Crystallization Pathways in Acoustically Levitated Droplets. Z. Krist.—Cryst. Mater. 2017, 232, 15–24. [Google Scholar] [CrossRef]
- Perrin, M.A.; Neumann, M.A.; Elmaleh, H.; Zaske, L. Crystal Structure Determination of the Elusive Paracetamol Form III. Chem. Commun. 2009, 3181–3183. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.; Day, G.M.; Price, S.L. The Prediction, Morphology, and Mechanical Properties of the Polymorphs of Paracetamol. J. Am. Chem. Soc. 2001, 123, 5086–5094. [Google Scholar] [CrossRef]
- Di Martino, P.; Conflant, P.; Drache, M.; Huvenne, J.P.; Guyot-Hermann, A.M. Preparation and Physical Characterization of Forms II and III of Paracetamol. J. Therm. Anal. 1997, 48, 447–458. [Google Scholar] [CrossRef]
- Szelagiewicz, M.; Marcolli, C.; Cianferani, S.; Hard, A.P.; Vit, A.; Burkhard, A.; von Raumer, M.; Hofmeier, U.C.; Zilian, A.; Francotte, E.; et al. In Situ characterization of polymorphic forms the Potential of Raman Techniques. J. Therm. Anal. Calorim. 1999, 57, 23–43. [Google Scholar] [CrossRef]
- Smith, S.J.; Bishop, M.M.; Montgomery, J.M.; Hamilton, T.P.; Vohra, Y.K. Polymorphism in Paracetamol: Evidence of Additional Forms IV and V at High Pressure. J. Phys. Chem. A 2014, 118, 6068–6077. [Google Scholar] [CrossRef]
- Haisa, B.Y.M.; Kashino, S.; Maeda, H. The Orthorhombic Form of P-Hydroxyacetanilide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 2510–2512. [Google Scholar] [CrossRef]
- Haisa, M.; Kashino, S.; Kawai, R.; Maeda, H. The Monoclinic Form of p-Hydroxyacetanilide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1976, 32, 1283–1285. [Google Scholar] [CrossRef]
- Naumov, D.Y.; Vasilchenko, M.A.; Howard, J.A.K.; IUCr. The Monoclinic Form of Acetaminophen at 150K. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 653–655. [Google Scholar] [CrossRef]
- Nicnols, G.; Frampton, C.S. Physicochemical Characterization of the Orthorhombic Polymorph of Paracetamol Crystallized from Solution. J. Pharm. Sci. 1998, 87, 684–693. [Google Scholar] [CrossRef]
- Welton, J.M.; McCarthy, G.J. X-Ray Powder Data for Acetaminophen (C8H9NO2). Powder Diffr. 1988, 3, 102–103. [Google Scholar] [CrossRef]
- Joiris, E.; Di Martino, P.; Berneron, C.; Guyot-Hermann, A.; Guyot, J. Compression Behavior of Orthorhombic Paracetamol. Pharm. Res. 1998, 15, 1122–1130. [Google Scholar] [CrossRef]
- Censi, R.; Di Martino, P. Polymorph Impact on the Bioavailability and Stability of Poorly Soluble Drugs. Molecules 2015, 20, 18759–18776. [Google Scholar] [CrossRef]
- Schlegel, M.C.; Wenzel, K.J.; Sarfraz, A.; Panne, U.; Emmerling, F. A Wall-Free Climate Unit for Acoustic Levitators. Rev. Sci. Instrum. 2012, 83, 2013–2016. [Google Scholar] [CrossRef]
- Boldyreva, E.V.; Shakhtshneider, T.P.; Vasilchenko, M.A.; Ahsbahs, H.; Uchtmann, H. Anisotropic Crystal Structure Distortion of the Monoclinic Polymorph of Acetaminophen at High Hydrostatic Pressures. Acta Crystallogr. Sect. B Struct. Sci. 2000, 56, 299–309. [Google Scholar] [CrossRef]
- Drebushchak, T.N.; Boldyreva, E.V. Variable Temperature (100–360 K) Single-Crystal X-Ray Diffraction Study of the Orthorhombic Polymorph of Paracetamol (p-Hydroxyacetanilide). Z. Krist.—Cryst. Mater. 2004, 219. [Google Scholar] [CrossRef]
- Dien, H.; Deane, C.M.; Knapp, B. Gro2mat: A Package to Efficiently Read Gromacs Output in MATLAB. J. Comput. Chem. 2014, 35, 1528–1531. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorph | I | II | III | IV | V | Polyamorphous I, II |
|---|---|---|---|---|---|---|
| Accessibility | Solvent choice, thermal | Solvent choice, thermal | Thermal only | Diamond-anvil cell | Diamond-anvil cell | Solvent dependent |
| Stability | Stable | Metastable | Unstable | Stable at 8 GPa | Stable at 11 GPa | Unstable |
| Time (picoseconds) | 0 | 1 | 10 | 15 | 20 | 25 | 30 | 35 | 40 | 50 | 60 | 80 | 100 | 110 |
| Temperature in Kelvin | 100 | 200 | 300 | 400 | 500 | 600 | 600 | 500 | 400 | 300 | 200 | 200 | 100 | 100 |
| Paracetamol Form I | Paracetamol Form II |
|---|---|
| Ethanol | Methanol |
| Chloroform | Water |
| Solvent | Number of Hydrogen Bonds |
|---|---|
| Chloroform | 16,376 |
| Ethanol | 2199 |
| Methanol | 1009 |
| Water | 523 |
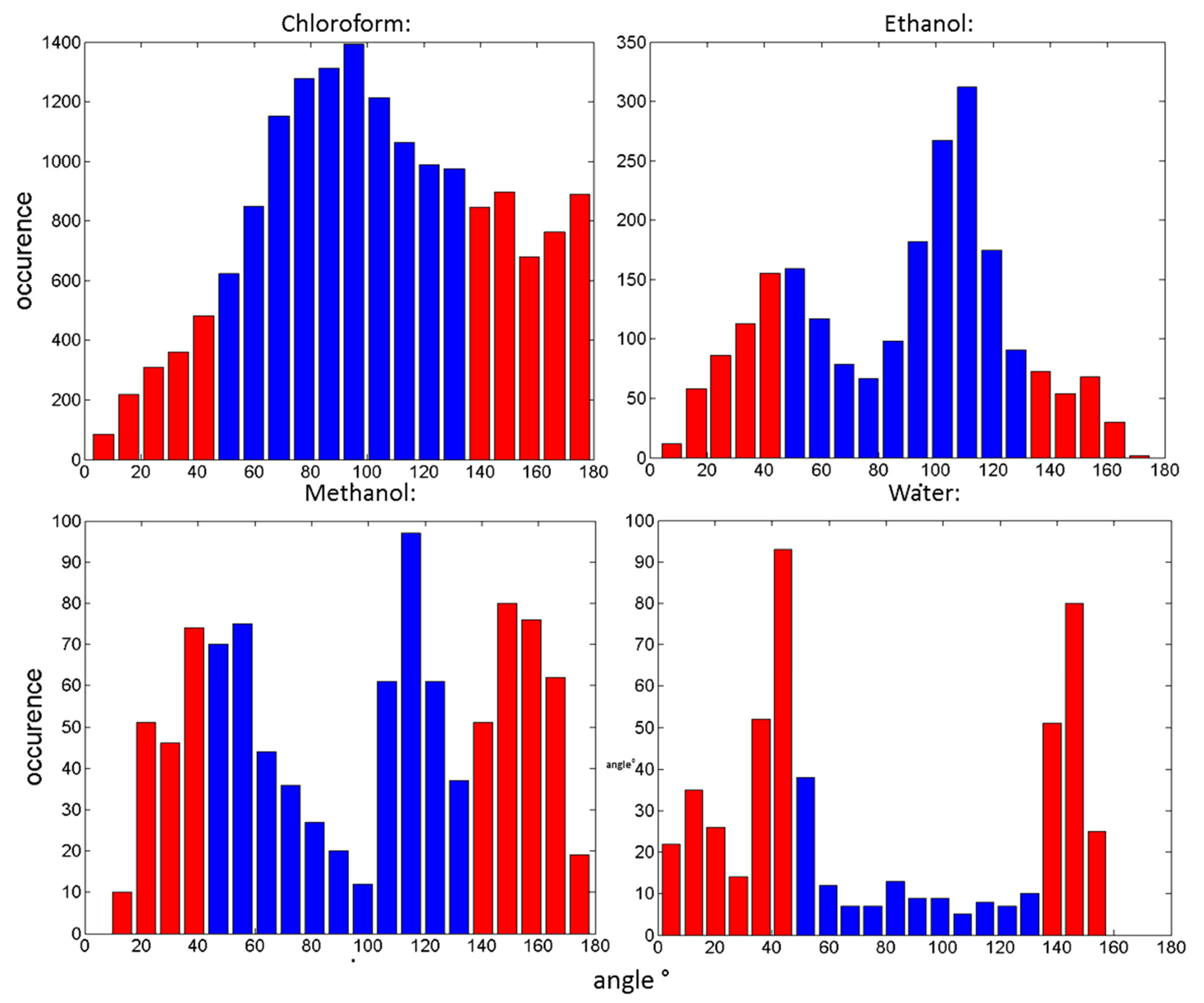
| Solvent | Percentage Distribution within the Range of 45–135° |
|---|---|
| Chloroform | 66.66 |
| Ethanol | 64.26 |
| Methanol | 46.28 |
| Water | 14.14 |
| Percentage of Ethanol | Percentage of Methanol | Type of Polymorph Observed |
|---|---|---|
| 90 | 10 | Form I: 90% Form II: 10% |
| 50 | 50 | Form I: 95% Form II: 5% |
| 20 | 80 | Form I: 100% Form II: NA |
| 5 | 95 | Form I: 55% Form II: 45% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chewle, S.; Emmerling, F.; Weber, M. Effect of Choice of Solvent on Crystallization Pathway of Paracetamol: An Experimental and Theoretical Case Study. Crystals 2020, 10, 1107. https://doi.org/10.3390/cryst10121107
Chewle S, Emmerling F, Weber M. Effect of Choice of Solvent on Crystallization Pathway of Paracetamol: An Experimental and Theoretical Case Study. Crystals. 2020; 10(12):1107. https://doi.org/10.3390/cryst10121107
Chicago/Turabian StyleChewle, Surahit, Franziska Emmerling, and Marcus Weber. 2020. "Effect of Choice of Solvent on Crystallization Pathway of Paracetamol: An Experimental and Theoretical Case Study" Crystals 10, no. 12: 1107. https://doi.org/10.3390/cryst10121107
APA StyleChewle, S., Emmerling, F., & Weber, M. (2020). Effect of Choice of Solvent on Crystallization Pathway of Paracetamol: An Experimental and Theoretical Case Study. Crystals, 10(12), 1107. https://doi.org/10.3390/cryst10121107