Probing Lithium-Ion Battery Electrolytes with Laboratory Near-Ambient Pressure XPS



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Near-Ambient Pressure X-ray Photoelectron Spectroscopy (NAP-XPS)
2.3. Sample Preparation
2.3.1. Lithium Hexafluorophosphate (LiPF6) in EC/DMC (LP30)
2.3.2. Lithium Bis(Trifluoromethane) Sulfonimide in PC
3. Results and Discussion
3.1. LiPF6 (Commercial) Electrolyte—Vapor Only and Vapor-Liquid Mix
3.2. LiPF6 (Commercial) Electrolyte-V2O5 Single Crystal in Solution
3.3. LiPF6 (Commercial) Electrolyte-V2O5 Crystal after Cleaning
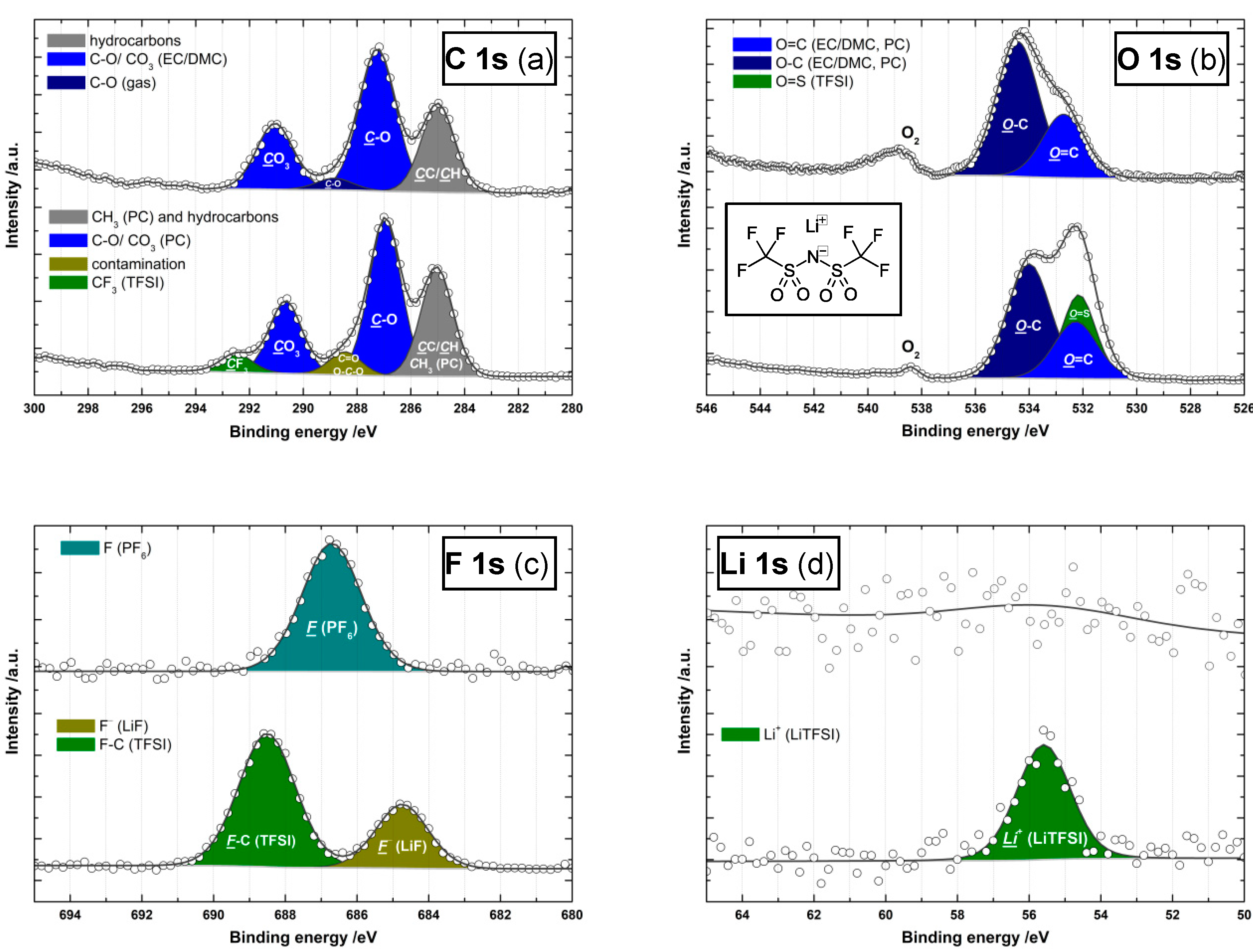
3.4. Comparison of LiTFSI (Model) and LiPF6 (Commercial) Electrolyte
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salmeron, M.; Schlögl, R. Ambient pressure photoelectron spectroscopy: A new tool for surface science and nanotechnology. Surf. Sci. Rep. 2008, 63, 169–199. [Google Scholar] [CrossRef]
- Bluhm, H. Photoelectron spectroscopy of surfaces under humid conditions. J. Electron Spectrosc. Relat. Phenom. 2010, 177, 71–84. [Google Scholar] [CrossRef]
- Brown, M.A.; Jordan, I.; Beloqui Redondo, A.; Kleibert, A.; Wörner, H.J.; van Bokhoven, J.A. In situ photoelectron spectroscopy at the liquid/nanoparticle interface. Surf. Sci. 2013, 610, 1–6. [Google Scholar] [CrossRef]
- Trotochaud, L.; Head, A.R.; Karslıoğlu, O.; Kyhl, L.; Bluhm, H. Ambient pressure photoelectron spectroscopy: Practical considerations and experimental frontiers. J. Phys. Condens. Matter 2016, 29, 053002. [Google Scholar] [CrossRef]
- Arble, C.; Jia, M.; Newberg, J.T. Lab-based ambient pressure X-ray photoelectron spectroscopy from past to present. Surf. Sci. Rep. 2018, 73, 37–57. [Google Scholar] [CrossRef]
- Palomino, R.M.; Hamlyn, R.; Liu, Z.; Grinter, D.C.; Waluyo, I.; Rodriguez, J.A.; Senanayake, S.D. Interfaces in heterogeneous catalytic reactions: Ambient pressure XPS as a tool to unravel surface chemistry. J. Electron Spectrosc. Relat. Phenom. 2017, 221, 28–43. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Bluhm, H.; Liu, Z. In situ investigation of electrochemical devices using ambient pressure photoelectron spectroscopy. J. Electron Spectrosc. Relat. Phenom. 2013, 190, 84–92. [Google Scholar] [CrossRef]
- Dou, J.; Sun, Z.; Opalade, A.A.; Wang, N.; Fu, W.; Tao, F. Operando chemistry of catalyst surfaces during catalysis. Chem. Soc. Rev. 2017, 46, 2001–2027. [Google Scholar] [CrossRef]
- Starr, D.E.; Favaro, M.; Abdi, F.F.; Bluhm, H.; Crumlin, E.J.; van de Krol, R. Combined soft and hard X-ray ambient pressure photoelectron spectroscopy studies of semiconductor/electrolyte interfaces. J. Electron Spectrosc. Relat. Phenom. 2017, 221, 106–115. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Hong, W.T.; Crumlin, E.J.; Bluhm, H.; Shao-Horn, Y. Insights into Electrochemical Reactions from Ambient Pressure Photoelectron Spectroscopy. Acc. Chem. Res. 2015, 48, 2976–2983. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for Rechargeable Li Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- An, S.J.; Li, J.; Daniel, C.; Mohanty, D.; Nagpure, S.; Wood, D.L. The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling. Carbon 2016, 105, 52–76. [Google Scholar] [CrossRef]
- Peled, E.; Menkin, S. Review—SEI: Past, Present and Future. J. Electrochem. Soc. 2017, 164, A1703. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novák, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Wu, Q.-H.; Thißen, A.; Jaegermann, W. Photoelectron spectroscopic study of Li intercalation into V2O5 thin films. Surf. Sci. 2005, 578, 203–212. [Google Scholar] [CrossRef]
- Maibach, J.; Källquist, I.; Andersson, M.; Urpelainen, S.; Edström, K.; Rensmo, H.; Siegbahn, H.; Hahlin, M. Probing a battery electrolyte drop with ambient pressure photoelectron spectroscopy. Nat. Commun. 2019, 10, 3080. [Google Scholar] [CrossRef]
- Kjærvik, M.; Schwibbert, K.; Dietrich, P.; Thissen, A.; Unger, W.E.S. Surface characterisation of Escherichia coli under various conditions by near-ambient pressure XPS. Surf. Interface Anal. 2018, 50, 996–1000. [Google Scholar] [CrossRef]
- Dietrich, P.M.; Bahr, S.; Yamamoto, T.; Meyer, M.; Thissen, A. Chemical surface analysis on materials and devices under functional conditions—Environmental photoelectron spectroscopy as non-destructive tool for routine characterization. J. Electron Spectrosc. Relat. Phenom. 2019, 231, 118–126. [Google Scholar] [CrossRef]
- Mocanu, A.; Isopencu, G.; Busuioc, C.; Popa, O.-M.; Dietrich, P.; Socaciu-Siebert, L. Bacterial cellulose films with ZnO nanoparticles and propolis extracts: Synergistic antimicrobial effect. Sci. Rep. 2019, 9, 17687. [Google Scholar] [CrossRef]
- Arinchtein, A.; Schmack, R.; Kraffert, K.; Radnik, J.; Dietrich, P.; Sachse, R.; Kraehnert, R. Role of Water in Phase Transformations and Crystallization of Ferrihydrite and Hematite. ACS Appl. Mater. Interfaces 2020, 12, 38714–38722. [Google Scholar] [CrossRef]
- Daubert, T.E.; Daubert, T.E.; Danner, R.P. Physical and Thermodynamic Properties of Pure Chemicals: Data Compilation; Taylor & Francis: Washington, DC, USA, 1989; ISBN 978-0-89116-948-2. [Google Scholar]
- Howard, P.H.; Meylan, W.M. Handbook of Physical Properties of Organic Chemicals; Lewis: Boca Raton, FL, USA, 1997; ISBN 978-1-56670-227-0. [Google Scholar]
- Nelson, R.F.; Adams, R.N. Propylene carbonate: A versatile solvent for electrochemistry and EPR. J. Electroanal. Chem. Interfacial Electrochem. 1967, 13, 184–187. [Google Scholar] [CrossRef]
- ISO 15472:2010 Surface Chemical Analysis—X-ray Photoelectron Spectrometers—Calibration of Energy Scales; International Organization for Standardization: Geneva, Switzerland, 2010.
- Avval, T.G.; Chatterjee, S.; Hodges, G.T.; Bahr, S.; Dietrich, P.; Meyer, M.; Thißen, A.; Linford, M.R. Oxygen gas, O2(g), by near-ambient pressure XPS. Surf. Sci. Spectra 2019, 26, 014021. [Google Scholar] [CrossRef]
- Patel, D.I.; Bahr, S.; Dietrich, P.; Meyer, M.; Thißen, A.; Linford, M.R. Ambient air, by near-ambient pressure XPS. Surf. Sci. Spectra 2019, 26, 024002. [Google Scholar] [CrossRef]
- Andersson, A.M.; Abraham, D.P.; Haasch, R.; MacLaren, S.; Liu, J.; Amine, K. Surface Characterization of Electrodes from High Power Lithium-Ion Batteries. J. Electrochem. Soc. 2002, 149, A1358. [Google Scholar] [CrossRef]
- Nie, M.; Abraham, D.P.; Seo, D.M.; Chen, Y.; Bose, A.; Lucht, B.L. Role of Solution Structure in Solid Electrolyte Interphase Formation on Graphite with LiPF6 in Propylene Carbonate. J. Phys. Chem. C 2013, 117, 25381–25389. [Google Scholar] [CrossRef]
- Nie, M.; Lucht, B.L. Role of Lithium Salt on Solid Electrolyte Interface (SEI) Formation and Structure in Lithium Ion Batteries. J. Electrochem. Soc. 2014, 161, A1001. [Google Scholar] [CrossRef]
- Hedman, J.; Hedén, P.-F.; Nordling, C.; Siegbahn, K. Energy splitting of core electron levels in paramagnetic molecules. Phys. Lett. A 1969, 29, 178–179. [Google Scholar] [CrossRef]
- Powell, C.J.; Seah, M.P. Precision, accuracy, and uncertainty in quantitative surface analyses by Auger-electron spectroscopy and x-ray photoelectron spectroscopy. J. Vac. Sci. Technol. A 1990, 8, 735–763. [Google Scholar] [CrossRef]
- Dietrich, P.M.; Streeck, C.; Glamsch, S.; Ehlert, C.; Lippitz, A.; Nutsch, A.; Kulak, N.; Beckhoff, B.; Unger, W.E.S. Quantification of Silane Molecules on Oxidized Silicon: Are there Options for a Traceable and Absolute Determination? Anal. Chem. 2015, 87, 10117–10124. [Google Scholar] [CrossRef]
- Avval, T.G.; Cushman, C.V.; Bahr, S.; Dietrich, P.; Meyer, M.; Thißen, A.; Linford, M.R. Dimethyl sulfoxide by near-ambient pressure XPS. Surf. Sci. Spectra 2019, 26, 014020. [Google Scholar] [CrossRef]
- Patel, D.I.; O’Tani, J.; Bahr, S.; Dietrich, P.; Meyer, M.; Thißen, A.; Linford, M.R. Ethylene glycol, by near-ambient pressure XPS. Surf. Sci. Spectra 2019, 26, 024007. [Google Scholar] [CrossRef]
- Shah, D.; Patel, D.I.; Bahr, S.; Dietrich, P.; Meyer, M.; Thißen, A.; Linford, M.R. Liquid water, by near-ambient pressure XPS. Surf. Sci. Spectra 2019, 26, 024003. [Google Scholar] [CrossRef]
- Shi, P.; Zheng, H.; Liang, X.; Sun, Y.; Cheng, S.; Chen, C.; Xiang, H. A highly concentrated phosphate-based electrolyte for high-safety rechargeable lithium batteries. Chem. Commun. 2018, 54, 4453–4456. [Google Scholar] [CrossRef] [PubMed]
- Rezqita, A.; Sauer, M.; Foelske, A.; Kronberger, H.; Trifonova, A. The effect of electrolyte additives on electrochemical performance of silicon/mesoporous carbon (Si/MC) for anode materials for lithium-ion batteries. Electrochim. Acta 2017, 247, 600–609. [Google Scholar] [CrossRef]
- Wu, Q.-H.; Thissen, A.; Jaegermann, W.; Liu, M. Photoelectron spectroscopy study of oxygen vacancy on vanadium oxides surface. Appl. Surf. Sci. 2004, 236, 473–478. [Google Scholar] [CrossRef]
- Laubach, S.; Schmidt, P.C.; Thißen, A.; Fernandez-Madrigal, F.J.; Wu, Q.-H.; Jaegermann, W.; Klemm, M.; Horn, S. Theoretical and experimental determination of the electronic structure of V2O5, reduced V2O5−x and sodium intercalated NaV2O5. Phys. Chem. Chem. Phys. 2007, 9, 2564–2576. [Google Scholar] [CrossRef]
- Donsanti, F.; Kostourou, K.; Decker, F.; Ibris, N.; Salvi, A.M.; Liberatore, M.; Thissen, A.; Jaegerman, W.; Lincot, D. Alkali ion intercalation in V2O5: Preparation and laboratory characterization of thin films produced by ALD. Surf. Interface Anal. 2006, 38, 815–818. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Component | Binding Energy Position (eV) 1 and Relative Peak Component Areas (%) 2 | |||
|---|---|---|---|---|
| Electrolyte Gas | Electrolyte Gas + Liquid | V2O5 in Electrolyte | V2O5 Cleaned and Dry | |
| CO3 (g) | 292.1 (32.9) | 292.2 (3.6) | 292.3 (0.9) | - |
| CO3 (l) | - | 291.4 (19.1) | 291.4 (17.6) | - |
| COO | - | - | - | 289.4 (4.5) |
| C-O (g) | 288.7 (67.1) | 289.0 (5.4) | 288.9 (3.6) | - |
| C-O (l) | - | 287.7 (44.8) | 287.6 (41.4) | 287.6 (15.8) 3 |
| CC/CH | - | 285.5 (27.2) | 285.4 (36.5) | 285.6 (79.7) |
| Peak Component | Binding Energy Position (eV) 1 and Relative Peak Component Areas (%) 2 | |||
|---|---|---|---|---|
| Electrolyte Gas | Electrolyte Gas + Liquid | V2O5 in Electrolyte | V2O5 Cleaned and Dry | |
| O2 (g) | 540.4 (16.1) | 540.4 (2.6) | 540.3 (6.6) | 540.3 (10.2) |
| 539.3 (31.9) | 539.3 (6.8) | 539.3 (15.3) | 539.3 (17.0) | |
| H-O-H (g) | 536.5 (5.2) | 537.0 (0.7) | - | - |
| O-C (g) | 535.6 (32.2) | 535.5 (17.9) | 535.3 (8.4) | 535.5 (3.9) 3 |
| O-C (l) | - | 534.7 (41.6) | 534.7 (25.7) | 534.4 (12.7) 3 |
| O=C (g) | 533.9 (14.7) | 533.8 (9.0) | 534.0 (4.2) | 533.5 (10.1) 4 |
| O=C (l) | - | 533.0 (21.4) | 532.9 (21.4) | 532.4 (10.6) 4 |
| O-V | - | - | 531.0 (18.5) | 531.1 (35.5) |
| Peak | Peak Component | Binding Energy Position (eV) 1 and Relative Peak Component Areas (%) 2 | |||
|---|---|---|---|---|---|
| LiPF6 | LiTFSI | ||||
| C 1s | CF3 | - | - | 292.5 | 4.9 |
| CO3 | 291.1 | 22.0 | 290.7 | 19.1 | |
| C-O (g)/C=O | 288.9 | 4.0 | 288.4 | 6.1 | |
| C-O (l) | 287.2 | 47.2 | 287.0 | 41.3 | |
| CC/CH | 285.0 | 26.7 | 285.0 | 28.6 | |
| O 1s | O=C | 532.9 | 31.4 | 532.3 | 24.2 |
| O–C | 534.5 | 68.6 | 534.0 | 48.3 | |
| O=S | 532.2 | 27.6 | |||
| F 1s | CF3 | - | - | 688.5 | 69.4 |
| PF6 | 686.7 | 100 | - | - | |
| F- | - | - | 684.7 | 30.6 | |
| Li 1s | Li+ | 55.6 | 100 | 55.6 | 100 |
| Sample | Element | Experimental Ratio | Stoichiometry |
|---|---|---|---|
| LiTFSI | N:S:F:Li | 1:3.5:6.25:14.5 | 1:2:6:1 |
| LiPF6 | P:F:Li | 1:2:4 | 1:6:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dietrich, P.M.; Gehrlein, L.; Maibach, J.; Thissen, A. Probing Lithium-Ion Battery Electrolytes with Laboratory Near-Ambient Pressure XPS. Crystals 2020, 10, 1056. https://doi.org/10.3390/cryst10111056
Dietrich PM, Gehrlein L, Maibach J, Thissen A. Probing Lithium-Ion Battery Electrolytes with Laboratory Near-Ambient Pressure XPS. Crystals. 2020; 10(11):1056. https://doi.org/10.3390/cryst10111056
Chicago/Turabian StyleDietrich, Paul M., Lydia Gehrlein, Julia Maibach, and Andreas Thissen. 2020. "Probing Lithium-Ion Battery Electrolytes with Laboratory Near-Ambient Pressure XPS" Crystals 10, no. 11: 1056. https://doi.org/10.3390/cryst10111056
APA StyleDietrich, P. M., Gehrlein, L., Maibach, J., & Thissen, A. (2020). Probing Lithium-Ion Battery Electrolytes with Laboratory Near-Ambient Pressure XPS. Crystals, 10(11), 1056. https://doi.org/10.3390/cryst10111056